
Abstract
The coronavirus disease-2019 (COVID-19) pandemic is likely to pose new challenges to the rheumatology community in the near and distant future. Some of the challenges, like the severity of COVID-19 among patients on immunosuppressive agents, are predictable and are being evaluated with great care and effort across the globe. A few others, such as atypical manifestations of COVID-19 mimicking rheumatic musculoskeletal diseases (RMDs) are being reported. Like in many other viral infections, severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection can potentially lead to an array of rheumatological and autoimmune manifestations by molecular mimicry (cross-reacting epitope between the virus and the host), bystander killing (virus-specific CD8 + T cells migrating to the target tissues and exerting cytotoxicity), epitope spreading, viral persistence (polyclonal activation due to the constant presence of viral antigens driving immune-mediated injury) and formation of neutrophil extracellular traps. In addition, the myriad of antiviral drugs presently being tried in the treatment of COVID-19 can result in several rheumatic musculoskeletal adverse effects. In this review, we have addressed the possible spectrum and mechanisms of various autoimmune and rheumatic musculoskeletal manifestations that can be precipitated by COVID-19 infection, its therapy, and the preventive strategies to contain the infection.
Similar content being viewed by others
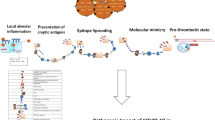

Introduction
The coronavirus disease-2019 (COVID-19) pandemic has taken a heavy toll on the healthcare system across the world. With a global incidence of over six million, the mortality rate so far has been approximately 6.5% [1]. Though a few countries have succeeded in containing the spread of the virus, the overall incidence of fresh infections is still on the rise. Moreover, with no vaccine or definitive treatment in sight [2], the health care systems across the world are strained and are staring at an uncertain future and a long-term coexistence of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) with the human host has been anticipated.
From the perspective of a rheumatologist, there are concerns regarding compelling actions and future preparedness for facing COVID-19 and its after effects. We have always considered rheumatic musculoskeletal diseases (RMDs) as conditions that are precipitated when a genetically susceptible host encounters specific environmental triggers. Our firsthand knowledge of the host-environment interaction in RMD until now was limited to epidemiological studies related to small infectious outbreaks like the Chikungunya epidemic which has given us insights into various post-infectious RMD manifestations. The COVID-19 pandemic, in contrast to these relatively small occurrences, is a mammoth event in the history of humankind. The infection has spread across all large countries affecting people from all age groups. Such an infection is likely to have long-term consequences, and RMDs could be a major fallout given the quantum of immune dysregulation the virus is known to cause [3]. There are already reports of post-COVID-19 autoimmunity with an increase in the incidence of Kawasaki disease [4] and Guillain-Barré syndrome [5, 6]. In a global attempt to curb this pandemic, a variety of drugs with varying mechanisms of action are being tried. While some of these drugs are known to cause adverse effects mimicking RMDs and other autoimmune diseases, there is a lack of information for the majority of them. There are also concerns regarding the right approach to treat existing rheumatic diseases and about the outcome of infection among those on immunosuppressive agents. This and many more challenges faced by patients with RMDs and their care providers during the COVID-19 pandemic are being addressed by the Global Alliance Registry and the COVID-19 database of the European League Against Rheumatism (EULAR); mining these data will serve as a guide in the future for the management of patients with RMDs [7].
The COVID-19 pandemic has undoubtedly uncovered the strengths of global collaborations, effective communication, and dissemination of information. Unless we prepare and foresee the likely new set of RMDs and the possible mimics of RMDs [8] due to this pandemic, we are going to fail in delivering care to our patients. We have recently learned the costs of being underprepared when challenged with the rheumatological manifestation [9] resulting from the use of immune checkpoint inhibitors in cancer treatment. The initial dilemma on the management plan and prognosis of these conditions set the rheumatology community on a backfoot [10]. Keeping this context in mind, it is time to pool all our existing knowledge and resources to prepare for any rheumatological fallout of COVID-19 and its treatment, which may have left behind its footprints even in the post-COVID-19 era.
Objective and search strategy
Our objective was to narrate various autoimmune and rheumatic manifestations that are associated with COVID-19 and the drugs used in its treatment. We conducted a literature search for articles since inception and published until 20 May 2020 in the English language using the Medline database. The search terms used in various combinations were: “coronavirus disease-19”, “COVID-19”, “SARS-CoV-2”, “rheumatic”, “autoimmune”, “musculoskeletal”, “clinical”, “manifestations”, “antivirals”, and “vaccines”. Additionally, we performed a focused literature search in the same database for articles on drugs used in COVID-19. We included original research articles, reviews, viewpoints, opinions, commentaries, case series, and case reports as relevant to our objective. The search strategy was used to obtain the titles and the abstracts of the relevant studies which were initially screened, and if necessary, the full text was retrieved to determine the suitability. The references and related citations for the resulting articles were also reviewed for pertinence. Finally, the information was synthesized in a logical sequence with expert inputs from the senior authors.
Clinical presentations of COVID-19
SARS-CoV-2 enters the cells through angiotensin-converting enzyme 2 (ACE2) receptors located on the nasopharyngeal and bronchial mucosa, and on alveolar pneumocytes in the lungs [11]. The smooth muscles of the arterial walls and endothelial cells of the veins in every organ of the body express ACE2 receptors explaining the ubiquitous distribution, and the possibility of multisystem involvement by the SARS-CoV-2 virus [12]. The spectrum of presentation of COVID-19 ranges from asymptomatic or mild symptoms to severe-critical illness which is categorized as mild, severe, and critical illness [13]. Patients with mild presentation have symptoms of fever, cough, nasal congestion, sore throat, malaise, headache, and myalgia without dyspnea. The severe presentation includes patients with respiratory distress as indicated by a respiratory rate of ≥ 30/min, SpO2 (blood oxygen saturation) of ≤ 93%, PaO2 (partial pressure of oxygen)/FiO2 (fraction of inspired oxygen) ratio of < 300, or presence of infiltrates in more than half of the lung fields in a radiograph or CT scan. Critical illness encompasses respiratory failure, septic shock, disseminated intravascular coagulation (DIC), and multiorgan dysfunction syndrome [13]. Besides respiratory symptoms, multiple cohorts have reported anosmia and dysgeusia, ranging from 34–87% of the patients [14, 15]. Gastrointestinal manifestations, including nausea, vomiting, and diarrhea have been reported in 11–24% of the cases and as presenting symptoms in a few reports of COVID-19 [16, 17]. In a case series from China, 32% of the patients had ophthalmological manifestations, mainly conjunctival redness and edema with increased eye discharge [18]. The other musculoskeletal, dermatological, neurological, and cardiovascular manifestations have been discussed in detail further in this review. The risk factors for severe disease and mortality include old age, immunocompromised status, and comorbidities, such as hypertension, diabetes mellitus, cardiovascular disease, chronic respiratory disorders, chronic kidney disease, liver diseases, cancer, and severe obesity [19]. Initial data from the COVID-19 Global Rheumatology Alliance provider registry suggests that the features and prognosis in patients with RMDs on immunosuppressive agents contracting SARS-CoV-2 are similar to those in the general population [20].
Spectrum of rheumatic musculoskeletal manifestations of SARS-CoV-2 infection and antiviral drugs
With scientific literature pouring in from different parts of the world on the clinical presentations and complications of SARS-CoV-2 infections, atypical clinical and laboratory manifestations mimicking RMDs have been reported (Table 1). Musculoskeletal manifestations reported with COVID-19 include arthralgia, myalgia, and proximal weakness with elevated creatine kinase (CK) level [21, 22]. Some of these manifestations preceded respiratory symptoms of COVID-19. For instance, a patient who presented with proximal weakness with muscle edema on magnetic resonance imaging (MRI), turned negative for myositis-specific and associated antibodies but progressed to develop the symptoms of COVID-19 [22]. The spectrum of dermatological manifestations mimicking RMDs includes COVID toes/pseudo-chilblain, transient urticarial or maculopapular rash, livedoid/necrotic lesions, punctiform or diffuse purpura, and erythema elevatum diutinum-like rash [23]. COVID toes/pseudo-chilblain is reported predominantly in children and young adults and seems to be a relatively late feature of COVID-19. On the other hand, livedoid/necrotic lesions are seen in elderly patients with severe COVID-19 with no association with the duration of infection. Cases of young stroke involving large vessels, commonly evaluated in rheumatology clinics, have also been reported with COVID-19 [24]. A couple of cases of Guillain-Barré syndrome (GBS) has been described as a consequence of post-viral autoimmune phenomenon. Out of the two reported cases, one succumbed to respiratory complications, and the other recovered with intravenous immunoglobulin (IVIg)/plasmapheresis [5, 6]. Myocarditis related to COVID-19 in young patients without any prior cardiac morbidity has been reported with early onset of cardiac symptoms. They present with intact coronaries in cardiac echo and MRI showing the classical features of myocarditis. Improvement of myocarditis was noticed following treatment with hydroxychloroquine, lopinavir/ritonavir, and intravenous methylprednisolone [25, 26]. An increase in the incidence of Kawasaki disease has been reported from Italy as a post-SARS-CoV-2 phenomenon. As compared to the classical Kawasaki disease, these children were older (mean age, 7.5 years) and reported to have more cardiac involvement, shock syndrome, and macrophage activation syndrome (MAS) [4]. Cytokine storm or secondary hemophagocytic lymphohistiocytosis (sHLH), yet another entity encountered in rheumatology practice, is reported with COVID-19. The clinical manifestations start 8–9 days after the onset of respiratory symptoms and range from unremitting fever, cytopenia, to hyperferritinemia ultimately resulting in multiorgan failure. Hyperferritinemia and elevated serum interleukin (IL)-6 are often associated with mortality in these patients [27]. There are reports of improvement with therapy targeting IL-1 and IL-6, and there are multiple ongoing clinical trials to evaluate the role of immunosuppressive therapy for this cytokine storm reported with COVID-19. In addition to these clinical manifestations mimicking RMDs, laboratory reports of positive antinuclear antibodies (ANA) [28], antiphospholipid antibodies, lupus anti-coagulant assay [29, 30] and increased level of D-dimer [31] have been reported with COVID-19. All these reports of COVID-19 mimicking or precipitating RMDs points towards a possibility of persisting intermediate to long-term immune dysregulation.
Based on evidence from in-vitro studies and experience from other viral infections, several antiviral therapies are currently in trial/practice in different parts of the world [32]. There are reports of rheumatic musculoskeletal adverse reactions following the use of these drugs. Table 2 depicts the list of the major anti-SARS-CoV-2 drugs with their mechanisms of action and the important rheumatological adverse events. Of these, some adverse events deserve special mention. Myopathy and neuromyopathy can rarely occur following long-term treatment with chloroquine and hydroxychloroquine [33]. Favipiravir can lead to hyperuricemia [34]. Lopinavir-ritonavir-related rheumatic adverse events include arthralgia, back pain, osteonecrosis, and vasculitis [35]. Ribavirin can cause arthralgia, back pain, myositis, and exacerbation of sarcoidosis [36]. Musculoskeletal pain and myalgia have been reported in up to half of the patients on interferon therapy. Additionally, in rare cases, interferon therapy can lead to drug-induced RMDS, such as rheumatoid arthritis, lupus, Sjogren syndrome, myositis, sarcoidosis, and vasculitis [37]. With the increased use of these drugs, there is a possibility of a rise in these adverse drug reactions necessitating active pharmacovigilance. Moreover, it must be noted that some of the frontrunner drugs like remdesivir have limited clinical data making it even more important to be vigilant.
Coagulopathy as a consequence of inflammation COVID-19 cases
One of the factors leading to mortality in COVID-19 patients is the presence of coagulopathy [38]. The autopsy findings of COVID-19 death from a majority of the cases show the presence of coagulopathy either in the form of deep venous thrombosis, pulmonary embolism, or multiple pulmonary thrombi coexisting with acute respiratory distress syndrome (ARDS) changes in the lungs [39]. The laboratory markers of COVID-19 coagulopathy include increased D-dimer level, borderline thrombocytopenia, and prolonged prothrombin time [40]. Though the exact mechanism underlying coagulopathy is unclear, a possibility of local lung-TMA as a coronavirus-associated hemostatic lung abnormality (CAHA) is proposed, linking inflammation and endothelial cell activation [41]. Endothelial cell activation results in the production of von Willebrand factor (vWF) in excess to the clearance capacity of ADAMTS-13. This leads to local thrombotic microangiopathy and perpetuates lung damage [42].
The association of netosis has also been proposed with coagulopathy in COVID-19 patients requiring further therapeutic exploration [43]. Moreover, the presence of lupus anti-coagulant and antiphospholipid antibodies may point towards the autoimmune contribution rather than just an epiphenomenon. Thus, linking inflammation to coagulopathy which is not a common occurrence in other viral infections suggests the role of immune activation at the vessel wall. This phenomenon warrants serious consideration of the anecdotal evidence supporting the role of immunosuppressants in addition to anticoagulants in severe COVID-19 cases [44].
SARS-CoV-2 infection and autoimmunity
The interplay of various genetic, hormonal, immunological, and environmental factors constitutes the mosaic of autoimmunity [45]. Viral infections play a substantial role in the development of several autoimmune diseases in individuals with underlying immune dysregulation [46]. Follow up data from survivors of viral outbreaks like influenza, Zika, Ebola, and Chikungunya have shown development of autoimmune phenomenon within weeks to months after recovery. While GBS, fulminant type1 diabetes, IgA vasculitis, APS have been observed after a previous outbreak of influenza [47], transverse myelitis, arthralgia, myalgia, and arthritis were reported following Zika [48], Chikungunya [49] and Ebola infections [50]. Besides overt clinical manifestations, long-term persistence of autoreactive cells and autoantibodies (against antiphospholipid with influenza and ds-DNA and heat shock protein-60 with Ebolavirus) have also been demonstrated with some of these infections [47, 49]. The mechanisms by which viruses disrupt self-tolerance include molecular mimicry, epitope spreading, bystander activation, persistence of the latent virus, and poly/oligoclonal immune activation in the background of autoimmunity mosaic [51]. Table 3 enlists known viral infections and associated autoimmune diseases with the possible mechanisms as elaborately described by Smatti et al. [52]. Similar mechanisms may lead to autoimmunity following SARS-CoV-2 infection. We herein describe some of the possible mechanism of autoimmunity following SARS-CoV-2 infection as follows:
Molecular mimicry
Molecular mimicry, as a result of the cross-reacting epitope between the virus and the host, leads to both humoral and cellular autoreactivity (Fig. 1a) [53]. This mechanism plays a vital role in the pathogenesis of prototype systemic rheumatological diseases such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), systemic sclerosis, and Sjogren syndrome [54]. SARS-CoV-2 proteins displayed at least one match with the human protein on a comparative peptidome analysis comprising of 37 viral proteins [55]. These similarities potentially can lead to loss of tolerance to self-peptides and result in autoimmunity. Recently, an epitope mapping analysis has identified immunogenic linear epitopes, 2′-O-ribose methyltransferase, RNA-dependent RNA polymerase and 3′-to-5′ exonuclease proteins from autoimmune dermatomyositis patients matching with the SARS-CoV-2 peptides [56].

Mechanism of autoimmunity through molecular mimicry and bystander activation of antigen-presenting cells and proinflammation. a Molecular mimicry: The processing of cross-reactive peptide and presentation via MHC1 and MHC2 to T cells lead to the generation of autoreactive cells. ACE2 angiotensin-converting enzyme 2, ER endoplasmic reticulum, ERAP endoplasmic reticulum aminopeptidase, MHC major histocompatibility. b Bystander activation of antigen-presenting cells and proinflammation: The cytoplasmic pattern recognition receptors after identifying viral RNA phosphorylates downstream IRF-3, IRF-7, and NFκB leading to the secretion of interferons as well as proinflammatory cytokines. CCL2 chemokine (C–C motif) ligand 2, CXCL8 C-X-C motif chemokine ligand 8, IL interleukin, IRF interferon regulatory transcription factor, ISRE interferon-stimulated response element, MDA-5 melanoma differentiation-associated protein-5, MVAS mitochondrial antiviral-signaling protein, MYD88 myeloid differentiation primary response-88, RIG-1 retinoic acid-inducible gene-I, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells), TLR toll-like receptor, TNF tumor necrosis factor
In addition to the presence of cross-reactive epitopes, activation of antigen-presenting cells (APCs) either by an adjuvant or infectious stimulus is essential to increase the expression of co-receptors [51]. Viral RNA of the SARS-CoV-2 activates dendritic cells through cytosolic RIG like receptors and endosomal TLRs, as well as by the release of interferon α/β and γ enhancing antigen-presenting capacity (Fig. 1b) [57]. Such activation by the virus can result in precipitation of inflammatory cascade in the presence of an already existing cross-reactive antigen. The clinical phenomenon of initial asymptomatic to mild symptoms followed by severe autoinflammatory syndrome in COVID-19 is proposed to be due to a similar autoreactive adaptive response with SARS-CoV-2 infection [3].
The role of genetic susceptibility should also be considered while evaluating the role of cross-reactive epitopes in precipitating autoimmunity. Human leukocyte antigen (HLA) susceptibility map for SARS-CoV-2 has shown that HLA-B*15:03 efficiently presents highly conserved SARS-CoV-2 peptides that are shared among the common human coronaviruses [58]. The association of HLA-B*15 with primary Sjogren’s syndrome [59] and Bechet’s diseases [60] is well documented, and the consequence of its association with SARS-CoV-2 peptide presentation needs to be carefully followed up.
Following molecular mimicry with the dominant epitope, diversification in epitope specificity commences resulting in the neo-epitopes presentation. Immune response to these neo-epitopes differs from that to the dominant epitope and involves newer targets for autoimmunity [61]. The sequential appearance of various autoantibodies in RA and SLE follows the theory of epitope spreading [62, 63]. In SLE, before clinical disease onset, autoantibodies are targeted against Ro, La, and phospholipid antigens. In contrast, the clinical manifestations commence with the appearance of autoantibodies to ds-DNA, Smith (Sm), and ribonuclear protein (RNP) antigens [64]. Although with SARS-CoV-2 infection, epitope diversification is not reported, it would be worthwhile to closely follow those patients with positive autoantibodies.
Bystander activation and damage
Pre-clinical studies of diabetes and experimental autoimmune encephalomyelitis suggest the role of bystander activation as one of the mechanisms predisposing to autoimmunity [65, 66]. The bystander damage starts with the virus-specific CD8+ T cells migrating to the infected target tissues and exerting perforin and granzyme-mediated cytotoxicity. The target cell death in the inflammatory milieu activates the surrounding macrophages to release reactive oxygen species and nitric oxide resulting in bystander killing of surrounding uninfected cells [67]. The CD4+ T cells contribute to this bystander damage through the release of proinflammatory cytokines and enhancing phagocytic activities of macrophage [68]. Ineffective clearance of these killed cells exposes autoantigen to antigen-presenting cells, resulting in the generation of autoreactive cells (Fig. 2) [51]. The association of hepatitis C virus and Sjogren syndrome serves a classical example among RMDs explaining the bystander activation and damage theory [69]. Hepatitis C, primarily a hepatotropic virus, also exhibits sialotropism and lymphotropism leading to bystander activation of the salivary gland epithelium and lymphocytes, respectively [70]. The activation of salivary epithelial cells and resultant antiviral defense in the salivary gland result in bystander damage manifesting as sicca symptoms and parotidomegaly. Bystander activation and dysregulated proliferation of the lymphocytes result in cryoglobulinemic vasculitis, generation of rheumatoid factor, autoantibodies (ANA, anti-SSA, anti-SSB), and low complements.

Bystander killing: virus-specific CD8+ T cells migrating to the infected target tissues and exerting perforin and granzyme-mediated cytotoxicity. The CD4+ T cells contribute to this bystander killing by the release of proinflammatory cytokines and enhancing phagocytic activities of the macrophages. The free oxygen radicals and cytokines secreted from the activated macrophages result in bystander killing off the surrounding non-infected cells. Ineffective clearance of these killed cells exposes autoantigen to the antigen-presenting cells, resulting in the generation of autoreactive cells in the presence of the costimulatory molecules
Lymphopenia due to reduced CD4+ T cells and CD8+ T cells is well documented in severe cases of SARS-CoV-2. The reduction is possibly secondary to functional exhaustion and preferential accumulation of the primed lymphocytes at the site of viral infection [71]. These preferentially accumulated lymphocytes exhibit increased activity as confirmed by the expression of HLA-DR, CD69, CD38, and CD44. Activated lymphocytes, along with activated macrophages at the target tissue lead to bystander killing of adjacent, non-infected healthy cells via proinflammatory cytokine and reactive oxygen species [72]. With SARS-CoV-2 infection, the multisystem inflammatory syndrome appears after the peak of viral load. This suggests a build-up of inflammatory cytokine and bystander activation of the macrophages by SARS-CoV-2 through pattern recognition receptors (PRR). It also suggests bystander killing of the cells devoid of ACE2 receptors by free oxygen radicals released in the inflammatory milieu [73]. Bystander damage can be one of the mechanisms responsible for the manifestations like ARDS, myocarditis, and neurological involvement reported with SARS-CoV-2 infection. Autopsy reports of COVID-19 patients show diffuse infiltration of lymphocytes in the lungs and focal infiltration in the heart, kidney, liver, pancreas, and adrenal gland, suggesting bystander damage by cytotoxic CD8+ T cells [74]. Besides, in patients surviving SARS-CoV-2 infection, bystander activation may lead to sequestered autoantigen presentation leading to the emergence of neoepitope and autoimmune manifestations, as seen in SLE [75].
Persistent immune activation
The third theory of persistent viral infection and oligo/polyclonal activation can be explained by the constant presence of viral antigens driving immune-proliferation resulting from the ineffective clearance of viruses [76]. Such mechanisms are commonly encountered with the Epstein-Barr virus which resides in autoreactive B cells imparting immortality to the cells. These long-living autoreactive B cells cause lymphoproliferation and polyclonal activation culminating in chronic autoimmunity [77]. As the possibility of the persistence of the SARS-CoV-2 virus cannot be completely ruled out, this may add to an additional mechanism of autoimmunity. On a brighter side, complete clearance is reported in cases of other members of the coronavirus family following the development of specific antibodies [78], making the possibility of persistent immune activation due to SARS-CoV-2 chronicity less likely.
Netosis
Knight et al. noted the formation of neutrophil extracellular traps (NETs) by demonstrating elevated serum levels of cell-free DNA, myeloperoxidase-DNA complexes, and citrullinated histone H3 among patients with COVID-19 compared to healthy controls [79]. NETs consist of extracellular webs of nuclear chromatin materials and supporting histones along with antibacterial proteins and oxidant enzymes from neutrophilic granules. The principal role of this transient phenomenon is to trap the microorganisms to resolve the infection. However, sustained netosis may beget inflammation and thrombosis [80]. In the past decade, researchers have established the role of netosis in autoimmune disease including but not limited to, SLE, RA, anti-neutrophil cytoplasmic antibody-associated vasculitis (AAV), antiphospholipid antibody syndrome (APS), and an autoinflammatory syndrome, deficiency of adenosine deaminase-2 (DADA-2) [81].
In COVID-19, a rise in neutrophil counts during early infection is a poor prognostic factor. The migration of these neutrophils towards virally infected sites follows the IL-8 gradient [82]. The production of free oxygen radicals and IL-1β by macrophages and pyroptosis of virally infected cells stimulate and maintain netosis of the migrated neutrophils [83]. Exposure of chromatin, histones, and neutrophil granules with sustained netosis, serve as a source of autoantigens. This may lead to the recognition of self-peptides by antigen-presenting cells and result in the expansion of autoreactive cells (Fig. 3).

NETosis: Neutrophils recruited to the target tissues following chemokine IL-8 gradient gets activated by IL-1β and free oxygen radicals leading to a sustained generation of NETosis. NETs carrying autoantigen, which gets recognized by dendritic cells, leads to the activation of autoreactive T cells. IL interleukin, NET neutrophil extracellular traps
Population behavioral modification with COVID-19
In addition to the interaction of virus and host, the environmental factors play an important role in susceptibility and protection to autoimmune disease [45]. A paradigm shift has taken place in the human behavior with advocacy of social distancing, handwashing, use of mask and gloves, and movement restrictions in an attempt to contain the spread of COVID-19 [21]. This shift is a double-edged sword in the mosaic of autoimmunity. The positive side is reflected by a reduction in airborne infectious disease with a documented shortening of the influenza season in the northern hemisphere by about six weeks [84]. This may reduce the occurrence of subsequent effects of influenza infection on RMDs. Diseases like acute rheumatic fever, where overcrowding plays an essential role in familial predisposition may also reduce with these hygienic measures [85]. At the negative spectrum of these hygienic measures, an increase in autoimmune disease should be born in mind as proposed by the proponents of the hygiene hypothesis. During the second half of the twentieth century, with the improvement in lifestyle especially in the developed world, the burden of infectious disease reduced with a parallel increase in autoimmune diseases like type1 diabetes mellitus, inflammatory bowel disease, and multiple sclerosis [86].
One of the key influencers to hygiene hypothesis is the microbiome of an individual which plays a protective role against autoimmune diseases. The mechanisms conferring protections by microbiome include antigenic competition, the role of lymphocyte homeostasis against pathogens, and the effect on immune regulatory pathway favoring the anti-inflammatory milieu with IL-10 and TGF-β secretion from the regulatory cells and TLR mediated receptor desensitization [87]. Disturbance in this microbiome is evident in the pathogenesis of autoimmune disorders including RA, SLE, and inflammatory bowel disease [88]. Over practice of distancing and antimicrobial sanitizers may lead to dysbiosis triggering autoimmunity. Additionally, the psychological stress caused by isolation, the anxiety of contracting the infection, and the economic burden along with its numerous adverse short- and long-term socio-economic consequences can turn as a trigger of various autoimmune diseases [89]. In summary, understanding the immune consequences of SARS-Cov-2 interaction with the host along with the environmental changes may explain the basis of rheumatic musculoskeletal manifestations.
Preventive therapy against SARS-CoV-2 and the risk of autoimmunity
Currently, a multitude of vaccines for the prevention of SARS-CoV-2 infection is under investigation [90]. Except for the nucleocapsid, all immunogenic epitopes have been reported to have at least one match with human proteins [55]. This homology between the human and viral proteins is an established factor in vaccine-induced autoimmunity with the mechanism of molecular mimicry, as discussed above. Furthermore, there is a theoretical possibility of the involvement of pathogenic priming in re-infection by COVID-19, triggering the release of proinflammatory cytokines leading to cytokine storm [55]. Similar instances have been experienced with H1N1 influenza [91], MERS [92], and SARS [93]. Thus, while developing vaccines, these epitopes should be carefully excluded to minimize unintended autoimmunity due to the risk of pathogenic priming.
Targeted immunosuppressive therapy for SARS-CoV-2 infection
The clinical outcome of the patients relies on the fate of interaction between the SARS-CoV-2 virus and immune cells of the host [94]. Modulation of this virus-host cell interaction and its aftereffects can be possibly achieved with immunomodulatory therapies repurposed from drugs used in autoimmune diseases. The potential drug targets for COVID-19 has been reviewed extensively by Misra et al. [8]. In the early asymptomatic or mild symptomatic stage, antiviral therapy is likely to have maximum efficacy, and the addition of interferon therapy at this stage may theoretically benefit from augmenting the innate antiviral response. In the next stage of pulmonary and systemic hyper-inflammation, early initiation of immunosuppressant, including IVIg, corticosteroids, IL-6, or IL-1 inhibitors may help to halt the immune-mediated damage [95]. We have summarized the plausible role of targeted immunosuppressive therapy in Fig. 4. The role of IL-1 and IL-6 in particular is worth noticing as they are significant drivers of proinflammation in the cytokine release syndrome (CRS) of COVID-19. The IL-1 receptor antagonist, anakinra, has proven its beneficial effects in the MAS in rheumatic diseases, as well as in chimeric antigen receptor T cell (CAR-T)-mediated severe CRS [96, 97]. Data from uncontrolled or historically controlled case series are encouraging for anakinra showing its safety and benefit on mortality, especially in critically ill patients with COVID-19 [98]. The high level of IL-6 in COVID-19 patients results from the secretion through viral infected respiratory cells, as well as from the infiltrating lymphocytes and monocytes [99]. The anti-IL-6 treatment strategy has shown efficacy in a similar CRS-like phenomenon observed with MAS in rheumatic diseases and has become an attractive target for COVID-19 critical cases [100]. To date, the supporting evidence for the beneficial effects of tocilizumab (a monoclonal antibody against the IL-6 receptor) is limited to observational studies [101]. The results of ongoing randomized controlled trials with IL-1 and IL-6 targeted therapy may clarify the role of tocilizumab in COVID-19. The downstream effects of IL-6 and its receptor are mediated via cytosolic Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling, a common pathway for multiple proinflammatory cytokines. Trials with baricitinib, which targets JAK-STAT signaling, are also ongoing in critically ill patients with COVID-19 [102].

Targeted immunosuppressive therapy for COVID-19: Macrophages play a significant role in cytokine release syndrome associated with SARS-CoV-2 infection. The cells get activated directly by viruses as well as bystander activation with autocrine and paracrine actions of the proinflammatory cytokines mainly derived from the macrophages, NK cells, and T cells. The target of immunosuppressive therapy is denoted by the numbered boxes as described below. The red color box denotes the drugs for which clinical trials are ongoing and the grey color box denotes the drugs for which there is no ongoing clinical trial registered at present for the treatment of COVID-19. (1) TLR7 mediated viral signaling at the endosomal level—> chloroquine and hydroxychloroquine. (2) TLR4-TRIF signaling—> plausible therapeutic target, no approved drug. (3) IRAK4 inhibitor—> PF-06650833, CA-4948. (4) (a) Anti IL-1β—> canakinumab, (b) IL-1 receptor antagonist- > anakinra. (5) TNF inhibitors—> infliximab, adalimumab, etanercept. (6) GM-CSF signaling inhibition—> lenzilumab. (7) (a) Anti IL-6—> siltuximab, clazakizumab (b) Anti IL-6 receptor > tocilizumab, sarilumab. (8) Anti IFN γ—> emapalumab. (9) JAK inhibitor—> baricitinib, ruxolitinib, tofacitinib (multi-cytokine targeted therapy). CCL2 chemokine (C–C motif) ligand 2, CXCL8 C-X-C motif chemokine ligand 8, ACE2 angiotensin-converting enzyme 2, GM-CSF granulocyte–macrophage colony-stimulating factor, IFN interferon, IL interleukin, IRAK4 interleukin-1 receptor-associated kinase 4, IRF interferon regulatory transcription factor, ISRE interferon-stimulated response element, JAK Janus kinase, MDA-5 melanoma differentiation-associated protein-5, MVAS mitochondrial antiviral-signaling protein, MYD88 myeloid differentiation primary response-88, RIG-1 retinoic acid-inducible gene-I, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells, STAT signal transducer and activator of transcription, TLR toll-like receptor, TNF tumor necrosis factor
Conclusion
In this review, we primarily aimed at understanding the potential pathways and manifestations leading to autoimmunity and other RMD-like illnesses that could be triggered due to COVID-19 or the treatment for the same. This information may help the rheumatology community to tackle the threat of novel RMDs, RMD mimics, and other manifestations, including cytokine storm, Kawasaki disease, and coagulopathy. The immune consequences of SARS-CoV-2-host interaction along with the environmental changes, explain the basis of rheumatic musculoskeletal manifestations of COVID-19. There is a need for preparedness for a possible surge in diverse autoimmune diseases following the pandemic. The rheumatology community is already joining hands to treat, support, and inform our existing patient partners during this pandemic with the advantage of accessibility to cutting edge technology at our disposal. With a likely long-term coexistence of SARS-CoV-2 and the human host and the use of numerous therapeutic strategies, this preparedness may help in the effective management of the rheumatic manifestations of SARS-CoV-2 infection in the post-COVID-19 era.
Abbreviations
- AAV:
-
Anti-neutrophil cytoplasmic antibody-associated vasculitis
- ACE2:
-
Angiotensin-converting enzyme 2
- ANA:
-
Antinuclear antibodies
- AIDS:
-
Acquired immunodeficiency syndrome
- APCs:
-
Antigen-presenting cells
- APS:
-
Antiphospholipid antibody syndrome
- ARDS:
-
Acute respiratory distress syndrome
- CAHA:
-
Coronavirus-associated hemostatic lung abnormality
- CAR-T:
-
Chimeric antigen receptor T cell
- CCL2:
-
Chemokine (C–C motif) ligand 2
- CK:
-
Creatine kinase
- COVID-19:
-
Coronavirus disease-2019
- CSF:
-
Cerebrospinal fluid
- CRS:
-
Cytokine release syndrome
- CXCL8:
-
C-X-C motif chemokine ligand 8
- DADA-2:
-
Deficiency of adenosine deaminase-2
- DIC:
-
Disseminated intravascular coagulation
- ER:
-
Endoplasmic reticulum
- ERAP:
-
Endoplasmic reticulum aminopeptidase
- EULAR:
-
European league against rheumatism
- GBS:
-
Guillain–Barré syndrome
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- HLA:
-
Human leukocyte antigen
- IFN:
-
Interferon
- IL:
-
Interleukin
- IRAK4:
-
Interleukin-1 receptor-associated kinase 4
- IRF:
-
Interferon regulatory transcription factor
- ISRE:
-
Interferon-stimulated response element
- IVIg:
-
Intravenous immunoglobulin
- JAK:
-
Janus kinase
- LAC:
-
Lupus anti-coagulant
- MAA:
-
Myositis-associated autoantibodies
- MAS:
-
Macrophage activation syndrome
- MRI:
-
Magnetic resonance imaging
- MDA-5:
-
Melanoma differentiation-associated protein-5
- MHC:
-
Major histocompatibility
- MSA:
-
Myositis-specific autoantibodies
- MVAS:
-
Mitochondrial antiviral-signaling protein
- MYD88:
-
Myeloid differentiation primary response-88
- NETs:
-
Neutrophil extracellular traps
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NIH-SS:
-
National Institute of Health Stroke Score
- PRR:
-
Pattern recognition receptors
- RA:
-
Rheumatoid arthritis
- RIG-1:
-
Retinoic acid-inducible gene-I
- RMDs:
-
Rheumatic musculoskeletal diseases
- RNA:
-
Ribonucleic acid
- RNP:
-
Ribonuclear protein
- PCR:
-
Polymerase chain reaction
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus-2
- SLE:
-
Systemic lupus erythematosus
- Sm:
-
Smith
- SJS-TEN:
-
Steven Johnson syndrome-toxic epidermal necrolysis
- STAT:
-
Signal transducer and activator of transcription
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- vWF:
-
Von Willebrand factor
References
WHO Coronavirus Disease (COVID-19) Dashboard. https://covid19.who.int/?gclid=Cj0KCQjwwr32BRD4ARIsAAJNf_0jdmZQ1hkz7onImyjahwc-N0H4WTwXnVU7bgQObfk_Vct9yucybbkaAk0oEALw_wcB. Accessed 28 May 2020
El-Aziz TMA, Stockand JD (2020) Recent progress and challenges in drug development against COVID-19 coronavirus (SARS-CoV-2) - an update on the status. Infect Genet Evol. https://doi.org/10.1016/j.meegid.2020.104327
Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP (2020) The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. https://doi.org/10.1038/s41577-020-0311-8
Verdoni L, Mazza A, Gervasoni A et al (2020) An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: an observational cohort study. Lancet Lond Engl. https://doi.org/10.1016/S0140-6736(20)31103-X
Toscano G, Palmerini F, Ravaglia S et al (2020) Guillain-Barré Syndrome associated with SARS-CoV-2. N Engl J Med. https://doi.org/10.1056/NEJMc2009191
Alberti P, Beretta S, Piatti M et al (2020) Guillain-Barré syndrome related to COVID-19 infection. Neurol Neuroimmunol Neuroinflammation 7(4):e741. https://doi.org/10.1212/NXI.0000000000000741
The Lancet Rheumatology (2020) Unity amidst uncertainty: COVID-19 pandemic fosters collaboration in rheumatology community. Lancet Rheumatol. https://doi.org/10.1016/S2665-9913(20)30082-5
Misra DP, Agarwal V, Gasparyan AY, Zimba O (2020) Rheumatologists’ perspective on coronavirus disease 19 (COVID-19) and potential therapeutic targets. Clin Rheumatol. https://doi.org/10.1007/s10067-020-05073-9
Ramos-Casals M, Brahmer JR, Callahan MK et al (2020) Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primer 6(1):1–21. https://doi.org/10.1038/s41572-020-0160-6
Kostine M, Cappelli LC, Calabrese C et al (2019) Addressing immune-related adverse events of cancer immunotherapy: how prepared are rheumatologists? Ann Rheum Dis 78(6):860–862. https://doi.org/10.1136/annrheumdis-2018-214748
Batlle D, Wysocki J (1979) Satchell K (2020) Soluble angiotensin-converting enzyme 2: a potential approach for coronavirus infection therapy? Clin Sci Lond Engl 134(5):543–545. https://doi.org/10.1042/CS20200163
Hamming I, Timens W, Bulthuis M, Lely A, Navis G, van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203(2):631–637. https://doi.org/10.1002/path.1570
Wu Z, McGoogan JM (2020) Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 323(13):1239–1242. https://doi.org/10.1001/jama.2020.2648
Giacomelli A, Pezzati L, Conti F et al (2020) Self-reported olfactory and taste disorders in patients with severe acute respiratory coronavirus 2 infection: a cross-sectional study. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa330
Lechien JR, Chiesa-Estomba CM, Hans S, Barillari MR, Jouffe L, Saussez S (2020) Loss of smell and taste in 2013 European patients with mild to moderate COVID-19. Ann Intern Med. https://doi.org/10.7326/M20-2428
Jin X, Lian J-S, Hu J-H et al (2020) Epidemiological, clinical and virological characteristics of 74 cases of coronavirus-infected disease 2019 (COVID-19) with gastrointestinal symptoms. Gut 69(6):1002–1009. https://doi.org/10.1136/gutjnl-2020-320926
Goyal P, Choi JJ, Pinheiro LC et al (2020) Clinical characteristics of Covid-19 in New York City. N Engl J Med 382(24):2372–2374. https://doi.org/10.1056/NEJMc2010419
Wu P, Duan F, Luo C et al (2020) Characteristics of ocular findings of patients with coronavirus disease 2019 (COVID-19) in Hubei Province. China JAMA Ophthalmol 138(5):575–578. https://doi.org/10.1001/jamaophthalmol.2020.1291
CDC Coronavirus Disease 2019 (COVID-19) Centers for Disease Control and Prevention (2020). https://www.cdc.gov/coronavirus/2019-ncov/need-extra-precautions/people-with-medical-conditions.html
Gianfrancesco MA, Hyrich KL, Gossec L et al (2020) Rheumatic disease and COVID-19: initial data from the COVID-19 Global Rheumatology Alliance provider registries. Lancet Rheumatol 2(5):e250–e253. https://doi.org/10.1016/S2665-9913(20)30095-3
Overview of Public Health and Social Measures in the context of COVID-19. https://www.who.int/publications-detail-redirect/overview-of-public-health-and-social-measures-in-the-context-of-covid-19
Beydon M, Chevalier K, Tabaa OA et al (2020) Myositis as a manifestation of SARS-CoV-2. Ann Rheum Dis. https://doi.org/10.1136/annrheumdis-2020-217573
Casas CG, Català A, Hernández GC, et al (2020) Classification of the cutaneous manifestations of COVID-19: a rapid prospective nationwide consensus study in Spain with 375 cases. Br J Dermatol. 10.1111/bjd.19163
Oxley TJ, Mocco J, Majidi S et al (2020) Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med 382(20):e60. https://doi.org/10.1056/NEJMc2009787
Inciardi RM, Lupi L, Zaccone G et al (2020) Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. https://doi.org/10.1001/jamacardio.2020.1096
Kim I-C, Kim JY, Kim HA, Han S (2020) COVID-19-related myocarditis in a 21-year-old female patient. Eur Heart J 41(19):1859–1859. https://doi.org/10.1093/eurheartj/ehaa288
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395(10229):1033–1034. https://doi.org/10.1016/S0140-6736(20)30628-0
Gazzaruso C, Carlo Stella N, Mariani G et al (2020) High prevalence of antinuclear antibodies and lupus anticoagulant in patients hospitalized for SARS-CoV2 pneumonia. Clin Rheumatol. https://doi.org/10.1007/s10067-020-05180-7
Harzallah I, Debliquis A, Drénou B (2020) Lupus anticoagulant is frequent in patients with Covid-19. J Thromb Haemost. https://doi.org/10.1111/jth.14867
Zhang Y, Xiao M, Zhang S et al (2020) Coagulopathy and antiphospholipid antibodies in patients with Covid-19. N Engl J Med 382(17):e38. https://doi.org/10.1056/NEJMc2007575
Zhou F, Yu T, Du R et al (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet Lond Engl 395(10229):1054–1062. https://doi.org/10.1016/S0140-6736(20)30566-3
Esposito S, Noviello S, Pagliano P (2020) Update on treatment of COVID-19: ongoing studies between promising and disappointing results. Infez Med 28(2):198–211
Casado E, Gratacós J, Tolosa C et al (2006) Antimalarial myopathy: an underdiagnosed complication? Prospective longitudinal study of 119 patients. Ann Rheum Dis 65(3):385–390. https://doi.org/10.1136/ard.2004.023200
Hayden FG, Shindo N (2019) Influenza virus polymerase inhibitors in clinical development. Curr Opin Infect Dis 32(2):176–186. https://doi.org/10.1097/QCO.0000000000000532
Chandwani A, Shuter J (2008) Lopinavir/ritonavir in the treatment of HIV-1 infection: a review. Ther Clin Risk Manag 4(5):1023–1033
Naik GS, Tyagi MG (2012) A pharmacological profile of ribavirin and monitoring of its plasma concentration in Chronic Hepatitis C Infection. J Clin Exp Hepatol 2(1):42–54. https://doi.org/10.1016/S0973-6883(12)60090-5
Psarras A, Emery P, Vital EM (2017) Type I interferon–mediated autoimmune diseases: pathogenesis, diagnosis and targeted therapy. Rheumatology 56(10):1662–1675. https://doi.org/10.1093/rheumatology/kew431
Tang N, Li D, Wang X, Sun Z (2020) Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 18(4):844–847. https://doi.org/10.1111/jth.14768
Wichmann D, Sperhake J-P, Lütgehetmann M et al (2020) Autopsy findings and venous thromboembolism in patients With COVID-19. Ann Intern Med. https://doi.org/10.7326/M20-2003
Levi M, Thachil J, Iba T, Levy JH (2020) Coagulation abnormalities and thrombosis in patients with COVID-19. Lancet Haematol. https://doi.org/10.1016/S2352-3026(20)30145-9
Thachil J, Srivastava A (2020) SARS-2 coronavirus-associated hemostatic lung abnormality in COVID-19: is it pulmonary thrombosis or pulmonary embolism? Semin Thromb Hemost. https://doi.org/10.1055/s-0040-1712155
Stockschlaeder M, Schneppenheim R, Budde U (2014) Update on von Willebrand factor multimers: focus on high-molecular-weight multimers and their role in hemostasis. Blood Coagul Fibrinolysis Int J Haemost Thromb 25(3):206–216. https://doi.org/10.1097/MBC.0000000000000065
Zuo Y, Zuo M, Yalavarthi S et al (2020) Neutrophil extracellular traps and thrombosis in COVID-19. MedRxiv. https://doi.org/10.1101/2020.04.30.20086736
Tang N, Bai H, Chen X, Gong J, Li D, Sun Z (2020) Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost 18(5):1094–1099. https://doi.org/10.1111/jth.14817
Shoenfeld Y, Isenberg DA (1989) The mosaic of autoimmunity. Immunol Today 10(4):123–126. https://doi.org/10.1016/0167-5699(89)90245-4
Arleevskaya MI, Manukyan G, Inoue R, Aminov R (2017) Editorial: microbial and environmental factors in autoimmune and inflammatory diseases. Front Immunol. https://doi.org/10.3389/fimmu.2017.00243
Toplak N, Avčin T (2009) Influenza and autoimmunity. Ann N Y Acad Sci 1173(1):619–626. https://doi.org/10.1111/j.1749-6632.2009.04759.x
Acosta-Ampudia Y, Monsalve DM, Castillo-Medina LF et al (2018) Autoimmune neurological conditions associated with zika virus infection. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2018.00116
Tanay A (2017) Chikungunya virus and autoimmunity. Curr Opin Rheumatol 29(4):389–393. https://doi.org/10.1097/BOR.0000000000000396
Fausther-Bovendo H, Qiu X, McCorrister S et al (2017) Ebola virus infection induces autoimmunity against dsDNA and HSP60. Sci Rep 7:42147. https://doi.org/10.1038/srep42147
Fujinami RS, von Herrath MG, Christen U, Whitton JL (2006) Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev 19(1):80–94. https://doi.org/10.1128/CMR.19.1.80-94.2006
Smatti MK, Cyprian FS, Nasrallah GK, Al Thani AA, Almishal RO, Yassine HM (2019) Viruses and autoimmunity: a review on the potential interaction and molecular mechanisms. Viruses 11(8):762. https://doi.org/10.3390/v11080762
Kim B, Kaistha SD, Rouse BT (2006) Viruses and autoimmunity. Autoimmunity 39(1):71–77. https://doi.org/10.1080/08916930500484708
Cusick MF, Libbey JE, Fujinami RS (2012) Molecular mimicry as a mechanism of autoimmune disease. Clin Rev Allergy Immunol 42(1):102–111. https://doi.org/10.1007/s12016-011-8294-7
Lyons-Weiler J (2020) Pathogenic priming likely contributes to serious and critical illness and mortality in COVID-19 via autoimmunity. J Transl Autoimmun 3:100051. https://doi.org/10.1016/j.jtauto.2020.100051
Megremis S, Walker TDJ, He X et al (2020) Antibodies against immunogenic epitopes with high sequence identity to SARS-CoV-2 in patients with autoimmune dermatomyositis. Ann Rheum Dis. https://doi.org/10.1136/annrheumdis-2020-217522
Totura AL, Baric RS (2012) SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol 2(3):264–275. https://doi.org/10.1016/j.coviro.2012.04.004
Nguyen A, David JK, Maden SK et al (2020) Human leukocyte antigen susceptibility map for SARS-CoV-2. J Virol. https://doi.org/10.1128/JVI.00510-20
Charfi A, Mahfoudh N, Kamoun A et al (2020) Association of HLA Alleles with Primary Sjögren Syndrome in the South Tunisian Population. Med Princ Pract 29(1):32–38. https://doi.org/10.1159/000501896
Choukri F, Chakib A, Himmich H, Hüe S, Caillat-Zucman S (2001) HLA-B∗51 and B∗15 alleles confer predisposition to Behçet’s disease in Moroccan patients. Hum Immunol 62(2):180–185. https://doi.org/10.1016/S0198-8859(00)00249-4
Lehmann PV, Targoni OS, Forsthuber TG (1998) Shifting T-cell activation thresholds in autoimmunity and determinant spreading. Immunol Rev 164(1):53–61. https://doi.org/10.1111/j.1600-065X.1998.tb01207.x
Sokolove J, Bromberg R, Deane KD et al (2012) Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS ONE. https://doi.org/10.1371/journal.pone.0035296
Deshmukh US, Gaskin F, Lewis JE, Kannapell CC, Fu SM (2003) Mechanisms of autoantibody diversification to SLE-related autoantigens. Ann N Y Acad Sci 987(1):91–98. https://doi.org/10.1111/j.1749-6632.2003.tb06036.x
Arbuckle MR, McClain MT, Rubertone MV et al (2003) Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med 349(16):1526–1533. https://doi.org/10.1056/NEJMoa021933
Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvennick N (1998) Diabetes induced by Coxsackie virus: Initiation by bystander damage and not molecular mimicry. Nat Med 4(7):781–785. https://doi.org/10.1038/nm0798-781
McCoy L, Tsunoda I, Fujinami RS (2006) Multiple sclerosis and virus induced immune responses: autoimmunity can be primed by molecular mimicry and augmented by bystander activation. Autoimmunity 39(1):9–19. https://doi.org/10.1080/08916930500484799
Ando K, Hiroishi K, Kaneko T et al (1997) Perforin, Fas/Fas ligand, and TNF-alpha pathways as specific and bystander killing mechanisms of hepatitis C virus-specific human CTL. J Immunol 158(11):5283–5291
Wucherpfennig KW, Strominger JL (1995) Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 80(5):695–705. https://doi.org/10.1016/0092-8674(95)90348-8
Ramos-Casals M, Loustaud-Ratti V, De Vita S et al (2005) Sjögren syndrome associated with hepatitis C virus: a multicenter analysis of 137 cases. Medicine (Baltimore) 84(2):81–89. https://doi.org/10.1097/01.md.0000157397.30055.c9
Ramos-Casals M, García-Carrasco M, Cervera R, Font J (1999) Sjögren’s syndrome and hepatitis C virus. Clin Rheumatol 18(2):93–100. https://doi.org/10.1007/s100670050064
Zheng M, Gao Y, Wang G et al (2020) Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol 17(5):533–535. https://doi.org/10.1038/s41423-020-0402-2
Huang C, Wang Y, Li X et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China The Lancet 395(10223):497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
Kim JY, Ko JH, Kim Y et al (2020) Viral load kinetics of SARS-CoV-2 infection in first two patients in Korea. J Korean Med Sci 35(7):e86. https://doi.org/10.3346/jkms.2020.35.e86
Ehrenfeld M, Tincani A, Andreoli L et al (2020) Covid-19 and autoimmunity. Autoimmun Rev. https://doi.org/10.1016/j.autrev.2020.102597
Podolska MJ, Biermann MH, Maueröder C, Hahn J, Herrmann M (2015) Inflammatory etiopathogenesis of systemic lupus erythematosus: an update. J Inflamm Res 8:161–171. https://doi.org/10.2147/JIR.S70325
Matteucci D, Paglianti M, Giangregorio AM, Capobianchi MR, Dianzani F, Bendinelli M (1985) Group B coxsackieviruses readily establish persistent infections in human lymphoid cell lines. J Virol 56(2):651–654
Nanbo A, Inoue K, Adachi-Takasawa K, Takada K (2002) Epstein-Barr virus RNA confers resistance to interferon-alpha-induced apoptosis in Burkitt’s lymphoma. EMBO J 21(5):954–965. https://doi.org/10.1093/emboj/21.5.954
de Wit E, van Doremalen N, Falzarano D, Munster VJ (2016) SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14(8):523–534. https://doi.org/10.1038/nrmicro.2016.81
Zuo Y, Yalavarthi S, Shi H et al (2020) Neutrophil extracellular traps in COVID-19. JCI Insight. https://doi.org/10.1172/jci.insight.138999
Papayannopoulos V (2018) Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18(2):134–147. https://doi.org/10.1038/nri.2017.105
Wigerblad G, Kaplan MJ (2020) NETs spread ever wider in rheumatic diseases. Nat Rev Rheumatol 16(2):73–74. https://doi.org/10.1038/s41584-019-0352-1
Wang D, Hu B, Hu C et al (2020) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan. China JAMA 323(11):1061–1069. https://doi.org/10.1001/jama.2020.1585
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A (2010) Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 191(3):677–691. https://doi.org/10.1083/jcb.201006052
Chan KH, Lee P, Chan CY, Lam KBH, Ho P (2020) Monitoring respiratory infections in covid-19 epidemics. BMJ. https://doi.org/10.1136/bmj.m1628
Baker MG, Gurney J, Oliver J et al (2019) Risk factors for acute rheumatic fever: literature review and protocol for a case-control study in New Zealand. Int J Environ Res Public Health. https://doi.org/10.3390/ijerph16224515
Stiemsma LT, Reynolds LA, Turvey SE, Finlay BB (2015) The hygiene hypothesis: current perspectives and future therapies. Immunotargets Ther 4:143–157. https://doi.org/10.2147/ITT.S61528
Bach J-F (2018) The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat Rev Immunol 18(2):105–120. https://doi.org/10.1038/nri.2017.111
Manasson J, Blank RB, Scher JU (2020) The microbiome in rheumatology: where are we and where should we go? Ann Rheum Dis 79(6):727–733. https://doi.org/10.1136/annrheumdis-2019-216631
Stojanovich L, Marisavljevich D (2008) Stress as a trigger of autoimmune disease. Autoimmun Rev 7(3):209–213. https://doi.org/10.1016/j.autrev.2007.11.007
Le TT, Andreadakis Z, Kumar A et al (2020) The COVID-19 vaccine development landscape. Nat Rev Drug Discov 19(5):305–306. https://doi.org/10.1038/d41573-020-00073-5
Ahmed SS, Volkmuth W, Duca J et al (2015) Antibodies to influenza nucleoprotein cross-react with human hypocretin receptor 2. Sci Transl Med 7(294):294ra105. https://doi.org/10.1126/scitranslmed.aab2354
Agrawal AS, Tao X, Algaissi A et al (2016) Immunization with inactivated Middle East Respiratory Syndrome coronavirus vaccine leads to lung immunopathology on challenge with live virus. Hum Vaccines Immunother 12(9):2351–2356. https://doi.org/10.1080/21645515.2016.1177688
Deming D, Sheahan T, Heise M et al (2006) Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med 3(12):e525. https://doi.org/10.1371/journal.pmed.0030525
Li G, Fan Y, Lai Y et al (2020) Coronavirus infections and immune responses. J Med Virol 92(4):424–432. https://doi.org/10.1002/jmv.25685
Siddiqi HK, Mehra MR (2020) COVID-19 Illness in Native and Immunosuppressed States: a clinical-therapeutic staging proposal. J Heart Lung Transpl. https://doi.org/10.1016/j.healun.2020.03.012
Sönmez HE, Demir S, Bilginer Y, Özen S (2018) Anakinra treatment in macrophage activation syndrome: a single center experience and systemic review of literature. Clin Rheumatol 37(12):3329–3335. https://doi.org/10.1007/s10067-018-4095-1
Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ (2019) The other side of CAR T-Cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden. Am Soc Clin Oncol Educ Book 39:433–444. https://doi.org/10.1200/EDBK_238691
Huet T, Beaussier H, Voisin O et al (2020) Anakinra for severe forms of COVID-19: a cohort study. Lancet Rheumatol. https://doi.org/10.1016/S2665-9913(20)30164-8
McGonagle D, Sharif K, O’Regan A, Bridgewood C (2020) The role of cytokines including Interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev 19(6):102537. https://doi.org/10.1016/j.autrev.2020.102537
Watanabe E, Sugawara H, Yamashita T, Ishii A, Oda A, Terai C (2016) Successful tocilizumab therapy for macrophage activation syndrome associated with adult-onset still’s disease: a case-based review. Case Rep Med 2016:5656320. https://doi.org/10.1155/2016/5656320
Xu X, Han M, Li T et al (2020) Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci 117(20):10970–10975. https://doi.org/10.1073/pnas.2005615117
Cantini F, Niccoli L, Matarrese D, Nicastri E, Stobbione P, Goletti D (2020) Baricitinib therapy in COVID-19: a pilot study on safety and clinical impact. J Infect. https://doi.org/10.1016/j.jinf.2020.04.017
Funding
No funding or support was received for the work.
Author information
Authors and Affiliations
Contributions
SS, VSN, and DD have initially conceptualized the review; SS, CKG, SD, and AMB were involved in drafting and critically revising the review; SS prepared all the figures with inputs from CKG and VSN; VSN and DD have provided expert inputs and updated the final review. All authors have provided substantial contributions to the conception and design of the work along with the interpretation. All authors have substantially contributed in drafting the manuscript and revising it critically for important intellectual content. All authors have approved the final version of the manuscript. All authors agree to be accountable for all aspects of the work related to accuracy and integrity.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shah, S., Danda, D., Kavadichanda, C. et al. Autoimmune and rheumatic musculoskeletal diseases as a consequence of SARS-CoV-2 infection and its treatment. Rheumatol Int 40, 1539–1554 (2020). https://doi.org/10.1007/s00296-020-04639-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-020-04639-9