
Abstract
Background
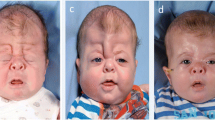
Apert syndrome is a rare form of syndromic craniosynostosis, also known as acrocephalosyndactyly, which is a disorder characterized by a unique set of craniofacial, hand, and foot abnormalities. Diagnosis is made through a genetic analysis, where the mutation of FGFR2, Ser252Trp, and Pro253Arg confirms the diagnosis.
Case presentation
Although craniosynostosis is the most common characteristic in clinical presentation, we present an atypical case of a one-and-a-half-year-old girl with Apert syndrome confirmed by genetic testing but without craniosynostosis.


Similar content being viewed by others


References
Lee DS, Chung KC (2010) Eugene Apert and his contributions to plastic surgery. Ann Plast Surg 64(3):362–365
Lajeunie E, Cameron R, El Ghouzzi V, de Parseval N, Journeau P, Gonzales M et al (1999) Clinical variability in patients with Apert’s syndrome. J Neurosurg 90(3):443–447
Cohen MM Jr, Kreiborg S, Lammer EJ, Cordero JF, Mastroiacovo P, Erickson JD et al (1992) Birth prevalence study of the Apert syndrome. Am J Med Genet 42(5):655–659
Moloney DM, Slaney SR, Oldridge M, Wall SA, Sahlin P, Stenman G, Wilkie AOM (1996) Exclusive paternal origin of new mutations in Apert syndrome. Nat Genet 13(1):48–53
Renier D, Lajeunie E, Arnaud E, Marchac D (2000) Management of craniosynostoses. Childs Nerv Syst 16(10–11):645–658
Wilkie AO, Johnson D, Wall SA (2017) Clinical genetics of craniosynostosis. Curr Opin Pediatr 29(6):622–628
Avantaggiato A, Carinci F, Curioni C (1996) Apert’s syndrome: cephalometric evaluation and considerations on pathogenesis. J Craniofac Surg 7(1):23–31
Cohen MM Jr, Kreiborg S (1990) The central nervous system in the Apert syndrome. Am J Med Genet 35(1):36–45
Bellus GA, McIntosh I, Smith EA, Aylsworth AS, Kaitila I, Horton WA, Greenhaw GA, Hecht JT, Francomano CA (1995) A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat Genet 10(3):357–359
Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet J-M, Maroteaux P, Merrer ML, Munnich A (1994) Mutations in the gene encoding fibroblast growth factor receptor 3 in achondroplasia. Nature 371(6494):252–254
Shiang R, Thompson LM, Zhu Y-Z, Church DM, Fielder TJ, Bocian M, Winokur ST, Wasmuth JJ (1994) Mutations in the transmembrane domain of FGFR3 cause the most common genetic form of dwarfism, achondroplasia. Cell 78(2):335–342
Orr-Urtreger A, Givol D, Yayon A, Yarden Y, Lonai P (1991) Developmental expression of two murine fibroblast growth factor receptors, flg and bek. Development 113(4):1419–1434
Coomaralingam S, Roth P (2012) Apert syndrome in a newborn infant without craniosynostosis. J Craniofac Surg 23(3):e209–ee11
Connolly JP, Gruss J, Seto ML, Whelan MF, Ellenbogen R, Weiss A, Buchman SR, Cunningham ML (2004) Progressive postnatal craniosynostosis and increased intracranial pressure. Plast Reconstr Surg 113(5):1313–1323
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
de Ângelis Ramos, D., Matushita, H., Cardeal, D.D. et al. Apert syndrome without craniosynostosis. Childs Nerv Syst 35, 565–567 (2019). https://doi.org/10.1007/s00381-019-04050-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-019-04050-1