
Abstract
Congenital diaphragmatic hernia (CDH) is a life-threatening anomaly with a mortality rate of approximately 40–50%, depending on case selection. It has been suggested that new therapeutic modalities such as nitric oxide (NO), high frequency oxygenation (HFO) and extracorporal membrane oxygenation (ECMO) might decrease mortality associated with pulmonary hypertension and the sequelae of artificial ventilation. When these new therapies indeed prove to be beneficial, a larger number of children with severe forms of CDH might survive, resulting in an increase of CDH-associated complications and/or consequences. In follow-up studies of infants born with CDH, many complications including pulmonary damage, cardiovascular disease, gastro-intestinal disease, failure to thrive, neurocognitive defects and musculoskeletal abnormalities have been described. Long-term pulmonary morbidity in CDH consists of obstructive and restrictive lung function impairments due to altered lung structure and prolonged ventilatory support. CDH has also been associated with persistent pulmonary vascular abnormalities, resulting in pulmonary hypertension in the neonatal period. Long-term consequences of pulmonary hypertension are unknown. Gastro-esophageal reflux disease (GERD) is also an important contributor to overall morbidity, although the underlying mechanism has not been fully understood yet. In adult CDH survivors incidence of esophagitis is high and even Barrett’s esophagus may ensue. Yet, in many CDH patients a clinical history compatible with GERD seems to be lacking, which may result in missing patients with pathologic reflux disease. Prolonged unrecognized GERD may eventually result in failure to thrive. This has been found in many young CDH patients, which may also be caused by insufficient intake due to oral aversion and increased caloric requirements due to pulmonary morbidity. Neurological outcome is determined by an increased risk of perinatal and neonatal hypoxemia in the first days of life of CDH patients. In patients treated with ECMO, the incidence of neurological deficits is even higher, probably reflecting more severe hypoxemia and the risk of ECMO associated complications. Many studies have addressed the substantial impact of the health problems described above, on the overall well-being of CDH patients, but most of them concentrate on the first years after repair and only a few studies focus on the health-related quality of life in CDH patients. Considering the scattered data indicating substantial morbidity in long-term survivors of CDH, follow-up studies that systematically assess long-term sequelae are mandatory. Based on such studies a more focused approach for routine follow-up programs may be established.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Congenital diaphragmatic hernia (CDH) is a life-threatening congenital anomaly, with an incidence of approximately 1:2,500 live births [1, 2]. Despite advances in antenatal diagnosis and postnatal management, mortality rate remains stable around 40–50% [1–4]. It has been shown that overall survival rate is mainly determined by the rate of antenatal termination, the incidence of CDH associated anomalies, degree of pulmonary hypoplasia and pulmonary hypertension in the neonatal period [1, 2, 5–7]. Reports of improved survival of CDH should be interpreted with caution, as variations in outcomes are more likely to be explained by case selection bias [2].
The incidence of malformations associated with CDH is 33–50% [1, 3, 8, 9]. Major anomalies, like cardiovascular defects, intestinal atresias and chromosomal anomalies [1, 3, 10] may all contribute to a fatal outcome [1–3, 8, 11–13]. Several non-life threatening anomalies such as atrial or ventricular septal defect, Meckel’s diverticulum, neurological abnormalities and genito-urinary abnormalities are also more frequently observed in CDH patients [3, 8].
In the past decades, new pathophysiologic concepts have been developed, leading to a change in therapeutic approach. Before 1985, surgical repair was performed on an emergency basis, because it was believed that removing the abdominal viscera from the chest allowed expansion of the lung on the involved side [14]. It was observed, however, that the clinical condition of children with CDH rarely, if ever, improved after emergency surgery; in fact, many children were significantly worse postoperatively [15]. This could be explained by the fact that pulmonary vascular resistance rapidly increases due to a variety of stimuli including endotracheal tube suctioning, loud ambient noise, pain, hypothermia and atelectasis. All of these stimuli are enhanced during repair of the diaphragm. Furthermore it was demonstrated that surgical reduction of the hernia does not improve lung mechanics and may even temporarily decrease the compliance of the chest wall [16, 17]. This can be explained by increased abdominal pressure associated with reduction of the viscera into the small abdominal cavity [16, 17].
When it was realized that surgical repair could further decrease lung compliance, which could be fatal in a patient already compromised by pulmonary hypoplasia and persistent pulmonary hypertension, repair was delayed and performed after patients were stabilized and adequately oxygenated with minimal ductal shunting on conventional ventilation [4, 18]. Delayed surgery is theorized to provide additional time for remodeling of the pulmonary vasculature, leading to a more stable infant, who is better able to tolerate a postoperative decrease in compliance [16, 19]. Although a significant change in survival after the introduction of delayed surgery was not observed [4, 20–22], a strategy of delaying correction of the defect for some period of time have been adopted by centers, with the majority of patients undergoing operation beyond 24 h of age [23, 24].
The introduction of ECMO in the early 1980s was expected to improve the outcome for infants with pulmonary hypertension refractory to maximal conventional medical management. Roughly 50% of infants with high-risk CDH are treated with ECMO [25, 26], the results, however, remain disappointing [2, 21, 27]. Some authors reported an improved survival rate when incorporating ECMO as a preoperative stabilization or postoperative rescue therapy [26, 28, 29], while others found a relatively unchanged mortality rate [2, 21, 22, 27, 30]. It is difficult to accurately compare results of those studies because each center varies in degree of illness, ventilatory management, timing of surgery, ECMO and selection criteria for ECMO. Another confounding feature of reports of survival in CDH patients treated with ECMO is the unreliability of historic controls [2, 26, 27]. Furthermore, most studies have a relatively short follow-up period, while significant late mortality has been found in CDH patients treated with ECMO [31]. If the use of ECMO has indeed a beneficial effect on survival, than it might be possible that a larger number of severely affected infants would nowadays survive [32]. It is likely that these infants will have more severe pulmonary hypoplasia and pulmonary hypertension [26, 31, 33] implying more serious long-term morbidity [31].
The last decade fetal endoscopic tracheal occlusion (TO) has been developed as an alternative strategy to promote fetal lung growth by preventing normal outflow of lung fluid. Occlusion of the fetal trachea was shown to stimulate fetal growth in a variety of animal models [34–40]. On the other hand, prematurity caused by the intervention itself can be detrimental. Results of studies regarding survival rate and TO are ambiguous and it is not yet clear whether survival rates will improve with this new technique [41]. Harrison et al. [42] showed in a (small) randomized controlled trial comparing fetal endoscopic TO to standard care that mortality is equal. Data concerning long-term outcomes of fetal endoscopic TO are very scarce [43, 44].
Traditionally most attention has been focused on therapies that reduce perinatal and neonatal mortality, while relatively few studies have addressed the chronic morbidity and long-term outcome. In follow-up of infants born with CDH, many complications have been described. In the following sections, we will describe the pulmonary, cardiovascular and gastro-intestinal morbidity secondary to CDH. We will also discuss failure to thrive, neurodevelopmental outcomes, musculoskeletal abnormalities and health-related quality of life in CDH patients.
Pulmonary morbidity
Structural lung abnormalities and the influence of treatment
One of the major characteristics of CDH is pulmonary hypoplasia and an altered lung structure, consisting of a reduced number of bronchi, a reduced number of alveoli and structural abnormalities of the pulmonary vascular bed. The latter will be discussed in the next section. The number of normal bronchi is reduced in the ipsilateral lung and to a lesser degree in the contralateral lung [45, 46]. Beals et al. [45] showed that the number of bronchi did not increase in time, which is compatible with Reid’s statement [47] that “bronchial development is complete by 16 weeks of life”. The alveolar number on the other hand, can increase over time. A significant increase in alveolar number in the contralateral lung, in contrast to the ipsilateral lung, was found in patients with CDH who deceased before they were 8 days old, between 8 and 21 days old and after they were 21 days old. [45]. Compared to normal lung tissue, there was still a reduction of the number of alveoli [6, 45] in the ipsilateral lung and to a lesser degree in the contralateral lung. In addition to the increase in alveolar number, it has been shown that size of alveoli may increase [46], resulting in overdistension of the alveoli to fill the hemithorax after repair [48]. In addition to structural lung abnormalities, treatment may also contribute to pulmonary morbidity. In the past, one of the treatment strategies has been the use of hyperventilation to achieve alkalosis. The principle of this hyperventilatory-induced alkalosis was used to reduce pulmonary vascular resistance and to reverse ductal shunting. This approach frequently requires high peak airway pressures and these may cause lung injury [4, 49]. Lung injury, secondary to mechanical ventilation can result in pulmonary edema and protein leak, also known as ventilator-induced lung injury [50], resulting in denaturation of surfactant and consequently lung damage [51]. This ventilator-induced lung injury due to high pressure ventilation appears to be one of the major contributors to mortality in CDH patients [4, 49, 52, 53] and therefore the strategy of high pressure ventilation has been abandoned. Since the introduction of “permissive hypercapnia” (i.e, allowing higher concentrations of carbon dioxide in the blood to prevent barotrauma), an improved survival rate has been documented [4, 19, 49, 52]. Nevertheless, barotrauma is still an important contributing factor to lung injury, which could possibly be further reduced by the use of ECMO [28]. But as stated before, the benefits of ECMO in CDH and the effect of ECMO in the long term are controversial. It has been demonstrated that neonatal ECMO survivors treated for meconium aspiration syndrome experience lung injury, such as hyperinflation, airway obstruction and lower-oxygen saturation with exercise, in later childhood [54]. This finding persisted when they were compared to non-ECMO treated survivors [55]. Others found that the pulmonary function of ECMO treated infants (non-CDH) appeared to be slightly better than that of infants treated conventionally [56].
Fetal tracheal occlusion
In animal experiments, fetal TO has been shown to accelerate lung growth [57], on the other hand, surfactant deficiency associated with TO could lead to deterioration in lung function with longer periods of ventilation [58]. Effects on pulmonary function of CDH patients treated with fetal TO are preliminary. Keller et al. [43] measured respiratory mechanics in the first 24 h after birth, before CDH repair, immediately after repair and before elective extubation. They found that fetal TO for severe CDH results only in modest improvements in neonatal pulmonary function. Cortes et al. [44] performed a follow-up study of 16 CDH survivors, 7 of them were treated with fetal TO. Percentages of patients needing oxygen supplementation after discharge and use of pulmonary medications were similar in both groups. Pulmonary function (n = 13) did not differ between the groups.
Respiratory symptoms
Several follow-up studies exploring respiratory symptoms in CDH survivors yield contradictory results. Symptoms like coughing or recurrent respiratory infections have been reported by 25–50% of affected children, especially in the first year of life [48, 59–61]. Bronchodilators and inhaled steroids were, at least transiently, used by the majority of CDH patients, during the first year of life and thereafter [61, 62]. In other studies, however, none of the participating children had significant respiratory symptoms [63, 64], suggesting that CDH patients were doing well, regarding long-term pulmonary outcome. Despite the subjective impression that CDH survivors older than 2 years generally were doing well, objective data are scarce, due to a limited number of included patients [62, 65].
Pulmonary function
Spirometric testing of CHD patients revealed evidence of obstructive airway disease [64] when compared with healthy controls [48, 62, 64, 66–69]. Some authors describe that one-third of the CDH patients have reduced forced expiratory volume in 1 s (FEV1) and forced vital capacity (FVC) [62, 69]. FEV1 is an index for assessing airway obstruction and is often performed in conjunction with FVC, i.e. the volume of air that can be forcibly and maximally exhaled out of the lungs. FEV1 and/or FEV1/FVC are decreased in patients with airway obstruction. Wischerman et al. [59] reported normal FEV1 in CDH patients, but found a tendency to obstruction of the smaller airways illustrated by reduction in the mid-expiratory flow rate and raised specific airway resistance. There was no correlation between the extent of pulmonary hypoplasia, assessed during the operation and by pre-and postoperative chest X-rays, and reduction of airflow rate [59]. Although FEV1 improves in approximately half of the patients after bronchodilator therapy [68, 69], it remains significantly lower when compared to healthy subjects [69]. Furthermore, there is evidence for more peripheral airway obstruction in CDH patients [69] possibly due to structural abnormalities in distal airways [69]. It has been speculated that there must be some degree of emphysema because of the finding of a high residual volume total lung capacity (RV/TLC) ratio, despite the lack of airway dysfunction [66]. Others suggest that the high RV/TLC ratio (index for the amount of trapped air after full exhalation) is associated with chest wall deformity [68]. Trachsel et al. demonstrated that CDH patients with a chest wall deformity (moderate to severe pectus excavatum) had a significantly higher RV/TLC ratio (40.4 vs. 27.9%). It has been known that pectus excavatum patients have slightly impaired pulmonary function, which may be due to decreased compliance of the chest wall [70].
It is hypothesized that reduced-lung tissue and consequent alveolar distension may result in early airway closure as found in the aging emphysematous lung, which could be an additional risk factor in the development of chronic obstructive disease on the longer term [48, 71]. Some long-term follow-up studies indicate a higher incidence of restrictive lung function impairment among CDH patients compared to the healthy population [63, 65] especially when there was a large diaphragmatic defect [65], while others cannot confirm this finding [59, 66]. Overall CDH patients appear not to have an important reduction of total lung capacity in their school-and adolescence years [66, 68, 69].
A negative correlation has been demonstrated between the duration of ventilation and pulmonary function at follow-up [63, 69], illustrated by the fact that CDH patients who had been ventilated for more than 7 days had significantly lower FEV1 and vital capacity (VC) than CDH patients who had been ventilated for less than 7 days [69]. This could reflect the severity of the pulmonary disease [63], but can also be the consequence of ventilator induced barotrauma. Sakurai et al. [49] found microscopic evidence of significant pulmonary injury related to the pressure effects of mechanical ventilation in deceased infants with CDH, implying destruction of the alveolar-capillary membrane. The diffusion capacity (i.e. rate at which a gas diffuses through the alveolar-capillary membrane) corrected for alveolar volume (DLCO/V′A) however, appears to be normal in CDH patients compared to healthy subjects [68, 69] suggesting that the alveolar membrane is not damaged [69].
Sport practice and cardiopulmonary exercise testing
CDH patients achieve normal aerobic capacity (V′O2max) (an index for maximal oxygen uptake during maximal exercise; it can determine an athlete’s capacity and is linked to aerobic endurance) when performing a maximal exercise test [66], which is compatible with the normal diffusion capacity that has been described [69, 72]. Nonetheless, Italian investigators report significantly lower V′O2max for CDH patients representing a decreased degree of physical fitness [67]. This could (partially) be explained by the fact that Italian law strictly regulates access to sports by requiring a thorough medical examination before the start of sports activity and that CDH patients may therefore not practice sport regularly. When comparing CDH patients active in sports and controls active in sports, the difference in V′O2max still remained [67]. All participating patients considered themselves healthy and they did not have any particular symptom. In the study from Marven et al. [66], CDH patients perceived their own fitness to be worse than the healthy control group. Other studies demonstrate that around 20% of interviewed CDH patients consider themselves less fit than their healthy peers [59, 65, 73]. It seems most likely that the decreased V′O2max found by Zaccara et al. is due to a lower degree of physical fitness. Fitness can be increased by regular motor activity and therefore they recommend active sports participation for CDH children.
It can be concluded that, although patients surviving CDH do not have many respiratory symptoms in their (pre-)school years, the majority of CDH survivors have some degree of obstructive and/or restrictive airway disease when carefully examined. The diffusion capacity appears to be normal but a significantly lower V′O2max has been demonstrated in CDH survivors possibly representing a lower degree of physical fitness, though the number of patients who were investigated in this study was rather small.
CDH survivors might be at risk for developing pulmonary emphysema due to consequent alveolar distension, but it remains unclear whether the alveolar membrane has been damaged. Also the introduction of more advanced interventions, such as fetal TO requires more research on long-term pulmonary function has to be done to further clarify CDH associated pulmonary sequelae.
Cardiovascular morbidity
Pulmonary vasculature
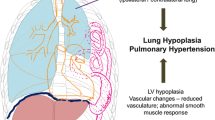
In addition to structural abnormalities of alveoli and airways in infants born with CDH, the pulmonary vascular bed also appears to be abnormal. Several studies have shown that under normal conditions, when approaching full term, pulmonary arteries show reduction in medial thickness [74–76]. Molecular basis underlying these changes in pulmonary vasculature during gestation are not known yet. The role of vascular smooth muscle cells appears to be crucial [77]. The physiologic reduction in medial thickness when approaching full term was not observed in CDH lungs [76–78]. Pulmonary vascular abnormalities in CDH consist of decreased number of pulmonary arteries per unit lung volume and an increase in the medial, adventitial and consequently total wall thickness of all arteries [4, 45, 76–81]. Due to failure of vascular remodeling, peripheral muscularization of pulmonary arterioles resulted in particularly medial and adventitial thickening [15, 78, 79, 81]. The arterioles are predominantly responsible for pulmonary vascular resistance [15, 79].
ECMO and pulmonary vasculature
There are differences observed between lungs of ECMO treated CDH patients and lungs of non-ECMO treated CDH patients. Whereas adventitial thickness and total-wall thickness appeared to be reduced in ECMO-treated CDH patients when compared to non-ECMO CDH patients, medial thickness remained increased [77, 82]. Although pulmonary arterial pressure decreases during 2 weeks of ECMO, the small acinar arteries remain thick-walled. Therefore ECMO may result in remodeling or muscle relaxation [82], ameliorating pulmonary hypertension.
Beals et al. [45] found fewer numbers of arteries with increased muscular wall thickness and a lower percentage of muscularization of arteries in deceased infants with CDH with increasing age suggesting that natural history may also play a role. A number of these infants had been treated with ECMO. These results are consistent with the experience that the tendency to pulmonary hypertension decreases over time [45]. Unfortunately, the time period over which these changes occur exceeds the current limitations of invasive support measures such as ECMO [45].
TO might be a promising alternative, since it has been shown that this intervention can reverse the pulmonary arterial structural changes that are seen in the nitrofen-induced fetal rat model of CDH, suggesting that TO might reduce pulmonary vascular reactivity [83]. More research in this area is mandatory.
Apart from the pulmonary vascular abnormalities observed in CDH patients, an altered expression of several factors may be involved in regulation of the vascular tone [84, 85]. All these factors together may predispose infants born with CDH for pulmonary hypertension [45, 76, 77].
Pulmonary hypertension and follow-up
In general, only few reports investigate natural course of persistent pulmonary hypertension of the newborn. In infants with pulmonary hypertension in the neonatal period due to chronic neonatal lung disease, echocardiographic evidence of pulmonary hypertension was found in almost 25% of these patients after 2–5 years, while none of them had clinical symptoms [86].
Most of the long-term consequences of pulmonary vascular abnormalities in CDH patients are unknown [62]. As stated before, vascular abnormalities might contribute to the development of pulmonary hypertension in CDH patients in the neonatal period. In the first 3 weeks of life, pulmonary hypertension resolves spontaneously in almost half of the CDH patients without the need for ECMO [87].
There are few studies describing pulmonary hypertension in CDH survivors beyond the neonatal period, in most of these, patients were treated with ECMO [33, 88, 89]. Iocono et al. [89] identified seven patients with pulmonary hypertension out of 40 CDH survivors at time of discharge, by echocardiographic measurements. One of them died, while the remaining six survived with normalization of pulmonary vascular pressure at an average age of around 400 days. Schwartz et al. [88] documented that 8 of 21 CDH patients (age 3.2 ± 1.4 years) in their study group met the criteria of pulmonary hypertension, such as right axis deviation or right ventricular hypertrophy. Only two of these seven patients desaturated mildly during the maximal exercise test (one patient was too young to perform the test). One of their patients, diagnosed with severe pulmonary hypertension, eventually needed lung transplantation [88].
Van Meurs et al. [33, 88] performed a complete cardiological evaluation in 14 CDH survivors. All children had normal auscultation and oxygen saturation. The electrocardiogram showed evidence for right ventricular hypertrophy in six of the 14 children; however, this could also be related to cardiac malposition due to the underlying pulmonary hypoplasia. In a group of 24 survivors of mild to moderate CDH, studied by Stefanutti et al. [63], no echocardiographic signs of pulmonary hypertension were found. Recently a nearly normal resting and exercise cardiorespiratory function has been reported in CDH survivors aged 10–16 years without evidence for exercise-induced pulmonary hypertension measured by echocardiography and maximal exercise testing [72]. So far there is only one study describing potential consequences in adulthood. Exercise capacity and gas exchange parameters were normal in all 12 adult CDH survivors, indicating these patients have a normal pulmonary vascular bed [90].
Lung perfusion
There are several studies investigating long-term lung perfusion of CDH survivors by performing pulmonary perfusion scintigraphy. It has been demonstrated that in survivors of left side hernia repair, mean perfusion of the ipsilateral lung was lower than that in healthy children [48, 62–65] and in the contralateral lung [48, 62, 71]. Patients who were ventilated for 4 days or more had poorer perfusion than patients who had required shorter ventilatory support [48]. Secondary lung injury may play an important role because ventilatory support and supplemental oxygen were required for a long period of time [71].
Presence of persistent pulmonary hypoperfusion on the side of the diaphragmatic defect suggests that an anatomic impairment of variable degree is still present several years after surgical repair in CDH patients and might reflect a limited ability of the ipsilateral lung to develop additional arterial branches [63, 71]. It has been speculated that, because of persistence of poorly perfused ventilated areas representing dead space, these patients have increased intrapulmonary shunting and hypoxemia possibly leading to pulmonary hypertension [62].
In summary, CDH has been associated with pulmonary vascular abnormalities which may result in pulmonary hypertension. Scintigraphic studies have shown that there is persistent hypoperfusion on the affected side. The long-term consequences are not clear.
Notably, all studies described above include a relatively small number of patients and most studies contain a heterogeneous group of patients.
It is still complex to diagnose pulmonary hypertension in an early stage because pulmonary hypertension may not become evident until complaints resulting from a significant degree of right heart failure become apparent. While there are techniques enabling detection of pulmonary hypertension in an early stage [91, 92], it is still unclear whether routine assessment of these techniques is justified as part of the long-term follow-up of CDH.
Gastro-intestinal morbidity
Gastro-esophageal reflux disease
Pathophysiology
Gastro-esophageal reflux disease (GERD) is a well-recognized consequence of CDH. The mechanisms responsible for GERD in CDH survivors have not been clarified and several theories have been proposed.
Firstly, it has been suggested that elevated intra-abdominal pressure, resulting from positioning the hernial contents into the abdominal cavity, in combination with reduced intra-thoracic pressure, increases the abdominal-thoracic pressure gradient, which could predispose to GERD [93, 94].
Secondly, abnormal anatomy of the gastro-esophageal junction caused by the rudimentary or absent left diaphragmatic crus, might contribute to occurrence of postoperative reflux [33, 95]. Especially when the stomach initially was positioned in the chest, the lack of an intra-abdominal esophageal segment following repair would adversely affect the angle of His [71, 96]. The combination of increased abdominal-intrathoracal pressure gradient and underdevelopment of the diaphragmatic crus may increase the strain on the crus and result in an insufficient anti-reflux barrier [97] and favorable conditions for hiatus hernia and GERD [96, 97].
Thirdly, abnormality of the esophageal dimensions might also contribute to development of GERD. The lesion might be congenital and may result (in part) from extrinsic pressure on the mediastinum and developing esophagus caused by the herniated viscera very early in gestation. Stolar et al. noted a mediastinal mass on plain chest radiographs obtained after repair of CDH in 20 of 21 patients. Subsequent upper gastro-intestinal (UGI) series showed it to be a dilated ectatic esophagus. After 5 years, esophageal contour was still abnormal, while in 9 of 13 patients pathologic reflux was diagnosed by pH studies. This phenomenon was also described by van Meurs et al. [33]. In retrospective analysis of chest radiographs, they identified a mega-esophagus in 40% of CDH survivors.
Incidence of GERD in the short term
According to most studies, the incidence of GERD in the first years after repair ranges between 20 and 72% [33, 93, 96, 98–100]. Even higher incidences have been reported [32]. The clinical presentation may vary, including recurrent vomiting or regurgitation, recurrent episodes of bradycardia and respiratory arrest, failure to thrive and recurrent pneumonia [96].
In the study by van Meurs et al. [33], symptoms consistent with GERD were present in 16 of 18 children (CDH patients treated with ECMO) during the initial hospitalization. At the time of discharge, 8 patients were receiving nasogastric or gastrostomy tube feedings. One child required a gastrostomy tube for severe failure to thrive. None required surgical anti-reflux procedure. Koot et al. [98] performed a prospective evaluation of GERD in patients treated for CDH. Six months after closure of the defect, 17 of the 31 patients had pathologic reflux on UGI series. Nine patients were treated conservatively by maintaining supine position and by frequent administration of small amounts of oral nutrition. Seventeen patients (with and without symptoms) received medical treatment. In three patients, who did not respond sufficiently to this treatment, Nissen fundoplication was performed. After surgery, clinical parameters improved and investigations confirmed disappearance of GERD [98]. In a study by Fasching et al. [93], although 20 of 25 children had no major clinical symptoms suggesting GERD in a follow-up period of 1–17 years, GERD was demonstrated with UGI series or 24-h pH studies in 8 of them. In contrast, of the 5 symptomatic children, only 2 actually had GERD, the other 3 having normal test results. Seven patients (28%) underwent a surgical anti-reflux-procedure [93]. In the retrospective study of Muratore et al. [99], 27 of the 45 CDH survivors appeared to have significant GERD confirmed by UGI series, 25 of them needing fundoplication. Again it is difficult to accurately compare results of those studies because each study varies regarding CDH severity, postoperative CDH management (ECMO or not) and follow-up period. Furthermore, different definitions for GERD have been used, ranging from symptoms suggestive for GERD to the need for antireflux surgery. Finally most studies contain only a small group of patients, which hampers the generalizability of the results.
Incidence of GERD in the long term
Reports concerning long-term gastro-intestinal morbidity in adults with repaired CDH are scarce. Vanamo et al. examined 60 of 107 CDH survivors aged 14–49 years. At time of follow-up, 38 of them reported symptoms suggestive of GERD, mainly heartburn and regurgitation. Endoscopy showed macroscopic esophageal pathology in 12 of the 41 recruited patients, an alarming finding being that Barrett’s esophagus was found in four patients. Recently the first CDH survivor with esophageal adenocarcinoma has been described, 22 years after CDH repair [101]. Vanamo et al. found that hiatus hernia was present in 30 patients, although as such this is not an uncommon finding in the adult population in the western world [102]. The overall prevalence of esophagitis was 54%, which is significantly higher than the expected 2% of endoscopically assessed esophagitis in the general adult population [103, 104]. Predictive value of symptoms for detection of esophagitis is poor: 6 of the 22 patients with esophagitis were completely asymptomatic and only 14 of the 26 patients with heartburn or regurgitation had esophagitis. Esophageal stricture was found in 2 of 11 patients with early postoperative GERD [105].
Prediction of GERD in CDH survivors
Several authors have attempted to define clinical variables that may predict GERD, with controversial outcomes. Koot et al. [98] could not identify a specific predictive variable, although they suggested a relation between duration of artificial ventilation and development of GERD [98, 106]. Vanamo et al. [105] also reported a significant relation between duration of ventilatory support and GERD. In addition, they found that early postoperative GERD was more common in patients with respiratory distress who had been symptomatic within 6 h after birth. Furthermore, postoperative intolerance of enteral feedings and prolonged hospitalization were found predominantly in patients with symptomatic GERD [105]. There might [79] or might not [87] be an association between GERD and preoperative thoracic position of the stomach.
Fasching et al. [93] found no significant relation between the size of the defect and occurrence of GERD, but they found severe GERD in all three examined patients with a patch. Yet, GERD appears to some extent to be related to size of the diaphragmatic defect, because it was more frequent and more severe in children requiring a prosthetic patch [100, 107] or ECMO [28, 32, 33]. On the other hand, use of a prosthetic patch could possibly diminish morbidity related to GERD by lowering the strain on the left crus [96, 106]. Furthermore, the diaphragmatic patch may reduce abdominal pressure and facilitate postoperative ventilation [96]. This contrasts with results of other studies reporting that requirement of a patch is an independent determinant that predicts occurrence of GERD [99, 107, 108].
While the mechanism responsible for GERD in CDH is still unclear, GERD as such is an important attribute of overall short term and long term morbidity of CDH [105].
Other gastro-intestinal problems
Intestinal malrotation is obligatory in babies born with CDH. It is assumed that this is related to abnormal positioning and fixation of the bowel in an abdomen that, although reduced in size, has a wide communication with the thorax. [109]. As a result, intestinal obstruction due to midgut volvulus or adhesions occurs in approximately 10–20% of CDH patients [32, 105, 110]. The most commonly performed second surgical intervention in CDH is re-exploration for small bowel obstruction [32, 110]. This percentage is considerably higher than those of postoperative intestinal obstruction in other patients who had laparotomy in the neonatal period, infancy and childhood (2.2–6%) and in patients who had Ladd’s procedure for malrotation (8.3%) [111–113], but comparable to patients operated for gastroschisis or omphalocele [114]. Several mechanisms may contribute to the increased susceptibility to intestinal obstruction among patients with diaphragmatic defects. The malrotation predisposes to volvulus of the intestine. The prolonged intestinal paralysis caused by critical illness in the neonatal period and its treatment may enhance adhesion formation. Increased intra-abdominal pressure may further interfere with peristalsis [105].
Finally, non-specific abdominal problems such as hiccups, vomiting, meteorism, and abdominal pain occurred in 13.3% of the children during follow-up [59].
Failure to thrive
Pathophysiology
Failure to thrive has been noted in many CDH survivors [32, 33, 60]. Pathophysiology of growth retardation in CDH is complex and includes catabolic stress in the neonatal period, oral aversion, GERD and an increased caloric requirement due to persistent pulmonary morbidity [108, 110].
Some authors reported that 20–30% of their patients remain at or below the 5th percentile for weight, despite optimization of caloric intake [108, 110]. Other studies show that nutritional and growth problems are affecting nearly half of the patients [32]. Several contributing factors have been suggested, such as increased work of breathing, prolonged (>4 months) oxygen dependency [33], poor sucking ability, and GERD [32]. It has been demonstrated that infants with broncho-pulmonary dysplasia also continue to have growth failure after hospital discharge, which might be due to increased energy utilization and decreased energy intake [115]. Inadequate tissue oxygenation may also play a role, since it has been demonstrated that infants with BPD and discharged with oxygen had less growth failure than infants who were discharged without oxygen [116], although this could not be confirmed for CDH patients [33, 99]. The role of GERD is debatable, because even after the reflux symptoms resolve, significant growth failure remains. Another predictor of growth failure is the need for ECMO [99, 110], CDH patients treated with ECMO were found to have significantly lower weight, length and weight/length percentiles when compared to other ECMO patients [33] suggesting that CDH plays a major role in growth failure.
Follow-up
Muratore et al. [99] reported that 56% of their population was below 25th percentile for height and weight during the first year of life, despite the fact that they were all born at full term. Contributing to the observed growth failure were feeding difficulties. Nearly one-quarter of their patients displayed behavior consistent with oral aversion. Duration of ventilation was the only independent determinant that predicted development of oral aversion. They speculated that prolonged endotracheal intubation might contribute to a delay in development of the swallowing reflex or the suckling mechanism. One-third of the patients in this study required gastrostomy tube placement for adequate caloric intake. Despite this, several patients still showed evidence of growth failure that appeared to be due to GERD [99]. On the other hand, according to Kamiyama et al. [106] early GERD does not seem to have a significant influence on physical development of these patients. They found no significant difference with respect to body weight between CDH patients with and without pathologic GERD in the early postoperative period [106]. Fluid restrictions as well as diuretics are required to prevent pulmonary vascular overload. In order to meet caloric needs in this situation, high energy density formulas are required, so the majority of these children have enriched feedings.
In the study by van Meurs et al. [33], 15 children with CDH treated with ECMO were compared with 15 ECMO-treated children with other diagnoses (controls). All were within normal (5–95) percentiles for weight, length and head circumference at birth, although 4 of the 15 CDH infants (27%) were less than the 5th percentile for weight-length ratio as compared to 2 of the control subjects. In the first 2 years of life, weight gain was significantly lower in the CDH group as compared to the ECMO treated, weight-matched control subjects without CDH. In the report by Davenport et al. [117], of 23 children born with CDH, only 1 was below the 10th centile for height while 6 were below the 10th centile for weight. They found a significant inverse correlation between attained weight centile and duration of ventilation as well as of hospital stay.
In summary, it appears that many CDH survivors experience growth failure. There are several attributes to growth failure, including persistent pulmonary morbidity resulting in increased caloric requirements, feeding problems due to a postoperative ileus, need for ECMO, GERD and oral aversion, especially when endotracheal intubation and prolonged ventilation are needed.
We conclude that all CDH patients should therefore undergo routine nutritional assessments with emphasis on weight, height, weight-for-height ratio, growth velocity and measurements of head circumference. Early intervention by nutritional specialists may avoid prolonged oral aversion, improve weight and length accretion and foster normal developmental milestones.
Neurodevelopmental outcomes
Neurocognitive defects
Although approximately half of the CDH cases occur as isolated malformations with normal brain development, neurodevelopmental outcome of these infants may be affected by acquired (e.g., hypoxia) neurologic insults secondary to CDH. For the 50% of infants with a co-existing anomaly, about 7% has anomalies of the central nervous system [8]. When these associated anomalies can be assigned to a syndromal diagnosis, neurodevelopmental outcome is generally poor.
Children born with CDH have high risk of hypoxemia at varying stages of their postnatal management due to the combination of pulmonary hypoplasia and persistent pulmonary hypertension. Persistent hypoxemia has a significant correlation with developmental delays [32, 118] possibly due to ischemia/reperfusion injury to the brain and loss of autoregulation of cerebral blood flow [119].
Neurological abnormalities in the neonatal period
Several neurological abnormalities in CDH survivors have been documented in the literature. Hunt et al. performed MR imaging at a median postnatal age of 25 days in 8 infants treated for CDH (non-ECMO). Abnormalities were detected in all 8 infants, in only 1 of these infants the abnormality had already been identified on cranial ultrasound. There were abnormalities of varying severity found in every infant who was already weaned from mechanical ventilation, including ventricular dilatation, abnormal signal in the white matter and basal ganglia, and abnormal myelination of posterior limb of the intern capsule. Van Meurs et al. [33] described that CT scans in 4 out of 17 CDH survivors treated with ECMO were normal. Ten children had minor abnormalities such as petechial hemorrhages, mild ventricular enlargement or widening of the interspheric fissure and three children had major abnormalities including cerebellar hemorrhages and hemorrhages in the basal ganglia. The ventricular dilation might reflect loss of white matter as a consequence of ischemia in the vulnerable immature periventricular white matter. It is hypothesized that inflammation might also play a role. Clearly surgery presents a significant inflammatory response [120], which may contribute to cerebral white matter injury [121]. The long-term implications of the abnormalities detected on MRI are unknown.
Long-term follow-up
Lund et al. [32] performed CT studies of the brain in all 33 CDH-survivors, showing marked bifrontal atrophy or ventricular dilation or a combination of both in 10 patients. All but one patient was treated with ECMO. During follow-up, head CT findings improved with growth. None of the children described in the study above had a syndromal diagnosis.
In patients treated with ECMO, including CDH-survivors, neurological deficits have been reported in 10–30% of patients [122–127]. Stolar et al. [128] investigated the role of CDH in ECMO-treated infants with CDH by comparing ECMO survivors with CDH to other ECMO survivors. They concluded that, diagnosis of CDH (among ECMO survivors) did not contribute independently to neurological risk. In contrast, a significant correlation has been found between CDH and a poorer cognitive outcome especially in boys. CDH infants appeared to be more unstable while on ECMO, have more complications, and they are the only infants treated with ECMO that require surgery [128], which contributes to an inflammatory response [120].
According to McGahren et al. [129] ECMO-treated newborns with CDH had a higher incidence of adverse neurological sequelae than non-ECMO treated neonates with CDH.
There is only one study concerning neurodevelopmental outcome of patients treated with fetal TO. Cortes et al. [44] studied 16 CDH survivors, 7 of them were treated with fetal TO. They found no differences between groups in neuromotor, cognitive or composite neurodevelopmental outcomes at 1 year or 2 years adjusted age.
Few studies report on cognitive development of CDH children not treated with ECMO. Bouman et al. studied 11 CDH-survivors (age 8–12 years) and found a mean IQ of 85, while almost half of the children obtained IQ scores around or more than 1 standard deviation below normal. They did not find differences in intelligence, school level, or adaptive functioning between high-risk (hospitalized within 1 day after birth because of progressive respiratory insufficiency and ventilated for more than 10 days) and low-risk (other) children. The children showed significantly more emotional and behaviorial problems than expected and more children than expected had depressive problems [130]. It has to be noted, however, that the number of participants (n = 11) in this study was rather small.
Davenport et al. [117] reported that neurological examinations (including hearing and vision) showed no abnormalities in 19 of 23 patients treated for CDH (non-ECMO) at a median age of 56 months. Of the remaining four infants, two had a major disability and two a minor disability. None of them had developmental delays according to the Griffith’s mental developmental scales, suggesting that potential improvement in survival with delayed surgery does not occur at the expense of poor neurological outcome or developmental delay in survivors.
Using the Bayley Scales or the Stanford-Binet intelligence scale, Van Meurs et al. [33] tested 7 of the 15 CDH survivors (age 8 months-5 years) treated with ECMO normal, three were suspected to be delayed (score <90) and two were definitively delayed (score <70). Three children were unavailable for testing without reason. Similar results were reported by Chen et al. [131].
Predictive factors of neurological outcome
Not only use of ECMO can affect neurological outcome in CDH patients [129], but also the ECMO modality that has been chosen can affect neurological outcome. Standard ECMO modality for rescue of CDH patients has been venoarterial (VA) ECMO because circulatory support can be continued during ECMO [132]. Recent studies however have shown that venovenous (VV) ECMO is as efficient in supporting CDH patients as VA ECMO, while the rate of adverse neurologic sequela is lower [133, 134]. A decreased right internal carotid artery blood flow in patients treated with VA ECMO [135] might explain the fact that these patients have more neurological complications such as seizures and cerebral infarction, when compared to VV ECMO treated patients [134].
Several characteristics of CDH, the presentation of CDH and the clinical course, such as the need for a patch closure or gastrostomy tube are also significantly associated with neurological outcome [129]. These factors may reflect the severity of the presentation of CDH [129]. This was confirmed by D’Agostino et al. [136], they found a difference between children (tested at the age of 1 year) who had a primary diaphragmatic repair and those who needed patch closure. Children with primary repairs had mean mental and motor skills in the average range, whereas mean scores for those required patch closure were in the abnormal range. They also showed that 7 of the 13 infants with CDH, treated with ECMO had normal mental and motor skills, 3 had normal mental functioning but delayed acquisition of motor milestones, and 3 had mental and motor delay at the age of 12 months [136]. Eleven children had muscle tone abnormalities [136]. This study also contained a small-sample size and it remains unclear whether these findings are related to CDH, ECMO or to general management.
In summary, evidence indicates that not only children with CDH requiring ECMO are at risk for cognitive problems. As children with CDH not treated with ECMO appear to have a reduced level of intellectual and school functioning, all children with CDH may be at risk for cognitive delay. This could be due to several factors, which can interfere with central nervous system development, such as perinatal and postnatal hypoxia, hypercapnia, and acidosis. These are the most frequent problems in children born with CDH either in the immediate postnatal period or during the course of stay in the intensive care unit. Most studies focus only on the first 5 years after CDH repair. In addition the small sample size and the disparate results indicate that there is a need for further follow-up studies of children with CDH, because serious problems in long-term psychosocial functioning may be expected.
Hearing loss
Hearing loss has been a troubling and frequent finding and may be due, at least in part, to side effects of treatment. Treatment of newborns with persistent pulmonary hypertension used to consist of hyperventilatory induced alkalosis. These infants were exposed to significantly higher pH values for longer periods and needed longer durations of mechanical ventilation resulting in approximately 35% of children requiring hearing aids [110, 137, 138]. Hypoxemia, induction of alkalosis and use of ototoxic medication are believed to be important causal factors, but the specific insult that leads to progressive hearing loss remains unclear [139]. In addition to ototoxic medication and ventilatory treatment, ECMO therapy is an additional risk factor for hearing loss in CDH survivors [110, 122]. Lund et al. [32] found that hearing aids were required in 21% of CDH survivors treated with ECMO. Another 21% had abnormal brain auditory evoked response (BAER) or latency intensity studies (LIS) results, but hearing loss could not be confirmed by audiometry. Six of the seven children that required hearing aids had been treated with ECMO. D’Agostino et al. [136] described that 1 of the 16 studied children with CDH suffered hearing loss that was detected by brainstem auditory evoked response testing at 11 months of age.
In the report from Rasheed et al. [140], CDH patients requiring ECMO before surgery had a higher incidence of hearing loss when compared with those requiring ECMO after CDH repair, which might be secondary to the prolonged period of hyperventilation or general intensive care.
Morini et al. [141] reported that 40 of the 82 CDH survivors (non-ECMO) had sensineural hearing loss (SHNL) at audiologic follow-up at a median age of 3.0 years. Patient’s age at follow-up was the only independent predictor for SNHL.
It is recommended for CDH patients, especially when treated with ECMO, to undergo regular hearing tests in their preschool years, particularly if there is any delay in speech or language skill development [110].
Musculoskeletal abnormalities
Chest wall deformities
Chest wall deformities are present in approximately 16–48% of the CDH survivors. The majority of patients have a pectus excavatum [32, 59, 64, 68, 142]. The close relationship between development of the lung, diaphragm, and thoracic cage is the reason that deformities of the chest wall are more common in patients with CDH [32]. Several factors contribute to thoracic deformities. First of all the thoracic cavity on the affected side may be reduced in size due to the smaller lung volume. Secondly, the increased work of breathing in these children may contribute to the development of a pectus abnormality [32, 110] because more negative intrapleural pressure is required to inflate the lungs [142]. Thirdly more negative intrathoracic pressure promotes retraction of the chest wall in its most compliant section, the cartilaginous anterior wall [142]. Postoperative empyema has been shown to increase the likelihood of postoperative thoracic deformity [143, 144]. Since chest wall deformities are more common among patients with anatomically large diaphragmatic defects, it is suggested that repair of a large defect causes tension, interfering with normal development of the thoracic cage and promoting asymmetry and/or flat chest [142].
It has been suggested that CDH patients with a clinically asymmetric chest and scoliosis are prone for pulmonary function impairment [65, 68]. In other CDH patients with pectus excavatum, a reduced lung function compared to normal CDH patients has been demonstrated [70, 145]. It is, however, controversial whether a Nuss procedure results in an improvement in pulmonary function [145–147].
Scoliosis
Mild to moderate thoracic scoliosis has been reported in 5–10% of CDH-survivors [59, 63, 110]. In most of the studies, only small numbers of patients are included [59, 63, 110]. Lund et al. [32] described that 4 of 33 patients had thoracic scoliosis concave towards the hernia side, only one of them required bracing. The only study focusing primarily on chest wall deformities and scoliosis in CDH patients came from Vanamo et al. [142] describing sixteen (27%) patients with significant radiological scoliosis (Cobb’s angle ≥10°). The mean curve was significantly greater in patients with a large defect. There was no significant difference in the incidence of scoliosis between patients with a diaphragmatic hernia and those with eventration.
It can be concluded that chest wall deformity and spinal deformities are common among patients with repaired diaphragmatic defects [142]. Scoliosis should particularly be kept in mind when determining how much tension to use when closing a hernia defect [32]. In the majority, deformities are mild and rarely have therapeutic consequences or impact on daily life. However, as more children with severe diaphragmatic defects are expected to survive with use of new treatment modalities, number of patients with severe thoraco-spinal deformities may increase in the future [142].
Overall health-related quality of life
Although the prevalence of co-morbidity is high among CDH survivors, little is known about the impact of long-term health problems on overall well being of these patients. There are only two studies describing health-related quality of life (HR-QoL) of CDH patients. Poley et al assessed HR-QoL in 111 CDH patients (4 treated with ECMO), 286 patients with anorectal malformations (ARM) and a healthy control group (age 1–52 years). In children aged 1–4 years, influence of symptoms on HR-QoL, assessed with the TAIQOL (quality of life questionnaire developed for children between ages of 1 and 4 years [148]), seemed less profound in CDH patients when compared to ARM patients in most domains. Statistically significant differences between the CDH group and the control group were found in five domains of the TAIQOL.
CDH patients aged 5–15 years scored relatively low in the domains “basic motor functioning” and “cognitive functioning” in the TACQOL (quality of life questionnaire developed for children between the ages of 5 and 15) [149]. Neither the 8 domains of the widely used Short Form 36 (SF-36) domains, used for patients aged 16 years and above, nor the physical summary measure discriminated between CDH patients and the general population [150].
Koivusalo et al. [73] studied 69 adult survivors of congenital diaphragmatic defects (CDD) consisting of survivors of CDH and diaphragmatic eventration (DE) in comparison with a healthy control group. None of the patients with DE underwent preoperative stabilization with medication or mechanical ventilation, and only 15% required mechanical ventilation for more than 1 week postoperatively. The frequency of high educational level was significantly lower in patients with CDD than in control subjects, but whether this difference was related to long-term sequelae of CDD remained unclear. A total of 17 of the 69 patients (25%) with CDD had low HR-QoL assessed in one or both of the summary measures Physical Health and Mental Health in SF-36 questionnaires. No statistical difference in quality of life was found between patients with CDH and DE. Incidence of GERD and recurrent intestinal obstruction was most often associated with low HR-QoL and Gastro-Intestinal Quality of Life (GIQLI) scores in patients with CDD. This study indicates that 75% of adult patients born with CDD have good HR-QoL similar to healthy subjects. Practically no or insignificant late sequelae associated with CDD were found. Low HR-QoL was found in 25% of the patients, exceeding the expected figure of 16% [73].
It is likely that, in the past, major selection took place in the first week of life due to high mortality in patients with severe pulmonary hypoplasia and therapy resistant pulmonary hypertension. ECMO nowadays might enable survival of patients with a relatively poor prognosis leading to iatrogenic morbidity [150]. While CDH is associated with considerable symptoms, so far available research in adults suggests a favorable long-term outlook for the vast majority of patients [73, 150], however, there are no studies describing HR-QoL in CDH survivors treated with ECMO.
Conclusion
Despite new therapeutic strategies including ECMO, inhalation of nitric oxide, high frequency oscillation and fetal TO, mortality rate of CDH remains high. Several studies have documented significant long-term morbidity.
Infants born with CDH have pulmonary vascular abnormalities, presumably causing pulmonary hypertension. There is a higher incidence of obstructive airway disease and restrictive lung function pattern among CDH survivors. Gastro-esophageal reflux disease sometimes in combination with failure to thrive is a well recognized complication and several patients require anti-reflux surgery. It is unknown whether GERD has an effect on the pulmonary function.
Pulmonary hypoplasia and persistent pulmonary hypertension predispose children born with CDH to a high risk of hypoxemia, which may result in neurodevelopmental delay.
Chest wall deformities and scoliosis are more common among CDH patients. Deformities are mild and surgery is rarely required.
Despite the significant morbidity due to CDH, limited numbers of quality of life studies in older patients suggest that the majority of patients apparently enjoy quite healthy lives.
It should be realized that, with the introduction of potential mortality reducing new therapies in the acute (neonatal) phase, the balance between decreasing early mortality and potential long-term outcome should be assessed.
It is therefore be, highly recommended that children born with CDH are evaluated periodically in a protocolized multidisciplinary setting to minimize short-term morbidity and to assess long-term morbidity. Particularly the study of long-term sequelae will enable to establish an adequate and focused program for routine follow-up of survivors of a severe congenital anomaly.
Abbreviations
- ARM:
-
Ano-rectal malformations
- BAER:
-
Brain auditory evoked response
- CDD:
-
Congenital diaphragmatic defect
- CDH:
-
Congenital diaphragmatic hernia
- CT:
-
Computed tomography
- DE:
-
Diaphragmatic eventration
- DLCO:
-
Diffusion capacity for carbon monoxide
- ECMO:
-
Extra corporal membrane oxygenation
- FEV1:
-
Forced expiratory volume in 1 s
- FVC:
-
Forced vital capacity
- GERD:
-
Gastro-esophageal reflux disease
- GIQLI:
-
Gastro-intestinal quality of life index
- HFO:
-
High-frequency oscillation
- HR-QoL:
-
Health-related quality of life
- IQ:
-
Intelligence quotient
- LIS:
-
Latency intensity studies
- MRI:
-
Magnetic resonance imaging
- NO:
-
Nitric oxide
- RV:
-
Residual volume
- SF-36:
-
Short form-36
- SHNL:
-
Sensineural hearing loss
- TACQoL:
-
TNO AZL children’s quality of life
- TLC:
-
Total lung capacity
- TO:
-
Tracheal occlusion
- UGI:
-
Upper gastro-intestinal
- VA ECMO:
-
Venoarterial extracorporal membrane oxygenation
- VA:
-
Alveolar volume
- VC:
-
Vital capacity
- VO2:
-
Peak oxygen consumption
- VV ECMO:
-
Venovenous extracorporal membrane oxygenation
References
Colvin J, Bower C, Dickinson JE, Sokol J (2005) Outcomes of congenital diaphragmatic hernia: a population-based study in Western Australia. Pediatrics 116(3):e356–e363. doi:10.1542/peds.2004-2845
Stege G, Fenton A, Jaffray B (2003) Nihilism in the 1990s: the true mortality of congenital diaphragmatic hernia. Pediatrics 112(3 Pt 1):532–535. doi:10.1542/peds.112.3.532
Vanamo K (1996) A 45-year perspective of congenital diaphragmatic hernia. Br J Surg 83(12):1758–1762. doi:10.1002/bjs.1800831231
Azarow K, Messineo A, Pearl R, Filler R, Barker G, Bohn D (1997) Congenital diaphragmatic hernia—a tale of two cities: the Toronto experience. J Pediatr Surg 32(3):395–400. doi:10.1016/S0022-3468(97)90589-3
Bos AP, Tibboel D, Koot VC, Hazebroek FW, Molenaar JC (1993) Persistent pulmonary hypertension in high-risk congenital diaphragmatic hernia patients: incidence and vasodilator therapy. J Pediatr Surg 28(11):1463–1465
Bohn D, Tamura M, Perrin D, Barker G, Rabinovitch M (1987) Ventilatory predictors of pulmonary hypoplasia in congenital diaphragmatic hernia, confirmed by morphologic assessment. J Pediatr 111(3):423–431. doi:10.1016/S0022-3476(87)80474-2
Rygl M, Pycha K, Stranak Z, Melichar J, Krofta L, Tomasek L, Snajdauf J (2007) Congenital diaphragmatic hernia: onset of respiratory distress and size of the defect: analysis of the outcome in 104 neonates. Pediatr Surg Int 23(1):27–31. doi:10.1007/s00383-006-1788-3
Fauza DO, Wilson JM (1994) Congenital diaphragmatic hernia and associated anomalies: their incidence, identification, and impact on prognosis. J Pediatr Surg 29(8):1113–1117. doi:10.1016/0022-3468(94)90290-9
Huddy CL, Boyd PA, Wilkinson AR, Chamberlain P (1999) Congenital diaphragmatic hernia: prenatal diagnosis, outcome and continuing morbidity in survivors. Br J Obstet Gynaecol 106(11):1192–1196
Bagolan P, Casaccia G, Crescenzi F, Nahom A, Trucchi A, Giorlandino C (2004) Impact of a current treatment protocol on outcome of high-risk congenital diaphragmatic hernia. J Pediatr Surg 39(3):313–318. doi:10.1016/j.jpedsurg.2003.11.009
Beresford MW, Shaw NJ (2000) Outcome of congenital diaphragmatic hernia. Pediatr Pulmonol 30(3):249–256. doi:10.1002/1099-0496(200009)30:3<249::AID-PPUL9>3.0.CO;2-9
(2001) Estimating disease severity of congenital diaphragmatic hernia in the first 5 minutes of life. The congenital diaphragmatic hernia study group. J Pediatr Surg 36(1):141–145. doi:10.1053/jpsu.2001.20032
Graziano JN (2005) Cardiac anomalies in patients with congenital diaphragmatic hernia and their prognosis: a report from the congenital diaphragmatic hernia study group. J Pediatr Surg 40(6):1045–1049. doi:10.1016/j.jpedsurg.2005.03.025
Langer JC, Filler RM, Bohn DJ, Shandling B, Ein SH, Wesson DE, Superina RA (1988) Timing of surgery for congenital diaphragmatic hernia: is emergency operation necessary? J Pediatr Surg 23(8):731–734. doi:10.1016/S0022-3468(88)80413-5
Geggel RL, Murphy JD, Langleben D, Crone RK, Vacanti JP, Reid LM (1985) Congenital diaphragmatic hernia: arterial structural changes and persistent pulmonary hypertension after surgical repair. J Pediatr 107(3):457–464. doi:10.1016/S0022-3476(85)80534-5
Sakai H, Tamura M, Hosokawa Y, Bryan AC, Barker GA, Bohn DJ (1987) Effect of surgical repair on respiratory mechanics in congenital diaphragmatic hernia. J Pediatr 111(3):432–438. doi:10.1016/S0022-3476(87)80475-4
Vacanti JP, O’Rourke PP, Lillehei CW, Crone RK (1988) The cardiopulmonary consequences of high-risk congenital diaphragmatic hernia. Pediatr Surg Int 3:1–5. doi:10.1007/BF00177071
Charlton AJ, Bruce J, Davenport M (1991) Timing of surgery in congenital diaphragmatic hernia. Low mortality after pre-operative stabilisation. Anaesthesia 46(10):820–823. doi:10.1111/j.1365-2044.1991.tb09592.x
Boloker J, Bateman DA, Wung JT, Stolar CJ (2002) Congenital diaphragmatic hernia in 120 infants treated consecutively with permissive hypercapnea/spontaneous respiration/elective repair. J Pediatr Surg 37(3):357–366. doi:10.1053/jpsu.2002.30834
Moyer V, Moya F, Tibboel R, Losty P, Nagaya M, Lally KP (2002) Late versus early surgical correction for congenital diaphragmatic hernia in newborn infants. Cochrane Database Syst Rev (3):CD001695
Wilson JM, Lund DP, Lillehei CW, O’Rourke PP, Vacanti JP (1992) Delayed repair and preoperative ECMO does not improve survival in high-risk congenital diaphragmatic hernia. J Pediatr Surg 27(3):368–372. doi:10.1016/0022-3468(92)90863-3
Nio M, Haase G, Kennaugh J, Bui K, Atkinson JB (1994) A prospective randomized trial of delayed versus immediate repair of congenital diaphragmatic hernia. J Pediatr Surg 29(5):618–621
Clark RH, Hardin WD Jr, Hirschl RB, Jaksic T, Lally KP, Langham MR Jr, Wilson JM (1998) Current surgical management of congenital diaphragmatic hernia: a report from the congenital diaphragmatic hernia study group. J Pediatr Surg 33(7):1004–1009. doi:10.1016/S0022-3468(98)90522-X
Hazebroek FW, Tibboel D, Bos AP, Pattenier AW, Madern GC, Bergmeijer JH, Molenaar JC (1988) Congenital diaphragmatic hernia: impact of preoperative stabilization. A prospective pilot study in 13 patients. J Pediatr Surg 23(12):1139–1146. doi:10.1016/S0022-3468(88)80330-0
(1999) Does extracorporeal membrane oxygenation improve survival in neonates with congenital diaphragmatic hernia? The congenital diaphragmatic hernia study group. J Pediatr Surg 34(5):720–724. doi:10.1016/S0022-3468(99)90363-9
Breaux CW Jr, Rouse TM, Cain WS, Georgeson KE (1991) Improvement in survival of patients with congenital diaphragmatic hernia utilizing a strategy of delayed repair after medical and/or extracorporeal membrane oxygenation stabilization. J Pediatr Surg 26(3):333–336. doi:10.1016/0022-3468(91)90512-R
Keshen TH, Gursoy M, Shew SB, Smith EO, Miller RG, Wearden ME, Moise AA, Jaksic T (1997) Does extracorporeal membrane oxygenation benefit neonates with congenital diaphragmatic hernia? Application of a predictive equation. J Pediatr Surg 32(6):818–822. doi:10.1016/S0022-3468(97)90627-8
West KW, Bengston K, Rescorla FJ, Engle WA, Grosfeld JL (1992) Delayed surgical repair and ECMO improves survival in congenital diaphragmatic hernia. Ann Surg 216(4):454–460. doi:10.1097/00000658-199210000-00009
Johnston PW, Bashner B, Liberman R, Gangitano E, Vogt J (1988) Clinical use of extracorporeal membrane oxygenation in the treatment of persistent pulmonary hypertension following surgical repair of congenital diaphragmatic hernia. J Pediatr Surg 23(10):908–912. doi:10.1016/S0022-3468(88)80382-8
Pusic AL, Giacomantonio M, Pippus K, Rees E, Gillis DA (1995) Survival in neonatal congenital hernia without extracorporeal membrane oxygenation support. J Pediatr Surg 30(8):1188–1190. doi:10.1016/0022-3468(95)90018-7
Schoeman L, Pierro A, Macrae D, Spitz L, Kiely EM, Drake DP (1999) Late death after extracorporeal membrane oxygenation for congenital diaphragmatic hernia. J Pediatr Surg 34(2):357–359. doi:10.1016/S0022-3468(99)90209-9
Lund DP, Mitchell J, Kharasch V, Quigley S, Kuehn M, Wilson JM (1994) Congenital diaphragmatic hernia: the hidden morbidity. J Pediatr Surg 29(2):258–262
Van Meurs KP, Robbins ST, Reed VL, Karr SS, Wagner AE, Glass P, Anderson KD, Short BL (1993) Congenital diaphragmatic hernia: long-term outcome in neonates treated with extracorporeal membrane oxygenation. J Pediatr 122(6):893–899
Alcorn D, Adamson TM, Lambert TF, Maloney JE, Ritchie BC, Robinson PM (1977) Morphological effects of chronic tracheal ligation and drainage in the fetal lamb lung. J Anat 123(Pt 3):649–660
Nardo L, Hooper SB, Harding R (1995) Lung hypoplasia can be reversed by short-term obstruction of the trachea in fetal sheep. Pediatr Res 38(5):690–696. doi:10.1203/00006450-199511000-00010
Wilson JM, DiFiore JW, Peters CA (1993) Experimental fetal tracheal ligation prevents the pulmonary hypoplasia associated with fetal nephrectomy: possible application for congenital diaphragmatic hernia. J Pediatr Surg 28(11):1433–1439. doi:10.1016/0022-3468(93)90426-L
DiFiore JW, Fauza DO, Slavin R, Peters CA, Fackler JC, Wilson JM (1994) Experimental fetal tracheal ligation reverses the structural and physiological effects of pulmonary hypoplasia in congenital diaphragmatic hernia. J Pediatr Surg 29(2):248–256. doi:10.1016/0022-3468(94)90328-X
Hedrick MH, Estes JM, Sullivan KM, Bealer JF, Kitterman JA, Flake AW, Adzick NS, Harrison MR (1994) Plug the lung until it grows (PLUG): a new method to treat congenital diaphragmatic hernia in utero. J Pediatr Surg 29(5):612–617
Wild YK, Piasecki GJ, De Paepe ME, Luks FI (2000) Short-term tracheal occlusion in fetal lambs with diaphragmatic hernia improves lung function, even in the absence of lung growth. J Pediatr Surg 35(5):775–779. doi:10.1053/jpsu.2000.6067
Luks FI, Wild YK, Piasecki GJ, De Paepe ME (2000) Short-term tracheal occlusion corrects pulmonary vascular anomalies in the fetal lamb with diaphragmatic hernia. Surgery 128(2):266–272. doi:10.1067/msy.2000.107373
Saura L, Castanon M, Prat J, Albert A, Caceres F, Moreno J, Gratacos E (2007) Impact of fetal intervention on postnatal management of congenital diaphragmatic hernia. Eur J Pediatr Surg 17(6):404–407. doi:10.1055/s-2007-989275
Harrison MR, Keller RL, Hawgood SB, Kitterman JA, Sandberg PL, Farmer DL, Lee H, Filly RA, Farrell JA, Albanese CT (2003) A randomized trial of fetal endoscopic tracheal occlusion for severe fetal congenital diaphragmatic hernia. N Engl J Med 349(20):1916–1924
Keller RL, Hawgood S, Neuhaus JM, Farmer DL, Lee H, Albanese CT, Harrison MR, Kitterman JA (2004) Infant pulmonary function in a randomized trial of fetal tracheal occlusion for severe congenital diaphragmatic hernia. Pediatr Res 56(5):818–825. doi:10.1203/01.PDR.0000141518.19721.D7
Cortes RA, Keller RL, Townsend T, Harrison MR, Farmer DL, Lee H, Piecuch RE, Leonard CH, Hetherton M, Bisgaard R, Nobuhara KK (2005) Survival of severe congenital diaphragmatic hernia has morbid consequences. J Pediatr Surg 40(1):36–45. doi:10.1016/j.jpedsurg.2004.09.037
Beals DA, Schloo BL, Vacanti JP, Reid LM, Wilson JM (1992) Pulmonary growth and remodeling in infants with high-risk congenital diaphragmatic hernia. J Pediatr Surg 27(8):997–1001. doi:10.1016/0022-3468(92)90546-J
Hislop A, Reid L (1976) Persistent hypoplasia of the lung after repair of congenital diaphragmatic hernia. Thorax 31(4):450–455
Reid LM (1984) Lung growth in health and disease. Br J Dis Chest 78(2):113–134. doi:10.1016/S0007-0971(84)80092-3
Falconer AR, Brown RA, Helms P, Gordon I, Baron JA (1990) Pulmonary sequelae in survivors of congenital diaphragmatic hernia. Thorax 45(2):126–129
Sakurai Y, Azarow K, Cutz E, Messineo A, Pearl R, Bohn D (1999) Pulmonary barotrauma in congenital diaphragmatic hernia: a clinicopathological correlation. J Pediatr Surg 34(12):1813–1817. doi:10.1016/S0022-3468(99)90319-6
Dreyfuss D, Basset G, Soler P, Saumon G (1985) Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. Am Rev Respir Dis 132(4):880–884
Ito Y, Veldhuizen RA, Yao LJ, McCaig LA, Bartlett AJ, Lewis JF (1997) Ventilation strategies affect surfactant aggregate conversion in acute lung injury. Am J Respir Crit Care Med 155(2):493–499
Wilson JM, Lund DP, Lillehei CW, Vacanti JP (1997) Congenital diaphragmatic hernia—a tale of two cities: the Boston experience. J Pediatr Surg 32(3):401–405. doi:10.1016/S0022-3468(97)90590-X
Ng GY, Derry C, Marston L, Choudhury M, Holmes K, Calvert SA (2008) Reduction in ventilator-induced lung injury improves outcome in congenital diaphragmatic hernia? Pediatr Surg Int 24(2):145–150. doi:10.1007/s00383-007-2051-2
Hamutcu R, Nield TA, Garg M, Keens TG, Platzker AC (2004) Long-term pulmonary sequelae in children who were treated with extracorporeal membrane oxygenation for neonatal respiratory failure. Pediatrics 114(5):1292–1296. doi:10.1542/peds.2003-1080-L
Boykin AR, Quivers ES, Wagenhoffer KL, Sable CA, Chaney HR, Glass P, Bahrami KR, Short BL (2003) Cardiopulmonary outcome of neonatal extracorporeal membrane oxygenation at ages 10–15 years. Crit Care Med 31(9):2380–2384. doi:10.1097/01.CCM.0000084856.00372.CE
Beardsmore C, Dundas I, Poole K, Enock K, Stocks J (2000) Respiratory function in survivors of the United Kingdom extracorporeal membrane oxygenation trial. Am J Respir Crit Care Med 161(4 Pt 1):1129–1135
Benachi A, Chailley-Heu B, Delezoide AL, Dommergues M, Brunelle F, Dumez Y, Bourbon JR (1998) Lung growth and maturation after tracheal occlusion in diaphragmatic hernia. Am J Respir Crit Care Med 157(3 Pt 1):921–927
Laberge JM (2008) Fetal tracheal occlusion for the treatment of congenital diaphragmatic hernia. World J Surg 32(10):2329
Wischermann A, Holschneider AM, Hubner U (1995) Long-term follow-up of children with diaphragmatic hernia. Eur J Pediatr Surg 5(1):13–18
Kamata S, Usui N, Kamiyama M, Tazuke Y, Nose K, Sawai T, Fukuzawa M (2005) Long-term follow-up of patients with high-risk congenital diaphragmatic hernia. J Pediatr Surg 40(12):1833–1838. doi:10.1016/j.jpedsurg.2005.08.022
Crankson SJ, Al Jadaan SA, Namshan MA, Al Rabeeah AA, Oda O (2006) The immediate and long-term outcomes of newborns with congenital diaphragmatic hernia. Pediatr Surg Int 22(4):335–340
Muratore CS, Kharasch V, Lund DP, Sheils C, Friedman S, Brown C, Utter S, Jaksic T, Wilson JM (2001) Pulmonary morbidity in 100 survivors of congenital diaphragmatic hernia monitored in a multidisciplinary clinic. J Pediatr Surg 36(1):133–140. doi:10.1053/jpsu.2001.20031
Stefanutti G, Filippone M, Tommasoni N, Midrio P, Zucchetta P, Moreolo GS, Toffolutti T, Baraldi E, Gamba P (2004) Cardiopulmonary anatomy and function in long-term survivors of mild to moderate congenital diaphragmatic hernia. J Pediatr Surg 39(4):526–531. doi:10.1016/j.jpedsurg.2003.12.006
Arena F, Baldari S, Centorrino A, Calabro MP, Pajino G, Arena S, Ando F, Zuccarello B, Romeo G (2005) Mid- and long-term effects on pulmonary perfusion, anatomy and diaphragmatic motility in survivors of congenital diaphragmatic hernia. Pediatr Surg Int 21(12):954–959
Vanamo K, Rintala R, Sovijarvi A, Jaaskelainen J, Turpeinen M, Lindahl H, Louhimo I (1996) Long-term pulmonary sequelae in survivors of congenital diaphragmatic defects. J Pediatr Surg 31(8):1096–1099. doi:10.1016/S0022-3468(96)90095-0
Marven SS, Smith CM, Claxton D, Chapman J, Davies HA, Primhak RA, Powell CV (1998) Pulmonary function, exercise performance, and growth in survivors of congenital diaphragmatic hernia. Arch Dis Child 78(2):137–142
Zaccara A, Turchetta A, Calzolari A, Iacobelli B, Nahom A, Lucchetti MC, Bagolan P, Rivosecchi M, Coran AG (1996) Maximal oxygen consumption and stress performance in children operated on for congenital diaphragmatic hernia. J Pediatr Surg 31(8):1092–1094. doi:10.1016/S0022-3468(96)90094-9
Trachsel D, Selvadurai H, Bohn D, Langer JC, Coates AL (2005) Long-term pulmonary morbidity in survivors of congenital diaphragmatic hernia. Pediatr Pulmonol 39(5):433–439. doi:10.1002/ppul.20193
Ijsselstijn H, Tibboel D, Hop WJ, Molenaar JC, de Jongste JC (1997) Long-term pulmonary sequelae in children with congenital diaphragmatic hernia. Am J Respir Crit Care Med 155(1):174–180
Koumbourlis AC, Stolar CJ (2004) Lung growth and function in children and adolescents with idiopathic pectus excavatum. Pediatr Pulmonol 38(4):339–343. doi:10.1002/ppul.20062
Nagaya M, Akatsuka H, Kato J, Niimi N, Ishiguro Y (1996) Development in lung function of the affected side after repair of congenital diaphragmatic hernia. J Pediatr Surg 31(3):349–356. doi:10.1016/S0022-3468(96)90737-X
Trachsel D, Selvadurai H, Adatia I, Bohn D, Schneiderman-Walker J, Wilkes D, Coates AL (2006) Resting and exercise cardiorespiratory function in survivors of congenital diaphragmatic hernia. Pediatr Pulmonol 41(6):522–529. doi:10.1002/ppul.20359
Koivusalo A, Pakarinen M, Vanamo K, Lindahl H, Rintala RJ (2005) Health-related quality of life in adults after repair of congenital diaphragmatic defects—a questionnaire study. J Pediatr Surg 40(9):1376–1381. doi:10.1016/j.jpedsurg.2005.05.037
Hislop A, Reid L (1973) Pulmonary arterial development during childhood: branching pattern and structure. Thorax 28(2):129–135
deMello DE, Sawyer D, Galvin N, Reid LM (1997) Early fetal development of lung vasculature. Am J Respir Cell Mol Biol 16(5):568–581
Shehata SM, Tibboel D, Sharma HS, Mooi WJ (1999) Impaired structural remodelling of pulmonary arteries in newborns with congenital diaphragmatic hernia: a histological study of 29 cases. J Pathol 189(1):112–118. doi:10.1002/(SICI)1096-9896(199909)189:1<112::AID-PATH395>3.0.CO;2-8
Shehata SM, Sharma HS, van der Staak FH, van de Kaa-Hulsbergen C, Mooi WJ, Tibboel D (2000) Remodeling of pulmonary arteries in human congenital diaphragmatic hernia with or without extracorporeal membrane oxygenation. J Pediatr Surg 35(2):208–215. doi:10.1016/S0022-3468(00)90011-3
Roubliova X, Verbeken E, Wu J, Yamamoto H, Lerut T, Tibboel D, Deprest J (2004) Pulmonary vascular morphology in a fetal rabbit model for congenital diaphragmatic hernia. J Pediatr Surg 39(7):1066–1072. doi:10.1016/j.jpedsurg.2004.03.049
Yamataka T, Puri P (1997) Pulmonary artery structural changes in pulmonary hypertension complicating congenital diaphragmatic hernia. J Pediatr Surg 32(3):387–390. doi:10.1016/S0022-3468(97)90587-X
Taira Y, Yamataka T, Miyazaki E, Puri P (1998) Adventitial changes in pulmonary vasculature in congenital diaphragmatic hernia complicated by pulmonary hypertension. j pediatr surg 33(2):382–387. doi:10.1016/S0022-3468(98)90468-7
Tenbrinck R, Gaillard JL, Tibboel D, Kluth D, Lachmann B, Molenaar JC (1992) Pulmonary vascular abnormalities in experimentally induced congenital diaphragmatic hernia in rats. J Pediatr Surg 27(7):862–865. doi:10.1016/0022-3468(92)90385-K
Thibeault DW, Haney B (1998) Lung volume, pulmonary vasculature, and factors affecting survival in congenital diaphragmatic hernia. Pediatrics 101(2):289–295. doi:10.1542/peds.101.2.289
Kanai M, Kitano Y, von Allmen D, Davies P, Adzick NS, Flake AW (2001) Fetal tracheal occlusion in the rat model of nitrofen-induced congenital diaphragmatic hernia: tracheal occlusion reverses the arterial structural abnormality. J Pediatr Surg 36(6):839–845. doi:10.1053/jpsu.2001.23950
Ijsselstijn H, Tibboel D (1998) The lungs in congenital diaphragmatic hernia: do we understand? Pediatr Pulmonol 26(3):204–218. doi:10.1002/(SICI)1099-0496(199809)26:3<204::AID-PPUL8>3.0.CO;2-K
Okazaki T, Sharma HS, McCune SK, Tibboel D (1998) Pulmonary vascular balance in congenital diaphragmatic hernia: enhanced endothelin-1 gene expression as a possible cause of pulmonary vasoconstriction. J Pediatr Surg 33(1):81–84. doi:10.1016/S0022-3468(98)90367-0
Fitzgerald D, Evans N, Van Asperen P, Henderson-Smart D (1994) Subclinical persisting pulmonary hypertension in chronic neonatal lung disease. Arch Dis Child Fetal Neonatal Ed 70(2):F118–F122
Dillon PW, Cilley RE, Mauger D, Zachary C, Meier A (2004) The relationship of pulmonary artery pressure and survival in congenital diaphragmatic hernia. J Pediatr Surg 39(3):307–312. doi:10.1016/j.jpedsurg.2003.11.010
Schwartz IP, Bernbaum JC, Rychik J, Grunstein M, D’Agostino J, Polin RA (1999) Pulmonary hypertension in children following extracorporeal membrane oxygenation therapy and repair of congenital diaphragmatic hernia. J Perinatol 19(3):220–226. doi:10.1038/sj.jp.7200141
Iocono JA, Cilley RE, Mauger DT, Krummel TM, Dillon PW (1999) Postnatal pulmonary hypertension after repair of congenital diaphragmatic hernia: predicting risk and outcome. J Pediatr Surg 34(2):349–353. doi:10.1016/S0022-3468(99)90207-5
Peetsold MG, Vonk-Noordegraaf A, Heij HH, Gemke RJ (2007) Pulmonary function and exercise testing in adult survivors of congenital diaphragmatic hernia. Pediatr Pulmonol 42(4):325–331. doi:10.1002/ppul.20579
Sun XG, Hansen JE, Oudiz RJ, Wasserman K (2001) Exercise pathophysiology in patients with primary pulmonary hypertension. Circulation 104(4):429–435
Yasunobu Y, Oudiz RJ, Sun XG, Hansen JE, Wasserman K (2005) End-tidal PCO2 abnormality and exercise limitation in patients with primary pulmonary hypertension. Chest 127(5):1637–1646. doi:10.1378/chest.127.5.1637
Fasching G, Huber A, Uray E, Sorantin E, Lindbichler F, Mayr J (2000) Gastro esophageal reflux and diaphragmatic motility after repair of congenital diaphragmatic hernia. Eur J Pediatr Surg 10(6):360–364. doi:10.1055/s-2000-12060
Nagaya M, Akatsuka H, Kato J (1994) Gastro esophageal reflux occurring after repair of congenital diaphragmatic hernia. J Pediatr Surg 29(11):1447–1451. doi:10.1016/0022-3468(94)90141-4
Sigalet DL, Nguyen LT, Adolph V, Laberge JM, Hong AR, Guttman FM (1994) Gastro esophageal reflux associated with large diaphragmatic hernias. J Pediatr Surg 29(9):1262–1265. doi:10.1016/0022-3468(94)90819-2
Kieffer J, Sapin E, Berg A, Beaudoin S, Bargy F, Helardot PG (1995) Gastro esophageal reflux after repair of congenital diaphragmatic hernia. J Pediatr Surg 30(9):1330–1333. doi:10.1016/0022-3468(95)90497-2
Qi B, Soto C, Diez-Pardo JA, Tovar JA (1997) An experimental study on the pathogenesis of gastro esophageal reflux after repair of diaphragmatic hernia. J Pediatr Surg 32(9):1310–1313. doi:10.1016/S0022-3468(97)90309-2
Koot VC, Bergmeijer JH, Bos AP, Molenaar JC (1993) Incidence and management of gastro esophageal reflux after repair of congenital diaphragmatic hernia. J Pediatr Surg 28(1):48–52. doi:10.1016/S0022-3468(05)80353-7
Muratore CS, Utter S, Jaksic T, Lund DP, Wilson JM (2001) Nutritional morbidity in survivors of congenital diaphragmatic hernia. J Pediatr Surg 36(8):1171–1176. doi:10.1053/jpsu.2001.25746
Su W, Berry M, Puligandla PS, Aspirot A, Flageole H, Laberge JM (2007) Predictors of gastro esophageal reflux in neonates with congenital diaphragmatic hernia. J Pediatr Surg 42(10):1639–1643. doi:10.1016/j.jpedsurg.2007.05.016
Steven MJ, Fyfe AH, Raine PA, Watt I (2007) Esophageal adenocarcinoma: a long-term complication of congenital diaphragmatic hernia? J Pediatr Surg 42(7):E1–E3. doi:10.1016/j.jpedsurg.2007.04.026
Petersen H, Johannessen T, Sandvik AK, Kleveland PM, Brenna E, Waldum H, Dybdahl JD (1991) Relationship between endoscopic hiatus hernia and gastro esophageal reflux symptoms. Scand J Gastroenterol 26(9):921–926. doi:10.3109/00365529108996243
Spechler SJ (1992) Epidemiology and natural history of gastro-oesophageal reflux disease. Digestion 51(Suppl 1):24–29
Wienbeck M, Barnert J (1989) Epidemiology of reflux disease and reflux esophagitis. Scand J Gastroenterol Suppl 156:7–13. doi:10.3109/00365528909091032
Vanamo K, Rintala RJ, Lindahl H, Louhimo I (1996) Long-term gastrointestinal morbidity in patients with congenital diaphragmatic defects. J Pediatr Surg 31(4):551–554. doi:10.1016/S0022-3468(96)90494-7
Kamiyama M, Kawahara H, Okuyama H, Oue T, Kuroda S, Kubota A, Okada A (2002) Gastro esophageal reflux after repair of congenital diaphragmatic hernia. J Pediatr Surg 37(12):1681–1684. doi:10.1053/jpsu.2002.36693
Diamond IR, Mah K, Kim PC, Bohn D, Gerstle JT, Wales PW (2007) Predicting the need for fundoplication at the time of congenital diaphragmatic hernia repair. J Pediatr Surg 42(6):1066–1070. doi:10.1016/j.jpedsurg.2007.01.046
Jaillard SM, Pierrat V, Dubois A, Truffert P, Lequien P, Wurtz AJ, Storme L (2003) Outcome at 2 years of infants with congenital diaphragmatic hernia: a population-based study. Ann Thorac Surg 75(1):250–256. doi:10.1016/S0003-4975(02)04278-9
Baoquan Q, Diez-Pardo JA, Tovar JA (1995) Intestinal rotation in experimental congenital diaphragmatic hernia. J Pediatr Surg 30(10):1457–1462. doi:10.1016/0022-3468(95)90407-7
Nobuhara KK, Lund DP, Mitchell J, Kharasch V, Wilson JM (1996) Long-term outlook for survivors of congenital diaphragmatic hernia. Clin Perinatol 23(4):873–887
Festen C (1982) Postoperative small bowel obstruction in infants and children. Ann Surg 196(5):580–583. doi:10.1097/00000658-198211000-00012
Janik JS, Ein SH, Filler RM, Shandling B, Simpson JS, Stephens CA (1981) An assessment of the surgical treatment of adhesive small bowel obstruction in infants and children. J Pediatr Surg 16(3):225–235. doi:10.1016/S0022-3468(81)80669-0
Choudhry MS, Grant HW (2006) Small bowel obstruction due to adhesions following neonatal laparotomy. Pediatr Surg Int 22(9):729–732. doi:10.1007/s00383-006-1719-3
van Eijck FC, Wijnen RM, van Goor H (2008) The incidence and morbidity of adhesions after treatment of neonates with gastroschisis and omphalocele: a 30-year review. J Pediatr Surg 43(3):479–483. doi:10.1016/j.jpedsurg.2007.10.027
Abrams SA (2001) Chronic pulmonary insufficiency in children and its effects on growth and development. J Nutr 131(3):938S–941S
Chye JK, Gray PH (1995) Rehospitalization and growth of infants with bronchopulmonary dysplasia: a matched control study. J Paediatr Child Health 31(2):105–111. doi:10.1111/j.1440-1754.1995.tb00756.x
Davenport M, Rivlin E, D’Souza SW, Bianchi A (1992) Delayed surgery for congenital diaphragmatic hernia: neurodevelopmental outcome in later childhood. Arch Dis Child 67(11):1353–1356
Bifano EM, Pfannenstiel A (1988) Duration of hyperventilation and outcome in infants with persistent pulmonary hypertension. Pediatrics 81(5):657–661
Stolar CJ, Reyes C (1988) Extracorporeal membrane oxygenation causes significant changes in intracranial pressure and carotid artery blood flow in newborn lambs. J Pediatr Surg 23(12):1163–1168. doi:10.1016/S0022-3468(88)80334-8
Kudoh A, Katagai H, Takazawa T, Matsuki A (2001) Plasma proinflammatory cytokine response to surgical stress in elderly patients. Cytokine 15(5):270–273
Hunt RW, Kean MJ, Stewart MJ, Inder TE (2004) Patterns of cerebral injury in a series of infants with congenital diaphragmatic hernia utilizing magnetic resonance imaging. J Pediatr Surg 39(1):31–36. doi:10.1016/j.jpedsurg.2003.09.005
Glass P, Miller M, Short BL (1989) Morbidity for survivors of extracorporal membrane oxygenation: neurodevelopmental outcome at 1 year of age. Pediatrics 83(1):72–78
Schumacher RE, Palmer TW, Roloff DW, LaClaire PA, Bartlett RH (1991) Follow-up of infants treated with extracorporeal membrane oxygenation for newborn respiratory failure. Pediatrics 87(4):451–457
Krummel TM, Greenfield LJ, Kirkpatrick BV, Mueller DG, Kerkering KW, Ormazabal M, Myer EC, Barnes RW, Salzberg AM (1984) The early evaluation of survivors after extracorporeal membrane oxygenation for neonatal pulmonary failure. J Pediatr Surg 19(5):585–590. doi:10.1016/S0022-3468(84)80110-4
Towne BH, Lott IT, Hicks DA, Healey T (1985) Long-term follow-up of infants and children treated with extracorporeal membrane oxygenation (ECMO): a preliminary report. J Pediatr Surg 20(4):410–414. doi:10.1016/S0022-3468(85)80230-X
Andrews AF, Nixon CA, Cilley RE, Roloff DW, Bartlett RH (1986) One- to three-year outcome for 14 neonatal survivors of extracorporeal membrane oxygenation. Pediatrics 78(4):692–698
(1998) The collaborative UK ECMO (extracorporeal membrane oxygenation) trial: follow-up to 1 year of age. Pediatrics 101(4):E1. doi:10.1542/peds.101.4.e1
Stolar CJ, Crisafi MA, Driscoll YT (1995) Neurocognitive outcome for neonates treated with extracorporeal membrane oxygenation: are infants with congenital diaphragmatic hernia different? J Pediatr Surg 30(2):366–371. doi:10.1016/0022-3468(95)90591-X
McGahren ED, Mallik K, Rodgers BM (1997) Neurological outcome is diminished in survivors of congenital diaphragmatic hernia requiring extracorporeal membrane oxygenation. J Pediatr Surg 32(8):1216–1220. doi:10.1016/S0022-3468(97)90685-0
Bouman NH, Koot HM, Tibboel D, Hazebroek FW (2000) Children with congenital diaphragmatic hernia are at risk for lower levels of cognitive functioning and increased emotional and behavioral problems. Eur J Pediatr Surg 10(1):3–7
Chen C, Friedman S, Butler S, Jeruss S, Terrin N, Tighiouart H, Ware J, Wilson JM, Parsons SK (2007) Approaches to neurodevelopmental assessment in congenital diaphragmatic hernia survivors. J Pediatr Surg 42(6):1052–1056. doi:10.1016/j.jpedsurg.2007.01.042
Rothenbach P, Lange P, Powell D (2005) The use of extracorporeal membrane oxygenation in infants with congenital diaphragmatic hernia. Semin Perinatol 29(1):40–44. doi:10.1053/j.semperi.2005.02.006
Kugelman A, Gangitano E, Pincros J, Tantivit P, Taschuk R, Durand M (2003) Venovenous versus venoarterial extracorporeal membrane oxygenation in congenital diaphragmatic hernia. J Pediatr Surg 38(8):1131–1136. doi:10.1016/S0022-3468(03)00256-2
Dimmitt RA, Moss RL, Rhine WD, Benitz WE, Henry MC, Vanmeurs KP (2001) Venoarterial versus venovenous extracorporeal membrane oxygenation in congenital diaphragmatic hernia: the extracorporeal life support organization registry, 1990–1999. J Pediatr Surg 36(8):1199–1204. doi:10.1053/jpsu.2001.25762
Lott IT, McPherson D, Towne B, Johnson D, Starr A (1990) Long-term neurophysiologic outcome after neonatal extracorporeal membrane oxygenation. J Pediatr 116(3):343–349. doi:10.1016/S0022-3476(05)82818-5
D’Agostino JA, Bernbaum JC, Gerdes M, Schwartz IP, Coburn CE, Hirschl RB, Baumgart S, Polin RA (1995) Outcome for infants with congenital diaphragmatic hernia requiring extracorporeal membrane oxygenation: the first year. J Pediatr Surg 30(1):10–15. doi:10.1016/0022-3468(95)90598-7
Hendricks-Munoz KD, Walton JP (1988) Hearing loss in infants with persistent fetal circulation. Pediatrics 81(5):650–656
Walton JP, Hendricks-Munoz K (1991) Profile and stability of sensorineural hearing loss in persistent pulmonary hypertension of the newborn. J Speech Hear Res 34(6):1362–1370
Lasky RE, Wiorek L, Becker TR (1998) Hearing loss in survivors of neonatal extracorporeal membrane oxygenation (ECMO) therapy and high-frequency oscillatory (HFO) therapy. J Am Acad Audiol 9(1):47–58
Rasheed A, Tindall S, Cueny DL, Klein MD, Aney-Black V (2001) Neurodevelopmental outcome after congenital diaphragmatic hernia: extracorporeal membrane oxygenation before and after surgery. J Pediatr Surg 36(4):539–544
Morini F, Capolupo I, Masi R, Ronchetti MP, Locatelli M, Corchia C, Bagolan P (2008) Hearing impairment in congenital diaphragmatic hernia: the inaudible and noiseless foot of time. J Pediatr Surg 43(2):380–384. doi:10.1016/j.jpedsurg.2007.10.048
Vanamo K, Peltonen J, Rintala R, Lindahl H, Jaaskelainen J, Louhimo I (1996) Chest wall and spinal deformities in adults with congenital diaphragmatic defects. J Pediatr Surg 31(6):851–854. doi:10.1016/S0022-3468(96)90152-9
Chetcuti P, Myers NA, Phelan PD, Beasley SW, Dickens DR (1989) Chest wall deformity in patients with repaired esophageal atresia. J Pediatr Surg 24(3):244–247. doi:10.1016/S0022-3468(89)80003-X
Freeman NV, Walkden J (1969) Previously unreported shoulder deformity following right lateral thoracotomy for esophageal atresia. J Pediatr Surg 4(6):627–636. doi:10.1016/0022-3468(69)90490-4
Lawson ML, Mellins RB, Tabangin M, Kelly RE Jr, Croitoru DP, Goretsky MJ, Nuss D (2005) Impact of pectus excavatum on pulmonary function before and after repair with the Nuss procedure. J Pediatr Surg 40(1):174–180. doi:10.1016/j.jpedsurg.2004.09.040
Quigley PM, Haller JA Jr, Jelus KL, Loughlin GM, Marcus CL (1996) Cardiorespiratory function before and after corrective surgery in pectus excavatum. J Pediatr 128(5 Pt 1):638–643. doi:10.1016/S0022-3476(96)80128-4
Morshuis W, Folgering H, Barentsz J, van Lier H, Lacquet L (1994) Pulmonary function before surgery for pectus excavatum and at long-term follow-up. Chest 105(6):1646–1652. doi:10.1378/chest.105.6.1646
TNO prevention and health, University of Leiden Pediatric Department, The TAIQOL, a quality of life questionnaire for children of 1–4 years (in Dutch). Leiden, TNO/LUMC; 1997
Fekkes M, Theunissen NC, Brugman E, Veen S, Verrips EG, Koopman HM, Vogels T, Wit JM, Verloove-Vanhorick SP (2000) Development and psychometric evaluation of the TAPQOL: a health-related quality of life instrument for 1–5-year-old children. Qual Life Res 9(8):961–972. doi:10.1023/A:1008981603178
Poley MJ, Stolk EA, Tibboel D, Molenaar JC, Busschbach JJ (2004) Short term and long term health related quality of life after congenital anorectal malformations and congenital diaphragmatic hernia. Arch Dis Child 89(9):836–841. doi:10.1136/adc.2002.016543
Acknowledgments
The authors thank Prof. D. Tibboel, Pediatric Surgical Intensive Care, Sophia Children’s Hospital, Erasmus University Medical Center, Rotterdam, for critical comments on earlier versions of this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Peetsold, M.G., Heij, H.A., Kneepkens, C.M.F. et al. The long-term follow-up of patients with a congenital diaphragmatic hernia: a broad spectrum of morbidity. Pediatr Surg Int 25, 1–17 (2009). https://doi.org/10.1007/s00383-008-2257-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-008-2257-y