
Abstract
Arrhythmogenic cardiomyopathy (AC) is an incurable genetic disease, whose pathogenesis is poorly understood. AC is characterized by arrhythmia, fibrosis, and cardiodilation that may lead to sudden cardiac death or heart failure. To elucidate AC pathogenesis and to design possible treatment strategies of AC, multiple murine models have been established. Among them, mice carrying desmoglein 2 mutations are particularly valuable given the identification of desmoglein 2 mutations in human AC and the detection of desmoglein 2 auto-antibodies in AC patients. Using two mouse strains producing either a mutant desmoglein 2 or lacking desmoglein 2 in cardiomyocytes, we test the hypothesis that inflammation is a major component of disease pathogenesis. We show that multifocal cardiomyocyte necrosis initiates a neutrophil-dominated inflammatory response, which also involves macrophages and T cells. Increased expression of Ccl2/Ccr2, Ccl3/Ccr5, and Cxcl5/Cxcr2 mRNA reflects the observed immune cell recruitment. During the ensuing acute disease phase, Mmp12+ and Spp1+ macrophages and T cells accumulate in scars, which mature from cell- to collagen-rich. The expression of Cx3cl1/Cx3cr1, Ccl2/Ccr2, and Cxcl10/Cxcr3 dominates this disease phase. We furthermore find that during chronic disease progression macrophages and T cells persist within mature scars and are present in expanding interstitial fibrosis. Ccl12 and Cx3cl1 are predominant chemokines in this disease phase. Together, our observations provide strong evidence that specific immune cell populations and chemokine expression profiles modulate inflammatory and repair processes throughout AC progression.
Similar content being viewed by others

Introduction
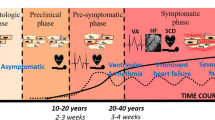
Arrhythmogenic cardiomyopathy (AC), previously referred to as arrhythmogenic right ventricular cardiomyopathy (ARVC), is a genetic disease that is characterized by arrhythmia and cardiac dilation. It is complicated by sudden cardiac arrest and leads to heart failure [10, 25]. A hallmark feature of late disease stages is the presence of excessive fibrofatty tissue within both ventricular walls [20]. The human disease can be separated into (i) a concealed preclinical phase with a risk of life-threatening arrhythmia, (ii) a phase with the onset of structural abnormalities and overt electrical disorders, and (iii) a chronic phase of progressive heart failure [62]. The pathogenic pathways leading to morphological disease onset, acute fibrotic scar formation and chronic deterioration of heart structure and function are still poorly understood. Mechanical dysfunction and complex tissue responses are discussed as factors determining the disease process [6, 8, 19, 39, 44]. Besides symptomatic treatment, a causal therapy is not available to date [64].
AC has been linked to mutations in genes encoding proteins of desmosomal cell–cell adhesions [29]. Among them, mutations of the desmosomal cadherin desmoglein 2 gene (denoted as Dsg2) have obtained particular attention given their severe phenotype [34] and the consistent and specific detection of desmoglein 2-autoantibodies in AC patients and AC animal models [13].
To better understand the pathomechanisms of AC and to explore possible therapies, multiple murine mouse models have been established [51]. To date, several mouse strains have been described that either overexpress mutant desmoglein 2 protein (denoted as DSG2) [52], lack DSG2 in cardiomyocytes [38] or constitutively produce mutant DSG2 [14, 42] reflecting situations encountered in human AC patients ranging between homo- and heterozygosity and between mutant and absent DSG2 [17, 18, 58]. An AC-like phenotype was reported in all transgenic mouse lines presenting with cardiomyocyte death, inflammation, fibrosis, and cardiac dysfunction [40, 42, 52]. Morphological disease onset with localized lesion formation was observed 2 weeks after birth. It is followed by the acute disease phase during which lesions are transformed into mature fibrous scars by the age of 10–12 weeks. At this time, the chronic disease phase sets in with progressive cardiac wall alterations in aging mice [31, 40].
It is has been reported that inflammation is a component of AC disease initiation and progression with multiple reports on the detection of immune cells in various disease stages [5, 8, 11, 15, 25, 39, 40, 59, 60]. A systematic study of the sequential responses in relation to the different disease stages and a thorough characterization of the disease onset, however, is still lacking. The goal of the current study was therefore to determine whether and how inflammation contributes to the different disease stages of murine AC and to dissect the cellular responses involved. To do this, special emphasis was on the examination of local responses by immunohistochemistry to capture the specific cell types and on the production of inflammatory cytokines and their receptors to elucidate mechanisms determining very early alterations and to connect the consecutive pathologies occurring during scar formation and the chronic phase.
Materials and methods
Animals and tissue sampling
Two different Dsg2 mutant mouse strains were used in this study. Homozygous Dsg2MT mice carry Dsg2 alleles lacking exons 4, 5, and 6 and encoding a truncated DSG2 protein of approximately 110 kDa which lacks parts of the extracellular domains 1 and 2 [42]. The mutant DSG2 still localizes to the intercalated discs but is less abundant than the wild-type protein [41, 42]. Additionally, Dsg2MT mice present a pronounced reduction of desmosomes at the intercalated discs [39, 40]. Approximately two-thirds of the Dsg2MT mice die during embryogenesis. Surviving Dsg2MT individuals are born apparently healthy and develop myocardial lesions around day 14 after birth [40, 42].
Cardiomyocyte-specific desmoglein 2 knock-out (Dsg2cKO) mice are homozygous carriers of a Dsg2 allele containing two loxP sites, which flank exons 4 and 5 of the Dsg2 gene (Dsg2flox(E4−5); [39]). Dsg2flox(E4−5) mice were then crossed with B6.FVB-Tg(Myh6-cre)2182Mds/J mice containing a Myh6-Cre transgene that is specifically expressed in cardiomyocytes after E10 [1]. The Cre recombinase-mediated excision of exons 4 and 5 leads to the mutant allele Dsg2cKO encoding a nonfunctional aminoterminal DSG2 polypeptide (for further details see [39]). Dsg2cKO mice show much less embryonic lethality than Dsg2MT mice probably because some wild-type DSG2 protein is still present during midgestation. After birth, however, DSG2 is no longer detectable in the myocardium of Dsg2cKO mice [39]. Dsg2cKO mice develop myocardial lesions slightly later than Dsg2MT mice around day 18. Besides these differences, the cardiac phenotype is the same for Dsg2MT and Dsg2cKO mice from 4 weeks onwards as determined by histomorphology, intercalated disc ultrastructure, electrocardiography, Cx43 distribution, and CD45 immunoreactivity [39]. Neither heterozygous Dsg2 mutant (Dsg2HT) mice nor homozygous Dsg2flox(E4−5) mice display a cardiac phenotype.
Mice were housed in the animal facility of the University Hospital of RWTH Aachen University. They received a standard rodent lab diet (Ssniff, Soest, Germany) and had free access to food and water. The experiments were conducted in accordance with the guidelines for the care and use of laboratory animals and were approved by the Ministry for Environment, Agriculture, Conservation and Consumer Protection of the State of North Rhine-Westphalia (reference number 84-02.04.2015.A190 and A4 notifications for killing animals for scientific purposes).
Animals were killed by cervical dislocation. The thoracic cavity was opened after cervical dislocation and gross heart morphology was documented in situ. The heart was then removed and dissected in two different ways for the ensuing analyses:
- i.
Hearts were divided into an apical and a basal part along the transverse plane so that both halves contained diseased myocardium. The tip of the heart was used for RNA isolation by homogenization in RNA isolation buffer (PeqLab Gold RNA isolation kit; VWR, Darmstadt, Germany). The homogenate was stored at − 80 °C until further processing. The remaining basal part of the heart was either chemically fixed in formaldehyde or cryofixed in liquid nitrogen.
- ii.
To obtain quantitative data on mRNA expression of the left and the right ventricular wall, the atria were first cut off. The right ventricular free wall was then removed, cleaned from adherent blood, and homogenized in RNA isolation buffer. The septum was inspected for fibrotic scar tissue or structural abnormalities during dissection. The remaining left ventricle was opened, blood clots were removed and the left ventricular myocardium was homogenized in RNA isolation buffer. All homogenates were stored at − 80 °C until RNA isolation.
RNA isolation, cDNA synthesis, and qRT-PCR
Total RNA isolation, cDNA synthesis, and qRT-PCR experiments were performed as described [31]. In brief, total RNA was isolated using the PeqGOLD Total RNA Kit. 1 µg of RNA was reverse transcribed using the Transcriptor First Strand kit (Roche, Mannheim, Germany) utilizing oligo dT-primer. The qRT-PCRs were performed with the help of the Light Cycler Taqman Master Kit, Universal Probe Library (UPL) probes (both Roche) and the primer pairs listed in Table 1.
Histology and immunohistochemistry
For paraffin embedding, dissected hearts were first fixed overnight by submersion in 4% neutrally buffered formaldehyde in phosphate-buffered saline (PBS). After rinsing in PBS for 1 h samples were dehydrated in an ascending isopropanol series (50%, 70%, 90%, 100%). After 1 h in 100% isopropanol at 60 °C, tissues were transferred to liquid paraffin (60 °C). After 2 h paraffin was exchanged and after overnight incubation tissues were embedded in paraffin blocks.
For immunohistochemical staining of fresh frozen samples hearts were cut into two halves along the transverse plane. The cut surface was placed on the bottom of a TissueTek cryomold and the tissue was then carefully covered with TissueTek OCT compound (Science Services, Munich, Germany). Tissue was shock frozen in liquid nitrogen and stored at − 40 °C until sectioning.
To assess the histology of mutant and healthy hearts 5 µm thick serial paraffin sections were prepared and stained with hematoxylin-eosin (HE), AZAN trichrome and von Kossa stain as described before [39, 40, 42].
For immunohistochemical staining of the CD44, CD3, CD45R, and myeloperoxidase (MPO), 5 µm thick paraffin sections were used. After deparaffinization including peroxidase blocking with 3% H2O2 in 70% ethanol sections were rehydrated in PBS. Prior to the incubation with the primary antibodies specific antigen retrieval was performed following different antigen-dependent regimen: (i) CD44 and TNC: 40 min incubation in 10 mM/L citrate buffer pH 6 at 94 °C followed by 20 min cool down to room temperature; (ii) CD3: 40 min incubation in boiling 10 mM/L citrate buffer pH 6; (iii) CD45R: 30 min incubation in 50 mM EDTA buffer at room temperature to remove calcium; (iv) MPO: 2 × 10 min boiling in 10 mM/L citrate buffer pH 6 followed by 20 min cool down to room temperature. The sections were then incubated with the first antibodies. Dilution and incubation times are provided in Table 2.
The presence and localization of monocyte/macrophage and T cell subsets were assessed on 10 µm thick cryostat section using antibodies against the CD11b, CD11c, CD4, CD8, F4/80 and CD206 antigens. After cutting, sections were allowed to adhere to the slides for 30 min at room temperature. Thereafter, sections were fixed for 10 min at − 20 °C in acetone that had been precooled to − 20 °C. After rehydration in PBS, the endogenous peroxidase was blocked by treating sections with 0.3% H2O2 in PBS for 10 min in the dark. After two washing steps in PBS (5 min each) the first antibody, which was either diluted in PBS or PBS containing 1.5% BSA, was applied. Details on dilution and incubation times are given in Table 2.
After incubation with the first antibodies, paraffin and cryostat sections were washed three times for 5 min with PBS. Thereafter antibody detection was accomplished by anti-rabbit and anti-rat polymer kits or in case of the CD11c and CD3e antibody by a goat anti-hamster IgG horseradish peroxidase conjugate (see Table 3 for incubation times and dilutions).
Two kinds of negative control experiments were performed by either omitting the first antibody or replacing the first antibody by a non-IgG control antibody of the same isotype and concentration. Spleen was used as a positive control.
Immunohistochemical intensity score
To compare and to quantify the density of CD11b-, CD11c-, CD3-, CD4- and F4/80-positive cells within myocardial scars of Dsg2MT mice, a semiquantitative score (0–4) was established (see Supplementary Fig. 1). Individual scars were scored and the mean value was calculated for each animal.
In situ hybridization
We used the type 1 probe sets for mouse Spp1 and Mmp12 mRNA (VB1-14479-VT and VB1-14709-VT, respectively; Thermo Fisher Scientific) and the ViewRNA ISH Tissue Assay Kit (1-plex; Cat# 19931, Thermo Fisher Scientific). 5 µm-thick sections of 4% formaldehyde-fixed and paraffin-embedded cardiac tissue (for details see above) were placed on Surgipath™ X-tra Adhesive pre-cleaned micro slides (Leica Biosystems, Wetzlar, Germany). Further processing was done as detailed in the protocols provided by the manufacturer. Negative controls were stained without the hybridization probe or with a scrambled probe to Mmp12.
Statistics
The mRNA expression data were analyzed using LightCycler 96 software 1.1 and are given as normalized ratio quantification (NRQ; [33]).
For comparisons of three groups (at disease onset at the age of 18–19 days in Dsg2cKO or day 14 in the Dsg2MT mice) the Kruskal Wallis test and selected Post hoc Dunn's comparison tests were used (Dsg2MT mice: Dsg2WT versus Dsg2MT and Dsg2WT vs Dsg2MT + Ph; in Dsg2cKO mice: Dsg2loxPversus Dsg2cKO and Dsg2loxP versus Dsg2cKO + Ph). When comparing Dsg2MT mice with Dsg2WT mice or Dsg2cKO mice with Dsg2loxP control mice in the other age groups (4–6 weeks, 12 weeks and 30–32 weeks), the non-parametric Mann Whitney test was applied. Statistics were performed by using GraphPad Prism Version 5.01 (GraphPad Software, Inc, USA).
Results
Cardiomyocyte necrosis triggers an inflammatory response during the early stage of cardiomyopathy
It has been suggested that cardiomyocyte necrosis is the key pathogenic event triggering an inflammatory response in murine Dsg2-related AC [40, 53]. To systematically study this early response, we collected Dsg2cKO hearts [39] at days 18 and 19 when the first macroscopic alterations, notably white scars and ventricular discoloration, became discernable in some but not all animals. Serial sectioning of inconspicuous hearts unveiled histological abnormalities in three out of three specimens. Figure 1a depicts a subepicardial lesion capturing an early stage of cardiomyocyte necrosis. The original size and shape of the cardiomyocytes were still maintained although the cytoplasm was amorphous and displayed strong eosinophilia. Furthermore, nuclei of the necrotic cells were either completely missing or pyknotic. Erythrocyte accumulations were seen in the vicinity of necrosis indicating enlarged capillaries or local hemorrhage (inset in Fig. 1a).

Cardiomyocyte-specific Dsg2 knockout (Dsg2cKO) mice develop cardiomyocyte necrosis and an inflammatory response 18–19 days after birth. a–c The micrographs show tissue sections that were obtained from Dsg2cKO mice without macroscopically detectable cardiac pathologies except for slight atrial dilation in the animal used for the sections shown in c. Serial sections were stained with hematoxylin–eosin (HE) or reacted with antibodies against CD44 to characterize the extent of the inflammatory infiltrate, MPO to identify neutrophil granulocytes, CD3 to detect T-lymphocytes and CD45R/B220 to identify B lymphocytes. Immunohistochemistry of the matricellular protein Tenascin C (TNC) indicates the onset of ventricular remodeling. a Note the presence of subepicardial necrosis (N) with adjacent enlarged blood vessels (demarcated by white broken line in the inset at left) and the detection of only very few CD44+ and MPO+ cells (arrowheads) with little to no detection of TNC indicating that an inflammatory response is just starting and that tissue remodeling has not occurred yet. b In contrast, the papillary muscle lesion 1 observed in the same animal presents with a strong inflammatory reaction as assessed by CD44 staining and commenced tissue remodeling (TNC positivity). The center of the lesion (L), which is delineated by a broken yellow line in the insert of the image at left is already free of cardiomyocyte remnants whereas necrotic cardiomyocytes (N) line the border of the lesion. The granular MPO staining (arrowheads) indicates the presence of neutrophil granulocytes, whereas the homogenous cytoplasmic MPO staining is indicative of macrophages. c The papillary muscle lesion 2 of another Dsg2cKO mouse also shows an extensive inflammatory infiltrate surrounded by necrotic cardiomyocytes. A considerable number of CD3+ T lymphocytes and some CD45R/B220+ B lymphocytes are present. d Tukey's whisker plots of ventricle-specific mRNA detection (normalized relative quantification; NRQ) show comparisons between the expression of chemotactic and inflammatory cytokines in Dsg2cKO (cKO) and Dsg2loxP (loxP) control mice 18–19 days after birth. The broken line indicates the mean expression of the loxP control. None of the animals had an overt cardiac phenotype. Left ventricle: n = 4–5 for Dsg2cKO, n = 4 for Dsg2loxP; right ventricle: n = 4–5 for Dsg2cKO, n = 3–4 for Dsg2loxP. The non-parametric Mann–Whitney test was applied: *p < 0.05. Detailed statistics in Supplementary Table 1
To test for the presence or absence of blood-borne immune cells, subsequent sections were reacted with anti-CD44 antibodies. Positive cells were not seen in the adjacent normal-appearing myocardium and only very few labeled cells were detected in the vicinity of necrotic cardiomyocytes. Based on their granular cytoplasmic MPO staining they could be classified as neutrophil granulocytes (Fig. 1a). CD3+ T cells were not enriched in this lesion and tenascin C (TNC), a sensitive indicator of tissue remodeling [37], was not expressed. Taken together and based on observations in murine myocardial infarction models [16], the depicted lesion and similar lesions can be classified as early nascent lesions.
In the same Dsg2cKO heart as well as in the other Dsg2cKO hearts without macroscopically visible alterations prominent changes such as myocardial necrosis and inflammation were noted in papillary muscles (Fig. 1b, c). The pathogenesis was obviously further progressed: large accumulations of inflammatory, i.e. CD44+ cells, had infiltrated the necrotic myocardium. The centers of the lesions were free of cardiomyocyte remnants, but necrotic cardiomyocytes lined the borders of the lesions. Homogenous cytoplasmic and granular MPO staining identified abundant macrophages and neutrophil granulocytes, respectively, in the infiltrates. Furthermore, a considerable number of CD3+ T cells and a few CD45R+ B cells were also detected. The observed removal of necrotic cardiomyocytes and the detection of immune cells belonging to the adaptive immune system support the conclusion that the papillary lesions shown in Fig. 1b, c represent stages subsequent to that shown in Fig. 1a.
To further characterize the early inflammatory response, we assessed the mRNA expression of the chemokines Ccl2, Ccl3, and their respective receptors Ccr2 and Ccr5 that are known to be involved in the recruitment of mononuclear cells in ischemic myocardium [23, 65]. Significant upregulation was observed for Ccl3 and Ccr5 in the right but not the left ventricle of 18–19 day-old Dsg2cKO hearts without an overt phenotype. In addition, a trend to elevated Ccl2 mRNA expression was noted in the right ventricles. The mRNA expression of Ccl2, Ccl3, Ccr2, and Ccr5 was also studied in the hearts of a second model of murine AC, i. e. in Dsg2MT mice, which develop a very similar phenotype (Fig. 2a; [42]). In Dsg2MT mice, disease onset occurred even earlier, namely 14 days after birth. We found that Ccl3 mRNA was already elevated in Dsg2MT hearts without macroscopically visible lesions. In hearts with visible surface scars Ccl2, Ccl3, Ccr2, and Ccr5 mRNAs were all upregulated. Together, our findings suggest that Ccl3 is one of the first upregulated cytokines during the onset of murine AC pathogenesis.

Chemokine and chemokine receptor mRNA expression increase at the onset of structural disease in the heart of Dsg2-mutant mice. Data are presented as Tukey's whisker plots as normalized relative quantification (NRQ). The broken line indicates the mean expression of the wild-type (WT) or Dsg2loxp (loxP) control. a mRNA expression (NRQ) of the classical chemokines Ccl2 and Ccl3 and their respective receptors Ccr2 and Ccr5 is shown for total hearts of 2 week-old Dsg2WT mice (WT; n = 3), Dsg2MT mice without visible cardiac lesions (MT; n = 6) and Dsg2MT mice with the overt phenotype (MT Ph + ; n = 5). At right the mRNA expression of the cytokine osteopontin (Spp1; WT: n = 3; MT: n = 3 and MT Ph + : n = 4) is depicted. The Kruskal Wallis test and selected post hoc Dunn's comparison tests were applied for WT versus MT and WT versus MT Ph + . *p < 0.05, **p < 0.01. Detailed statistics in Supplementary Table 2. b The plots show ventricle-specific mRNA expression of chemokines and chemokine receptors that are involved in the recruitment of monocytes/macrophages (Ccl7, Ccl12, Cx3cl1, Cx3cr1), T cells (Ccr3, Cxcl10, Cxcr3), neutrophils (Cxcl5, Cxcr2) and bone marrow-derived immune cells (Cxcr4) in the hearts of 18–19 day-old Dsg2cKO mice either without the overt disease (cKO; n = 9–10) or with the overt disease (cKO Ph + ; n = 4) and Dsg2loxP controls (loxP; n = 8–9). The Kruskal Wallis test and selected post hoc Dunn`s comparison tests were applied for loxP versus cKO and loxP versus cKO Ph + . *p < 0.05, **p < 0.01. Detailed statistics in Supplementary Tables 3 and 4
The mRNA expression of the inflammation-associated cytokines Tnfα and interleukin Il1β was highly variable in inconspicuous Dsg2cKO hearts and was therefore statistically not different from that in Dsg2loxP controls (Fig. 1d). Very minor differences in lesion age and size may account for the observed variability in Tnfα and Il1β mRNA production as suggested by the reported rapid changes in cytokine gene expression in ischemic murine hearts [23]. Most interestingly, anti-inflammatory cytokine Il10 mRNA expression was significantly upregulated in the right ventricles of Dsg2cKO mice.
The mRNA expression of chemokines and chemokine receptors orchestrating the recruitment of specialized immune cell subpopulations that are involved in the resolution of necrotic cells and the induction of tissue repair were then assessed in isolated left and right ventricles of Dsg2cKO mice without and with overt lesions at 18–19 days (Fig. 2b). The examined chemokines and corresponding chemokine receptors were selected based on studies in myocardial infarction [2, 30, 43, 50]. Consistent with early neutrophil recruitment was the detection of significant Cxcl5 mRNA upregulation in both ventricles and the detection of Cxcr2 mRNA in the right ventricles of phenotypic Dsg2cKO mice. Furthermore, the upregulation of Cxcl10 and Cxcr3 mRNA in both ventricles was taken as an indication of a beginning T cell response. Elevated Ccl7 mRNA expression in ventricles of Dsg2cKO mice in conjunction with increased Ccl2 and Ccr2 in Dsg2MT mice (see Fig. 2a) indicated the recruitment of CCR2+ inflammatory monocytes into the mutant heart that likely differentiate into macrophages. In sum, the chemokine profile correlated well with the composition of the immune cell infiltrate found during disease onset (Fig. 1b, c).
Inflammation is involved in the formation of purely fibrotic and calcifying scars
Tissue remodeling occurs after cardiomyocyte necrosis and initial recruitment of inflammatory cells in Dsg2MT and Dsg2cKO mice invoking two types of replacement fibrosis: predominantly collagenous scars (Fig. 3a–c) and scars containing calcified necrotic cardiomyocytes (Fig. 3d–f). In collagenous scars, necrotic cardiomyocytes are completely phagocytosed by infiltrating immune cells leaving only endomysial and perimysial connective tissue. Cardiac wall thinning is typically observed in these instances. On the other hand, calcification occurs when necrotic cardiomyocytes are not phagocytosed. The calcified regions elicit fulminant adjacent fibrosis leading to local cardiac wall thickening, which manifests as solid white scars. To extend previous reports of CD45+ immune cells in immature scars [39, 40] and to dissect the immune cell types associated with the different fibrotic responses, we decided to study the expression of specific immune cell markers during scar formation between 4 and 12 weeks.

Purely fibrotic or calcifying scars form during the acute disease phase in response to extensive cardiomyocyte necrosis at disease onset while interstitial fibrosis indicates ongoing myocardial remodeling during the chronic disease phase in Dsg2MT and Dsg2cKO mice. a, b, d, and e show immature scars of juvenile, 4 week-old Dsg2cKO mice with (a, b) and (d, e) being serial sections of the same animals, respectively. c, f show micrographs of mature scars from adult, 3 month-old Dsg2MT mice after AZAN trichrome staining. Calcified necrotic areas are denoted by N, black calcium deposits by arrows and fibrotic lesions by L. Note that the cardiomyocyte-free mature fibrotic lesion in c consists mainly of connective tissue (short arrows) that extends to the endo- and perimysium of the adjacent intact-appearing myocardium and is coupled to wall thinning, whereas the mature calcified lesion in f is associated with extremely collagen-rich thick connective tissue. The micrographs in g–i show sections of 30–40 week-old Dsg2MT hearts during the chronic disease phase. AZAN staining reveals that besides the scars depicted in c and f increasingly areas with loose (g) and dense (h) interstitial fibrosis are present. i depicts myocardium that is not yet affected
The majority of infiltrating cells consisted of CD11b+ and F4/80+ macrophages (Fig. 4a). In addition and in contrast to the incipient lesions, a considerable number of CD3+ T cells and a few round-shaped CD4+ cells, which are most likely T-helper cells, were present (Fig. 4a). CD11b+ and F4/80+ macrophages also resembled the dominant immune cell type in mature, purely collagenous and calcified scars of 12 week-old Dsg2MT mice (Fig. 4a). In comparison to the immature scars, fewer but still increased CD3+ and CD4+ cells were present. Only some of the CD4+ cells, however, fit the typical round-shaped T-helper cell phenotype whereas the majority was spindle-shaped most likely being dendritic cells (Fig. 4a) [63]. CD8a+ cells were detected only rarely in cardiac tissue sections without positional preference for scars and at a comparable frequency to wild-type controls (Fig. 4a). Of note, immune cell numbers within the structurally unaffected Dsg2MT myocardium did not deviate from those found in wild-type control myocardium (Fig. 4a).

Comparison of immature and mature scars in Dsg2MT mice identify macrophages as the predominant immune cell population in both and shows that increased expression of inflammation-associated cytokines is strongly reduced in mature scars. a The micrographs show tissue cryosections that were obtained from 4 and 12 week-old Dsg2MT mice (immature scars [n = 3] and mature scars [n = 4], respectively) and corresponding wild-type Dsg2WT mice (WT; n = 3–5) after reaction with antibodies directed against various immune cell-specific surface antigens. All images are shown at the same magnification (scale bar in the picture at the upper left corner). Note that CD11b+ and F4/80+ macrophages are the predominant immune cell type in immature and mature scars and that the number of CD3+ T cells is higher in immature than in mature scars. Typical round-shaped CD4+ T cells are present in immature scars, whereas primarily spindle-shaped CD4+ cells are detected in mature scars. CD8a+ cells were only rarely detected in Dsg2MT and control hearts (arrowheads). b Tukey's whisker plots show the results of qRT-PCR analyses assessing the mRNA expression (normalized relative quantification; NRQ) of inflammation-associated cytokines in total hearts of Dsg2MT (n = 5–7) and Dsg2WT mice (n = 5–7) at 4 and 12 weeks. They are significantly upregulated in Dsg2MT mice of both age groups compared to the wild-type controls (broken lines). However, their expression decreases with age. The non-parametric Mann Whitney test was applied to compare Dsg2WT and Dsg2MT expression within each age group. *p < 0.05 and **p < 0.01. More detailed statistic data are provided in Supplementary Table 5
To gain insight into inflammatory activity occurring during scar formation and maturation, we assessed the mRNA expression of pro- and anti-inflammatory cytokines (Fig. 4b). Tnfα, Il1β, and Il10 were significantly upregulated at 4 weeks and their expression remained significantly upregulated by 12 weeks albeit at reduced levels. The mRNA expression of galectin 3 (Lgals3), which exerts profibrotic effects and is produced by activated macrophages [56], was highly elevated in 4 and 12 week-old Dsg2MT mice, although expression was substantially lower at 12 weeks.
Inflammatory cells and cytokine production persist in the chronic disease phase
During the chronic phase of AC fibrotic and calcified scars persisted and increasing interstitial fibrosis was observed (Fig. 3g, h). In the absence of active replacement, we observed that an increased number of CD44+ immune cells remained in calcified and fibrotic scars (Fig. 5a). They encompassed CD11b+ macrophages and CD11c+ dendritic cells (Fig. 5a). Of note, macrophages were also elevated in interstitial fibrosis. In addition, increased CD3+ T cells were detected in the different lesion types (Fig. 5a). Anti-CD4 staining identified cells, some of which were not round as to be expected of typical T-helper cells but were rather spindle-shaped and probably resemble a subtype of dendritic cells (right panel in Fig. 5a). An increased number of CD8a+ cells could not be detected in the lesioned myocardium (lower right of Fig. 5a).

The inflammatory response persists during the chronic disease phase in Dsg2MT mice. a The images depict immunohistochemical stainings detecting distinct immune cells in the myocardium of wild-type (WT) and in fibrotic or calcified necrotic (N) scars of Dsg2MT (MT) mice at 30–32 weeks (n = 4–5 per genotype). Many CD44+ immune cells are detected. Note that the majority of the scar-associated immune cells are CD11b+ macrophages and CD11c+ macrophages/dendritic cells. A cluster of CD3+ T cells next to a vein is depicted in the upper right picture. Scattered, spindle-shaped CD4+ T cells are detectable in scar tissue whereas CD8a+ T cells are only very rarely detected in wild-type and non-lesioned mutant hearts. b Tukey's whisker plots show the mRNA expression in arbitrary units (ratio of target and housekeeping gene Hmbs) of the inflammatory cytokines Tnfα and Il1β in the right and left ventricles of 30–32 week-old Dsg2MT (MT) and wild-type control mice (WT; n = 4–5 per genotype). Mann Whitney U test was applied (*p < 0.05 and **p < 0.01)
The mRNA expression of the inflammatory cytokine Tnfα remained significantly increased in right and left ventricles during chronic disease progression. Additionally, Il1β was slightly elevated in the right ventricles (Fig. 5b).
Longitudinal assessment of immune cells and cytokine expression reveals different activity levels and activation types
To semiquantitatively compare the changing levels of phagocytic cells and T cells during tissue remodeling, the staining intensities of antibodies directed against the different cell populations were visually assessed in Dsg2MT hearts during the acute disease phase at 4, 6 and 12 weeks and the chronic disease phase at 32 weeks (summary in Fig. 6a; examples of immunostaining in Figs. 4a and 5a). As expected, the most intense reactions were observed for the macrophage markers CD11b and F4/80. A somewhat less intense reaction was detected for the dendritic cell marker CD11c, which peaked at 12 weeks. CD3+ T cells appeared to increase from 4 to 32 weeks. The population of CD4+ cells, which consists of few round-shaped T cells and many spindle-shaped dendritic cells [63], slightly decreased over time.

Profiling the immune response from the acute to the chronic stage of cardiomyopathy in Dsg2MT mice reveals persistent upregulation of macrophages together with dendritic cells and T cells as well as phase-specific chemokine/chemokine receptor upregulation. a The histogram depicts the results of a semiquantitative microscopical intensity score assessment of immunohistochemical immune cell detection in myocardial scars of Dsg2MT mice (n = 3–5 for each age group). b Tukey's whisker plots show the results of qRT-PCR (normalized relative quantification; NRQ) performed on RNA isolates from right and left ventricles of Dsg2MT and wild-type controls (dotted lines). Expression was assessed by using non-parametric Mann Whitney tests: *p < 0.05, **p < 0.01; §p > 0.05 and < 0.06. For details see Supplementary Table 6 and 7. The increase of Cd45 (immune cells), Cd68 and F4/80 (monocytes/macrophages), and Cd3e (T cells) mRNA supports the semiquantitative immunohistochemical results in a. Furthermore, the mRNA expression data suggest that the immune response is more pronounced in the right ventricle than in the left ventricle
In support of the immunohistochemical findings, the mRNA expression of the general immune cell marker Cd45, the T cell marker Cd3e, and the macrophage markers Cd68 and F4/80 (Emr1) were elevated to various degrees in right and left ventricles of different disease stages. The strongest upregulation was noted for the macrophage markers with peak expression preferentially in the right ventricle of 4 week-old Dsg2MT mice when scars were cell-rich and contained little collagen (Fig. 6b). Furthermore, increased expression of Cxcr4, which is expressed on bone marrow-derived immune cells, and of Ccr3, which is abundant in Th2 cells, was also detected (Fig. 6b).
Chemokine profiles were examined next since they dictate the composition of recruited blood-borne immune cells and tissue-specific macrophage differentiation [7, 43, 50, 57]. The results highlight the predominant involvement of the right ventricle in the inflammatory response. This is consistent with the increased immune cell marker expression in the right ventricles (top panel of Fig. 6b). In the following, we will therefore solely summarize the observations in the right ventricles or total hearts.
Specifically, Ccl2 mRNA and its cognate receptor Ccr2 mRNA, which are expressed in classical inflammatory monocytes, were upregulated during disease onset (Fig. 2a) and remained upregulated in the acute and chronic phases (Fig. 6b). The mouse-specific MCP-1-related CCL12 and CCL7 are other chemokines, which recruit CCR2+ cells. Ccl12 was significantly upregulated but only from the chronic phase transition onwards, and Ccl7 only at 12 weeks (Fig. 6b). The chemokine CX3CL1 attracts CX3CR1+ monocytes, which are involved in tissue repair. The mRNA of both was increased in the acute and chronic phases (for minor changes during disease onset see Fig. 2b).
Ccl3 and Ccr5, which are one of the earliest upregulated ligand-receptor pairs (see Figs. 1d, 2a), were still elevated at 4 weeks but declined thereafter. The mRNA expression of the chemokine Cxcl10 and its corresponding receptor Cxcr3, both of which were increased during disease onset (Fig. 2b), remained elevated at 4 and 12 weeks but returned to wild-type levels during the chronic disease stage.
Cxcl5, which encodes a chemokine that recruits CXCR2+ neutrophil granulocytes, was upregulated early on during disease initiation (Fig. 2b) but declined during the acute phase.
Distinct macrophage populations accumulate and differentiate in the hearts of AC mice
Since the microenvironment of a tissue impinges on macrophage differentiation [26, 57] macrophage phenotype and distribution were further characterized. Even healthy myocardium contains a considerable number of homogeneously distributed resident macrophages that express the macrophage-specific CD11b and the CD206 antigen, a marker for anti-inflammatory and reparative macrophages (lower panel of Fig. 7a). F4/80 immunolabelling revealed comparatively weak staining. All three macrophage markers were detected at increased levels in mature scars and interstitial fibrosis of Dsg2cKO mice (upper panels in Fig. 7a). CD206 and CD11b showed similar staining intensity, whereas the F4/80 staining was again fainter. We then studied the mRNA expression of osteopontin (Spp1), which is a marker for reparative macrophages [49, 55, 57], the matrix metalloproteinase 12, a macrophage-specific elastase expressed in pro-inflammatory macrophages [4], and the lectin YM1 (CHI3L3), which is an indicator for reparative or alternatively activated macrophages [45]. Spp1 mRNA stayed elevated and continued to be significantly increased in 8 and 12 week-old Dsg2MT mice (Fig. 7b). Mmp12 mRNA expression was elevated in the left and right ventricles throughout the acute and chronic disease phase. Ym1 mRNA expression was significantly elevated during the early acute phase and decreased toward the chronic stage.

Macrophage distribution and macrophage marker expression is lesion type-specific. a depicts the immunolocalization of the macrophage markers CD11b, F4/80 and CD206 in serial cardiac sections (from left to right) of Dsg2cKO mice with interstitial fibrosis and fibrotic or calcified scars and of Dsg2loxP control mice at 35 weeks (n = 3 and n = 2, respectively). SEF marks a subepicardial fibrotic scar, arrows point to areas with interstitial fibrosis. b Tukey's whisker plots show the quantification (normalized relative quantification; NRQ) of RNAs encoding the macrophage markers Spp1, Ym1, and Mmp12 in Dsg2MT and wild-type control mice (the dotted line indicating relative mean levels of the wildtype). Mann Whitney tests were applied to compare Dsg2WT and Dsg2MT mice at each time point: *p < 0.05, **p < 0.01. For more details see Supplementary Table 7. c Microscopy of in situ hybridizations detecting Mmp12 and Spp1 mRNA (red signal) and nuclei (blue) in paraffin sections of Dsg2cKO or Dsg2MT and corresponding control hearts at the age of 4 and 12 weeks. Sections of 3–4 Dsg2MT/cKO and 2–3 Dsg2WT/loxP hearts were assessed for each age group. Lesions are demarcated by broken lines
To examine the impact of the tissue microenvironment [26], we studied Mmp12 and Spp1 mRNA expression by in situ hybridization (Fig. 7c). Mmp12 and Spp1 mRNA-expressing macrophage-like cells were confined to scar tissue in Dsg2MT and Dsg2cKO hearts. Since the remote and healthy myocardium were negative, despite the presence of macrophages, we concluded that Mmp12- and Spp1-expressing macrophages are recruited to and/or differentiate exclusively in scar tissue.
Discussion
Disease stage-specific inflammatory responses occur in Dsg2 cKO and Dsg2 MT mice
The data presented in this paper provide overwhelming evidence for a contribution of inflammatory cells to all stages of murine AC, specifically:
Disease onset: We find that immune cells are even detectable in lesions that can only be identified microscopically. Necrotic cardiomyocytes are surrounded by CD44+ inflammatory cells. MPO+ neutrophil granulocytes were shown to be a major immune cell type, which is in line with previous observations of hematoxylin–eosin-stained sections in another Dsg2-related murine AC model [53]. The increase in Cxcl5/Cxcr2 mRNA expression also testifies to neutrophil granulocyte recruitment. A major function of these cells is most likely the removal of dying cardiomyocytes.
Acute disease progression: This disease stage is the most dynamic. It is characterized by the changing morphology of maturing scars with concomitant changes in immune cell infiltrates and chemokine signaling. Neutrophil granulocytes are gradually replaced by macrophages. This involves recruitment and differentiation of macrophages. Remarkably, Spp1 and Mmp12 are exclusively produced in macrophages that are localized in scar tissue. The recruitment and activation of different immune cell types are reflected in the upregulation of Ccl2-Ccl7-Ccl12/Ccr2, Ccl3/Ccr5, and Cx3cl1/Cx3cr1 and Cxcl10/Cxcr3 expression. Tnfα and Il1β upregulation are further signs of increased inflammation. Both cytokines are known to be produced primarily by inflammatory monocytes and macrophages [48]. On the other hand, tissue remodeling by fibrotic replacement and increased fibrosis are evidenced by an increase in Lgals3 mRNA and anti-Tenascin C staining. In parallel to the non-specific immune response, a limited adaptive immune cell response kicks in, which is reflected by a minor increase in CD45R+ B cells and considerable upregulation of CD3+ T cells including CD4+ but not CD8a+ cells.
Chronic disease progression: Ongoing interstitial fibrosis and increasing cardiodilation are the main morphologic features of this stage. We now provide substantial evidence that inflammation persists albeit at a reduced level in comparison to the acute phase. Thus, the classical inflammation marker Tnfα remains increased and a slight elevation of Il1β is still detectable. Furthermore, distinct macrophage populations are distinguishably attesting to the ongoing inflammatory remodeling. T cells persist in the chronic disease phase and likely modulate disease progression.
The immune response reported for another Dsg2 knockout model [14, 15] differs considerably from our observations. The most likely explanation for the difference is that the onset of the disease occurs only when mice reach adulthood, i.e. after 8 weeks, and appears to be less severe in that genetic constellation.
Early stages of arrhythmogenic cardiomyopathy share features with ischemic myocardial infarction
When looking at the sequence of tissue alterations and inflammation, similarities to myocardial infarction are readily apparent, which have been characterized in much detail using different murine ischemia models [43, 54]. The pathogenesis is triggered by cardiomyocyte necrosis followed by inflammation, scar formation and maturation, and variable long-term responses. The inflammatory reaction sets in within a few hours of ischemia. Neutrophil granulocytes together with inflammatory Ly6Chi M1 monocytes and T cells are the first to invade the ischemic regions. At the same time, multiple chemokines are upregulated including Ccl2, Ccl3, Ccl7 and Cxcl10 [3], which are either slightly or significantly increased at disease onset in Dsg2 mutants. Our analyses of AC mice furthermore revealed that the corresponding chemokine receptors Ccr2, Ccr5, and Cxcr3 are also elevated. Of particular note, the initial increase in Ccl3/Ccr5 and Il10 that we have noted at AC disease onset has also been observed in myocardial infarction [23]. It was shown to be due to the recruitment of regulatory CD4+ T cells, which limit the inflammatory response. These Treg cells are attracted by CCL3/CCR5 interaction and produce IL10 [21].
The next phase of post-ischemia starts within 4 days and lasts 2–4 weeks. It is characterized by changes in the inflammatory cell repertoire [54]. Neutrophil granulocytes die by apoptosis while an increasing number of Ly6Clo, CX3CR1+, non-classical monocytes is recruited to the lesion. Increased Ym1, Cd206, Il10 and Spp1 mRNA indicate that lesion-associated monocytes differentiate into M2 macrophages [57]. The increased expression of Ym1, Il 10, Spp1, and Cx3cl1/Cx3cr1 in the AC model at 4 weeks suggest that comparable inflammatory mechanisms are at work. The change in immune cell composition is flanked by collagen production by myofibroblasts in the myocardial infarct model [16]. Similar phenomena are observed during the acute phase of Dsg2-related AC. Thus, the pro-inflammatory cytokines Tnfα and Il1β were detected together with the mRNA of the pro-fibrotic cytokines Lgals3 and, as previously described, also Tgfβ1 and Tgfβ3 [38]. T cells persist in the myocardial infarction and AC models. Of particular interest are beside CD4+ Treg cells and CD4−/CD8− T cells, which are involved in organ repair [21].
Subsequent scar maturation is reflected by collagen fiber bundling and reorganization accompanied by a reduction in immune cells and chemokine production in the myocardial infarction model [3, 16, 57]. We find, in contrast, a persistent enrichment of macrophages and T cells and upregulation of mRNAs coding for the chemokines and chemokine receptors Ccl2, Ccl7/Ccr2 and Cxcl10/Cxcr3. In parallel, pathologic hypertrophy occurs in the chronic disease phase [31]. Together, these findings point to fundamental differences between lesions formed in myocardial infarct and AC.
It has been reported that Ly6Chi monocytes, M1 macrophages, dendritic cells, and T cells re-appear in chronic ischemic heart failure within weeks or even months after infarct healing [54]. Maybe, this situation is comparable to the continuing presence of CD11b+ and CD206+ macrophages, CD11c+ cells, as well as CD3+ T cells that we observed in myocardial scars of AC mice at 30–32 weeks. The reduction in Ym1 and the continuously high Mmp12 expression supports a shift towards an M1-like phenotype within the fibrosed myocardium. On the other hand, most if not all cardiac macrophages express the M2 phenotype-associated CD206 antigen in Dsg2-mutant mice so that the pathophysiological role of macrophages in AC needs further investigation.
Inflammation impacts on the onset and progression of arrhythmogenic cardiomyopathy
Our observations that lesions consisting of necrotic cardiomyocytes are the first structural indication of disease onset and that an inflammatory, phagocyte-dominated infiltrate appears only next to necrotic cardiomyocyte but not in normal-appearing myocardium are in full agreement with the sequence of events first proposed by Pilichou and coworkers for another murine AC model [53]. Our observations further show, that cardiomyocyte necrosis is triggered during the most intense postnatal maturation and growth phase of the murine heart, i.e. between 2 and 6 weeks [22, 32, 61], and consistently activates a stereotypical inflammatory repair program, which also includes adverse effects on myocardial structure and function. We, therefore, interpret the finding of necrotic cardiomyocytes next to regions, where the cellular debris had already been cleared by neutrophils and inflammatory macrophages, as a consequence of the damaging effects of effector proteases and reactive oxygen species released by inflammatory cells. The chronic nature of this response is reflected by the continued presence of macrophages, dendritic cells, and T cells in scar tissue. Furthermore, the previously reported connexin 43 redistribution in cardiomyocytes next to immune cell-enriched scars, alterations of actin distribution and actin isoform expression as well as pathological cardiomyocyte hypertrophy in murine Dsg2 mutants [31, 38, 39] are known to be induced by the macrophage-derived cytokines TNFα, IL1β, TGFβ and also CX3CL1 [24, 27, 28, 36, 47, 66]. These cytokines are also likely involved in the formation of interstitial fibrosis that spreads from the established scars and may thereby contribute to the formation of arrhythmogenic substrates [27]. Finally, the replacement and functional modulation of resident cardiac macrophages, which are known to support electrical excitation propagation [35], by infiltrating macrophages may further contribute to the arrhythmogenic phenotype. The almost inescapable conclusion from all these observations together with the reports on other murine models [15, 51, 53] and human AC patients [5, 8, 11, 46] is that inflammation is a major "driver" of AC progression.
What are the implications of our findings for human AC patients?
Using murine AC models allows temporal and spatial resolution of disease pathogenesis that cannot be achieved in human patients. Topologically-restricted and continuously recurring lesions are further obstacles in tracking histopathological alterations in human AC patients. The murine AC models are particularly useful to dissect early disease stages, which are most relevant for pediatric AC patients [46, 62]. Their clinical phenotype is especially severe and the observed lesions have been compared to those formed in myocardial infarction [9]. In adult AC patients inflammatory immune cell infiltrates consisting either of neutrophils, macrophages, T lymphocytes, and mast cells or primarily of lymphocytes are detected in severely affected hearts [8, 12]. Similar to our AC mouse models infiltrates are confined to areas of extensive fibro-fatty replacement and necrotic cardiomyocytes.
Availability of data and material
Primary data will be made available upon request.
References
Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD (1997) Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest 100:169–179. https://doi.org/10.1172/JCI119509
Altara R, Mallat Z, Booz GW, Zouein FA (2016) The CXCL10/CXCR3 axis and cardiac inflammation: implications for immunotherapy to treat infectious and noninfectious diseases of the heart. J Immunol Res 2016:4396368. https://doi.org/10.1155/2016/4396368
Alves GD, Pazzine M, de Macedo G, Braga LM, Irigoyen MC, De Angelis K, Ikuta N, Camassola M, Nardi NB (2012) Molecular mapping of the regenerative niche in a murine model of myocardial infarction. Int J Mol Med 29:479–484. https://doi.org/10.3892/ijmm.2011.850
Aristorena M, Gallardo-Vara E, Vicen M, de Las C-E, Ojeda-Fernandez L, Nieto C, Blanco FJ, Valbuena-Diez AC, Botella LM, Nachtigal P, Corbi AL, Colmenares M, Bernabeu C (2019) MMP-12, secreted by pro-inflammatory macrophages, targets endoglin in human macrophages and endothelial cells. Int J Mol Sci. https://doi.org/10.3390/ijms20123107
Asimaki A, Tandri H, Duffy ER, Winterfield JR, Mackey-Bojack S, Picken MM, Cooper LT, Wilber DJ, Marcus FI, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, Stevenson WG, McKenna WJ, Gautam S, Remick DG, Calkins H, Saffitz JE (2011) Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol 4:743–752. https://doi.org/10.1161/CIRCEP.111.964890
Austin KM, Trembley MA, Chandler SF, Sanders SP, Saffitz JE, Abrams DJ, Pu WT (2019) Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat Rev Cardiol 16:519–537. https://doi.org/10.1038/s41569-019-0200-7
Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, Epelman S, Kreisel D, Liu Y, Itoh A, Shankar TS, Selzman CH, Drakos SG, Lavine KJ (2018) The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 24:1234–1245. https://doi.org/10.1038/s41591-018-0059-x
Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M (1996) Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation 94:983–991. https://doi.org/10.1161/01.cir.94.5.983
Bauce B, Rampazzo A, Basso C, Mazzotti E, Rigato I, Steriotis A, Beffagna G, Lorenzon A, De Bortoli M, Pilichou K, Marra MP, Corbetti F, Daliento L, Iliceto S, Corrado D, Thiene G, Nava A (2011) Clinical phenotype and diagnosis of arrhythmogenic right ventricular cardiomyopathy in pediatric patients carrying desmosomal gene mutations. Heart Rhythm 8:1686–1695. https://doi.org/10.1016/j.hrthm.2011.06.026
Bennett RG, Haqqani HM, Berruezo A, Della Bella P, Marchlinski FE, Hsu CJ, Kumar S (2019) Arrhythmogenic cardiomyopathy in 2018–2019: ARVC/ALVC or both? Heart Lung Circ 28:164–177. https://doi.org/10.1016/j.hlc.2018.10.013
Campian ME, Verberne HJ, Hardziyenka M, de Groot EA, van Moerkerken AF, van Eck-Smit BL, Tan HL (2010) Assessment of inflammation in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur J Nucl Med Mol Imaging 37:2079–2085. https://doi.org/10.1007/s00259-010-1525-y
Campuzano O, Alcalde M, Berne P, Castro V, Guzzo G, Iglesias A, Alonso-Pulpon L, Garcia-Pavia P, Brugada J, Brugada R (2012) Genetic testing of candidate genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur J Med Genet 55:225–234. https://doi.org/10.1016/j.ejmg.2012.02.007
Chatterjee D, Fatah M, Akdis D, Spears DA, Koopmann TT, Mittal K, Rafiq MA, Cattanach BM, Zhao Q, Healey JS, Ackerman MJ, Bos JM, Sun Y, Maynes JT, Brunckhorst C, Medeiros-Domingo A, Duru F, Saguner AM, Hamilton RM (2018) An autoantibody identifies arrhythmogenic right ventricular cardiomyopathy and participates in its pathogenesis. Eur Heart J 39:3932–3944. https://doi.org/10.1093/eurheartj/ehy567
Chelko SP, Asimaki A, Andersen P, Bedja D, Amat-Alarcon N, DeMazumder D, Jasti R, MacRae CA, Leber R, Kleber AG, Saffitz JE, Judge DP (2016) Central role for GSK3beta in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight. https://doi.org/10.1172/jci.insight.85923
Chelko SP, Asimaki A, Lowenthal J, Bueno-Beti C, Bedja D, Scalco A, Amat-Alarcon N, Andersen P, Judge DP, Tung L, Saffitz JE (2019) Therapeutic modulation of the immune response in arrhythmogenic cardiomyopathy. Circulation 140:1491–1505. https://doi.org/10.1161/CIRCULATIONAHA.119.040676
Chen B, Frangogiannis NG (2016) Macrophages in the Remodeling failing heart. Circ Res 119:776–778. https://doi.org/10.1161/CIRCRESAHA.116.309624
Chen X, Peng H, Zheng C, Zhang H, Yan C, Ma H, Dai X, Li X (2019) Two pedigrees with arrhythmogenic right ventricular cardiomyopathy linked with R49H and F531C mutation in DSG2. Hum Genome Var 6:38. https://doi.org/10.1038/s41439-019-0069-3
Christensen AH, Andersen CB, Wassilew K, Svendsen JH, Bundgaard H, Brand SM, Schmitz B (2019) Rare non-coding Desmoglein-2 variant contributes to arrhythmogenic right ventricular cardiomyopathy. J Mol Cell Cardiol 131:164–170. https://doi.org/10.1016/j.yjmcc.2019.04.029
Corrado D, Basso C, Judge DP (2017) Arrhythmogenic cardiomyopathy. Circ Res 121:784–802. https://doi.org/10.1161/CIRCRESAHA.117.309345
Corrado D, Basso C, Nava A, Thiene G (2001) Arrhythmogenic right ventricular cardiomyopathy: current diagnostic and management strategies. Cardiol Rev 9:259–265. https://doi.org/10.1097/00045415-200109000-00005
D'Alessio FR, Kurzhagen JT, Rabb H (2019) Reparative T lymphocytes in organ injury. J Clin Invest 129:2608–2618. https://doi.org/10.1172/JCI124614
DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, Hinson JT, Homsy J, Gray J, Pu W, Bruneau BG, Seidman JG, Seidman CE (2016) Single-cell resolution of temporal gene expression during heart development. Dev Cell 39:480–490. https://doi.org/10.1016/j.devcel.2016.10.001
Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG (2010) CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol 176:2177–2187. https://doi.org/10.2353/ajpath.2010.090759
Duncan DJ, Yang Z, Hopkins PM, Steele DS, Harrison SM (2010) TNF-alpha and IL-1beta increase Ca2+ leak from the sarcoplasmic reticulum and susceptibility to arrhythmia in rat ventricular myocytes. Cell Calcium 47:378–386. https://doi.org/10.1016/j.ceca.2010.02.002
Elliott PM, Anastasakis A, Asimaki A, Basso C, Bauce B, Brooke MA, Calkins H, Corrado D, Duru F, Green KJ, Judge DP, Kelsell D, Lambiase PD, McKenna WJ, Pilichou K, Protonotarios A, Saffitz JE, Syrris P, Tandri H, Te Riele A, Thiene G, Tsatsopoulou A, van Tintelen JP (2019) Definition and treatment of arrhythmogenic cardiomyopathy: an updated expert panel report. Eur J Heart Fail 21:955–964. https://doi.org/10.1002/ejhf.1534
Epelman S, Lavine KJ, Randolph GJ (2014) Origin and functions of tissue macrophages. Immunity 41:21–35. https://doi.org/10.1016/j.immuni.2014.06.013
Francis Stuart SD, De Jesus NM, Lindsey ML, Ripplinger CM (2016) The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J Mol Cell Cardiol 91:114–122. https://doi.org/10.1016/j.yjmcc.2015.12.024
Frangogiannis NG (2019) Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med 65:70–99. https://doi.org/10.1016/j.mam.2018.07.001
Fressart V, Duthoit G, Donal E, Probst V, Deharo JC, Chevalier P, Klug D, Dubourg O, Delacretaz E, Cosnay P, Scanu P, Extramiana F, Keller D, Hidden-Lucet F, Simon F, Bessirard V, Roux-Buisson N, Hebert JL, Azarine A, Casset-Senon D, Rouzet F, Lecarpentier Y, Fontaine G, Coirault C, Frank R, Hainque B, Charron P (2010) Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: spectrum of mutations and clinical impact in practice. Europace 12:861–868. https://doi.org/10.1093/europace/euq104
Frodermann V, Nahrendorf M (2018) Macrophages and cardiovascular health. Physiol Rev 98:2523–2569. https://doi.org/10.1152/physrev.00068.2017
Gercek M, Gercek M, Kant S, Simsekyilmaz S, Kassner A, Milting H, Liehn EA, Leube RE, Krusche CA (2017) Cardiomyocyte hypertrophy in arrhythmogenic cardiomyopathy. Am J Pathol 187:752–766. https://doi.org/10.1016/j.ajpath.2016.12.018
Harrer JM, Haghighi K, Kim HW, Ferguson DG, Kranias EG (1997) Coordinate regulation of SR Ca(2+)-ATPase and phospholamban expression in developing murine heart. Am J Physiol 272:H57–66. https://doi.org/10.1152/ajpheart.1997.272.1.H57
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J (2007) qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8:R19. https://doi.org/10.1186/gb-2007-8-2-r19
Hermida A, Fressart V, Hidden-Lucet F, Donal E, Probst V, Deharo JC, Chevalier P, Klug D, Mansencal N, Delacretaz E, Cosnay P, Scanu P, Extramiana F, Keller DI, Rouanet S, Charron P, Gandjbakhch E (2019) High risk of heart failure associated with desmoglein-2 mutations compared to plakophilin-2 mutations in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur J Heart Fail 21:792–800. https://doi.org/10.1002/ejhf.1423
Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wulfers EM, Seemann G, Courties G, Iwamoto Y, Sun Y, Savol AJ, Sager HB, Lavine KJ, Fishbein GA, Capen DE, Da Silva N, Miquerol L, Wakimoto H, Seidman CE, Seidman JG, Sadreyev RI, Naxerova K, Mitchell RN, Brown D, Libby P, Weissleder R, Swirski FK, Kohl P, Vinegoni C, Milan DJ, Ellinor PT, Nahrendorf M (2017) Macrophages facilitate electrical conduction in the heart. Cell 169(510–522):e520. https://doi.org/10.1016/j.cell.2017.03.050
Husberg C, Nygard S, Finsen AV, Damas JK, Frigessi A, Oie E, Waehre A, Gullestad L, Aukrust P, Yndestad A, Christensen G (2008) Cytokine expression profiling of the myocardium reveals a role for CX3CL1 (fractalkine) in heart failure. J Mol Cell Cardiol 45:261–269. https://doi.org/10.1016/j.yjmcc.2008.05.009
Imanaka-Yoshida K, Hiroe M, Nishikawa T, Ishiyama S, Shimojo T, Ohta Y, Sakakura T, Yoshida T (2001) Tenascin-C modulates adhesion of cardiomyocytes to extracellular matrix during tissue remodeling after myocardial infarction. Lab Invest 81:1015–1024. https://doi.org/10.1038/labinvest.3780313
Kant S, Freytag B, Herzog A, Reich A, Merkel R, Hoffmann B, Krusche CA, Leube RE (2019) Desmoglein 2 mutation provokes skeletal muscle actin expression and accumulation at intercalated discs in murine hearts. J Cell Sci. https://doi.org/10.1242/jcs.199612
Kant S, Holthofer B, Magin TM, Krusche CA, Leube RE (2015) Desmoglein 2-dependent arrhythmogenic cardiomyopathy is caused by a loss of adhesive function. Circ Cardiovasc Genet 8:553–563. https://doi.org/10.1161/CIRCGENETICS.114.000974
Kant S, Krull P, Eisner S, Leube RE, Krusche CA (2012) Histological and ultrastructural abnormalities in murine desmoglein 2-mutant hearts. Cell Tissue Res 348:249–259. https://doi.org/10.1007/s00441-011-1322-3
Kant S, Krusche CA, Gaertner A, Milting H, Leube RE (2016) Loss of plakoglobin immunoreactivity in intercalated discs in arrhythmogenic right ventricular cardiomyopathy: protein mislocalization versus epitope masking. Cardiovasc Res 109:260–271. https://doi.org/10.1093/cvr/cvv270
Krusche CA, Holthofer B, Hofe V, van de Sandt AM, Eshkind L, Bockamp E, Merx MW, Kant S, Windoffer R, Leube RE (2011) Desmoglein 2 mutant mice develop cardiac fibrosis and dilation. Basic Res Cardiol 106:617–633. https://doi.org/10.1007/s00395-011-0175-y
Liehn EA, Postea O, Curaj A, Marx N (2011) Repair after myocardial infarction, between fantasy and reality: the role of chemokines. J Am Coll Cardiol 58:2357–2362. https://doi.org/10.1016/j.jacc.2011.08.034
Lombardi R, Chen SN, Ruggiero A, Gurha P, Czernuszewicz GZ, Willerson JT, Marian AJ (2016) Cardiac fibro-adipocyte progenitors express desmosome proteins and preferentially differentiate to adipocytes upon deletion of the desmoplakin gene. Circ Res 119:41–54. https://doi.org/10.1161/CIRCRESAHA.115.308136
Martinez FO, Gordon S (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 6:13. 10.12703/P6-13
Martins D, Ovaert C, Khraiche D, Boddaert N, Bonnet D, Raimondi F (2018) Myocardial inflammation detected by cardiac MRI in Arrhythmogenic right ventricular cardiomyopathy: a paediatric case series. Int J Cardiol 271:81–86. https://doi.org/10.1016/j.ijcard.2018.05.116
Monnerat G, Alarcon ML, Vasconcellos LR, Hochman-Mendez C, Brasil G, Bassani RA, Casis O, Malan D, Travassos LH, Sepulveda M, Burgos JI, Vila-Petroff M, Dutra FF, Bozza MT, Paiva CN, Carvalho AB, Bonomo A, Fleischmann BK, de Carvalho ACC, Medei E (2016) Macrophage-dependent IL-1beta production induces cardiac arrhythmias in diabetic mice. Nat Commun 7:13344. https://doi.org/10.1038/ncomms13344
Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML (2018) Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol 113:26. https://doi.org/10.1007/s00395-018-0686-x
Murry CE, Giachelli CM, Schwartz SM, Vracko R (1994) Macrophages express osteopontin during repair of myocardial necrosis. Am J Pathol 145:1450–1462
Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ (2007) The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 204:3037–3047. https://doi.org/10.1084/jem.20070885
Padron-Barthe L, Dominguez F, Garcia-Pavia P, Lara-Pezzi E (2017) Animal models of arrhythmogenic right ventricular cardiomyopathy: what have we learned and where do we go? Insight for therapeutics. Basic Res Cardiol 112:50. https://doi.org/10.1007/s00395-017-0640-3
Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, Frigo G, Vettori A, Valente M, Towbin J, Thiene G, Danieli GA, Rampazzo A (2006) Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation 113:1171–1179. https://doi.org/10.1161/CIRCULATIONAHA.105.583674
Pilichou K, Remme CA, Basso C, Campian ME, Rizzo S, Barnett P, Scicluna BP, Bauce B, van den Hoff MJ, de Bakker JM, Tan HL, Valente M, Nava A, Wilde AA, Moorman AF, Thiene G, Bezzina CR (2009) Myocyte necrosis underlies progressive myocardial dystrophy in mouse dsg2-related arrhythmogenic right ventricular cardiomyopathy. J Exp Med 206:1787–1802. https://doi.org/10.1084/jem.20090641
Prabhu SD, Frangogiannis NG (2016) The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res 119:91–112. https://doi.org/10.1161/CIRCRESAHA.116.303577
Schuch K, Wanko B, Ambroz K, Castelo-Rosa A, Moreno-Viedma V, Grun NG, Leitner L, Staffler G, Zeyda M, Stulnig TM (2016) Osteopontin affects macrophage polarization promoting endocytic but not inflammatory properties. Obesity (Silver Spring) 24:1489–1498. https://doi.org/10.1002/oby.21510
Sharma UC, Pokharel S, van Brakel TJ, van Berlo JH, Cleutjens JP, Schroen B, Andre S, Crijns HJ, Gabius HJ, Maessen J, Pinto YM (2004) Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 110:3121–3128. https://doi.org/10.1161/01.CIR.0000147181.65298.4D
Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, Adachi H, Yashiro K, Suzuki K (2016) Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest 126:2151–2166. https://doi.org/10.1172/JCI85782
Syrris P, Ward D, Asimaki A, Evans A, Sen-Chowdhry S, Hughes SE, McKenna WJ (2007) Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: a genotype-phenotype characterization of familial disease. Eur Heart J 28:581–588. https://doi.org/10.1093/eurheartj/ehl380
Tabib A, Loire R, Chalabreysse L, Meyronnet D, Miras A, Malicier D, Thivolet F, Chevalier P, Bouvagnet P (2003) Circumstances of death and gross and microscopic observations in a series of 200 cases of sudden death associated with arrhythmogenic right ventricular cardiomyopathy and/or dysplasia. Circulation 108:3000–3005. https://doi.org/10.1161/01.CIR.0000108396.65446.21
Thiene G, Corrado D, Nava A, Rossi L, Poletti A, Boffa GM, Daliento L, Pennelli N (1991) Right ventricular cardiomyopathy: is there evidence of an inflammatory aetiology? Eur Heart J 12(Suppl D):22–25. https://doi.org/10.1093/eurheartj/12.suppl_d.22
Tiemann K, Weyer D, Djoufack PC, Ghanem A, Lewalter T, Dreiner U, Meyer R, Grohe C, Fink KB (2003) Increasing myocardial contraction and blood pressure in C57BL/6 mice during early postnatal development. Am J Physiol Heart Circ Physiol 284:H464–474. https://doi.org/10.1152/ajpheart.00540.2002
Turrini P, Basso C, Daliento L, Nava A, Thiene G (2001) Is arrhythmogenic right ventricular cardiomyopathy a paediatric problem too? Images Paediatr Cardiol 3:18–37
Vremec D, Pooley J, Hochrein H, Wu L, Shortman K (2000) CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J Immunol 164:2978–2986. https://doi.org/10.4049/jimmunol.164.6.2978
Wang W, James CA, Calkins H (2019) Diagnostic and therapeutic strategies for arrhythmogenic right ventricular dysplasia/cardiomyopathy patient. Europace 21:9–21. https://doi.org/10.1093/europace/euy063
Willenborg S, Lucas T, van Loo G, Knipper JA, Krieg T, Haase I, Brachvogel B, Hammerschmidt M, Nagy A, Ferrara N, Pasparakis M, Eming SA (2012) CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood 120:613–625. https://doi.org/10.1182/blood-2012-01-403386
Xuan W, Liao Y, Chen B, Huang Q, Xu D, Liu Y, Bin J, Kitakaze M (2011) Detrimental effect of fractalkine on myocardial ischaemia and heart failure. Cardiovasc Res 92:385–393. https://doi.org/10.1093/cvr/cvr221
Acknowledgements
Open Access funding provided by Projekt DEAL. We thank Claudia Schmitz, Sabine Eisner, Marina Lürkens-Weber and Christine Eherer for expert technical support, Andreas Faissner (Ruhr-University, Bochum) for antibodies, Philipp Krull for Spp1 qRT-PCR, Mustafa Gerçek for Ccl2 and Ccl3 qRT-PCR and Adam Breitscheidel for graphics. This work was funded by the Interdisciplinary Center for Clinical Research within the Faculty of Medicine at Rheinisch-Westfälische Technische Hochschule Aachen University grants T9-2 and K7-4 (REL and CAK), START 30/18 (SK) and the Kaltenbach-Stipendium of the Deutsche Herzstiftung to SG. Some of the reported results are part of the MD theses of NL, SG and MG.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by NL, SG, MG, SK and CAK. The first draft of the manuscript was written by CAK and REL and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding authors state that there is no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lubos, N., van der Gaag, S., Gerçek, M. et al. Inflammation shapes pathogenesis of murine arrhythmogenic cardiomyopathy. Basic Res Cardiol 115, 42 (2020). https://doi.org/10.1007/s00395-020-0803-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-020-0803-5