
Abstract
The aging immune system (immunosenescence) has been implicated with increased morbidity and mortality in the elderly. Of note, T cell aging and low-grade inflammation (inflammaging) are implicated with several age-related conditions. The expansion of late-differentiated T cells (CD28−), regulatory T cells, increased serum levels of autoantibodies, and pro-inflammatory cytokines were implicated with morbidities during aging. Features of accelerated immunosenescence can be identified in adults with chronic inflammatory conditions, such as rheumatoid arthritis, and are predictive of poor clinical outcomes. Therefore, there is an interplay between immunosenescence and age-related diseases. In this review, we discuss how the aging immune system may contribute to the development and clinical course of age-related diseases such as neurodegenerative diseases, rheumatoid arthritis, cancer, cardiovascular, and metabolic diseases.
Similar content being viewed by others

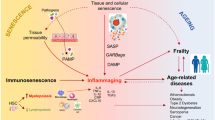

Introduction
Aging is a major risk factor for the higher incidence and prevalence of chronic conditions, such as cardiovascular diseases, metabolic diseases, and neurodegenerative diseases. Chronic systemic sterile inflammation is crucially involved with the etiology and progression of these conditions [1]. Several features of premature aging have been reported in young adults or adults with these chronic conditions (Fig. 1). In the elderly, these conditions are often presented with multimorbidity and may finally lead to organ failure and death. With the advance of immunosenescence (aging of the immune system), older adults also become more susceptible to infectious diseases and cancer. Elderly population is at increased risk for developing and dying from influenza and coronavirus disease 2019 (COVID-19). Of note, the adults with chronic (inflammatory) conditions are the ones with heightened risk for developing severe COVID-19 and dying [2]. Therefore, there is an interplay between immunosenescence and age-related diseases. In this way, it is important to intervene more quickly and multidimensionally with novel preventive and therapeutic approaches. The study of immunosenescence can bring viable solutions for the prevention and treatment of these diseases as well as to increase the healthspan of elderly populations.

Multiple mechanisms of accelerated aging are similarly found in age-related diseases. Abbreviations: CMV, cytomegalovirus; SASP, senescence-associated secretory phenotype
First of all, it is important to differentiate acute from chronic inflammatory processes. Acute inflammation is a transient and useful process aiming the elimination of pathogens and tissue regeneration, orchestrated by cells of the innate immunity. It is a self-regulated process with alarm, leukocyte mobilization and resolution phases. But aging starts a chronic inflammatory process, known as “inflammaging” [3], with persistent and non-resolved production of pro-inflammatory mediators (cytokines, chemokines, and acute phase proteins) that increases the risk for age-related morbidity and mortality. Some lifestyle factors, including smoking, obesity, and lack of exercise, are known to be associated with persistent inflammation. Although there are many sources of inflammaging, some evidence indicates the presence of overt infections during life to fuel inflammaging [4]. Age-related intrinsic factors may also contribute to the inflammaging. When cells reach senescence, they produce cytokines, chemokines, growth factors, proteases, and angiogenic factors that characterize a senescence-associated secretory phenotype (SASP) [5]. As senescent cells accumulate during aging, SASP may also contribute to inflammaging. Inflammaging can be therefore interpreted as the complex result of the interplay between SASP, lifestyle factors, and of dysregulated innate immune cell functions with aging.
In this review, we will discuss how the aging immune system may contribute to the development and clinical course of age-related (chronic) diseases such as neurodegenerative diseases, rheumatoid arthritis, cancer, cardiovascular, and metabolic diseases.
Neurodegenerative diseases
Immunosenescence (of note inflammaging) has been repeatedly implicated with cognitive processes and neurodegenerative diseases. The most common age-related neurodegenerative diseases include Alzheimer’s disease (AD) and Parkinson’s disease (PD).
Inflammatory mechanisms (inflammaging) have been implicated to cognitive decline and dementia during aging. Peripheral inflammatory mediators, especially C-reactive protein (CRP), have been largely related to cognitive impairment in the elderly [6]. Also, the presence of high plasma CRP levels can predict, in 12 years, future memory impairments [7]. In addition to the contribution to cognitive impairments in non-pathological conditions, the presence of CRP is related to the formation of β-amyloid plaques in AD, as well as increased plasma levels are observed in individuals with PD whose mental faculties are reduced [8]. A cohort study involving 873 non-demented elderly (70–90 years) found that elevated levels of TNFα, IL-1β, IL-6, IL-10, and IL-12 were associated with poorer cognitive performance—adjusting for age, sex, education, and obesity [9]. In a 5-year prospective observational study with participants with metabolic syndrome and high inflammation (i.e., CRP and IL-6 levels), they were more likely than those without metabolic syndrome to develop cognitive impairment. Interestingly, subjects with metabolic syndrome but low inflammation did not have an increased likelihood of cognitive impairment, suggesting an important role for inflammation in metabolic syndrome-related cognitive impairment [10]. The underlying mechanisms of inflammation-induced cognitive impairment are known. The excessive and/or prolonged release of pro-inflammatory cytokines in the CNS leads to reduced brain-derived neurotrophic factor (BDNF) levels, associated with neurogenesis, glutamatergic activation (excitotoxicity), oxidative stress, and induction of apoptosis [11], which are all involved with cognitive decline. These are the main neuroinflammatory and neurodegenerative mechanisms involved in the development of dementia. Neuroinflammation is known to impair various brain functions, such as inhibition of hippocampal neurogenesis and impairing cognitive abilities [12]. However, it should be noted that pro-inflammatory cytokines, under physiological conditions, are beneficial for providing trophic support to neurons and strengthening neurogenesis, contributing to cognitive function [13].
Changes in T cell senescence have also been implicated with cognitive decline. Different stages of T cell differentiation can be determined based on the cell-surface expression of the costimulatory molecules CD27 and CD28: early-differentiated (CD27+CD28+), intermediate-differentiated (CD27−CD28+), and late-differentiated or aged T cells (CD27−CD28−). The CD28− T cells have shortened telomeres and display differential features (cytotoxic, immunosupressive, or regulatory) in various conditions [14]. Dramatic fluctuations in T cell subsets were identified in peripheral blood of AD patients: decreased percentages of naïve T cells, elevated memory cells, and great expansion in late-differentiated CD28− T cells (in both CD4+ and CD8+ populations) as compared to healthy young or older adults [15]. In non-pathological conditions, better cognitive performance was associated with lower numbers of effector memory CD4+ T cells and higher numbers of naive CD8+ T and B cells [16]. A recent study performed mass cytometry of peripheral blood mononuclear cells of AD and discovered an immune signature that consists of increased numbers of CD8+ T effector memory CD45RA+ (TEMRA) cells [17]. Interestingly, these cells were found in the brain adjacent to Aβ plaques and were found negatively associated with cognition. The CD8+ TEMRA cells were clonally expanded to Epstein–Barr virus (EBV) antigens, although there is no causal link between EBV infectivity and AD. The mechanism by which senescent T cells regulate cognition is largely unknown, but there is some evidence that they may promote neuroinflammation. Senescent effector memory T cells were shown to activate microglia, endowing with pro-inflammatory features [18]. Microglia participate in neuroprotection by clearing debris, suppressing inflammation, restoring blood-brain barrier (BBB) integrity and inducing cortical neurogenesis by stripping inhibitory synapses [19]. It should be noted, however, that some T cells are necessary to maintain cognition. Indeed, mice deficient in T cells are cognitively impaired, and repopulation with T cells from wild-type donors can reverse this defect [20]. Further studies are necessary to better understand the interplay between senescent T cell subsets, their specific cytokine profiles, and cognitive performance.
In addition, older adults identified with immune deficits and accelerated immunosenescence had poor cognition. In studies with European 80+ older adults, an immunological risk profile (IRP) was identified and characterized by a CD4:CD8 ratio of less than 1, expansion of late-differentiated T cells (CD28−), and increased CMV serology. In healthy adults, the CD4:CD8 ratio is around 2:1 [21], and the inversion of this index has been associated with early immunosenescence. The IRP+ older adults had higher mortality rates, which was greatly increased if the elderly person was both IRP+ and cognitively impaired [22]. Therefore, it appears that changes in immunosenescence and cognition have a synergistic effect that impacts the life expectancy of the elderly. In a previous cohort study with 362 healthy older adults (mean age 69 years), we identified a subgroup (16%) IRP+ [23]. This subgroup was identified with cognitive impairment, more functional dependence, and an elevated IgG serology for cytomegalovirus (CMV), another feature of immunosenescence. Together, these data indicate that changes in the peripheral innate and adaptive immune responses are related to cognition in both health and pathological conditions.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic inflammatory disease, associated with symmetrical and destructive inflammation in joints and other tissues. It affects 1% of the world’s population and is more frequent in women than in men (3:1 F/M ratio) [24]. Aging is associated with increased incidence of autoimmune diseases. For example, the incidence of RA significantly increases with aging, going from 7.3/100,000 in the 18–34 years age group to 107.3/100,000 in the 75–84 years age group [25]. Furthermore, RA has been considered a model of premature senescence because as the disease progresses, there is a higher prevalence of age-related diseases. The most frequent comorbidities observed in RA may include cancer, cardiovascular disease, lung disease, osteoporosis, and neuropsychiatric disorders [26]. Immunosenescence may have a special importance on the development of these co-morbidities, as they are all immune-mediated conditions.
Several features of premature immunosenescence have been observed in RA. These alterations include decreased thymic functionality, expansion of late-differentiated effector T cells, increased telomeric attrition, and increased production of pro-inflammatory cytokines (senescence-associated secretory phenotype) [27]. A significant expansion of late-differentiated T cells (CD4+CD28− and CD8+CD28−) in RA was reported several years ago [28, 29], as similarly observed during healthy aging [30]. Interestingly, RA patients with manifested extra-articular manifestations, had increased frequencies of these aged T cells [31]. The subset of CD28− T cells includes terminally differentiated memory cells re-expressing CD45RA (TEMRA), which may contribute to inflammaging and further aggravate RA by increased production of TNFα, IL-1β, IL-6, and interferon (IFN)γ under stimulation [32]. Furthermore, RA is also associated with accumulation of other senescent cells, including senescent synovial fibroblasts (p16INK4a+) that display an enhanced inflammatory phenotype [33]. More than just epiphonema, these changes have been implicated in poor disease outcomes.
The expansion of late-differentiated or aged CD28− T cells was linked with disease severity in RA [34]. Indeed, the aged CD4+ T cells were associated with worsening of inflammatory responses due to their strong capacity of Th1-type cytokine production [35]. The expansion of CD28− T cell was particularly observed in patients with extra-articular manifestations [31]. Treatment with anti-TNF was capable of significantly reducing CD8+CD28− T cells (but not CD4+CD28− T cells), and it was associated with better clinical response, as assessed by DAS28 scoring and CRP [36].
The RA progression has been linked with the development of age-related co-morbidities, including cardiovascular diseases, osteoporosis, and cognitive impairment. Patients with RA are at greater risk of developing cardiovascular complications [37]. The risk rates for developing myocardial infarction and coronary artery disease is two to three times higher in RA and can be observed even in the early stages of the disease. Changes in the cellular composition in the heart as well as in the atherosclerotic plaque may contribute to the early onset and progression of cardiovascular diseases. Previous studies have linked the expansion of senescent T cells with cardiovascular disease. Interestingly, the high numbers of CD4+CD28− T cells have been described in subjects with angina, myocardial infarction, acute coronary syndrome, abdominal aortic aneurysms, and, chronic heart failure [38,39,40]. Increased telomeric erosion was associated to the development of atherosclerotic plaque and cardiovascular disease in RA [41]. In addition, the telomere shortening was associated with increased autoreactivity to self-antigens and loss of immunological tolerance, indicating a potential age-related risk factor for the development of RA [42].
The early development of cognitive impairment in RA is another shared characteristic with aging. RA patients are indeed cognitively impaired in several cognitive dimensions when compared with controls, including deficits in attention and working memory, processing speed and executive functions, inhibitory functions, and verbal declarative memory [29, 43]. On average, patients with RA have cognitive performance reduced in 50% as compared with age-matched healthy controls. Of note, patients with active disease had worsened cognitive performance as compared with those in remission [43]. Several mechanisms are potentially underlying the cognitive dysfunction in RA. Persistent inflammation could be a mechanism, which is known to be involved with cognitive decline during healthy aging [7, 9]. The expansion of senescent cells constitutes another potential mechanism of cognitive dysfunction reported in RA. Indeed, we have shown that expansion of late-differentiated CD8+CD28− T cells was negatively correlated with the memory functions of RA patients [29]. In contrast, patients with higher numbers of memory T cells (CD45RO+) had better cognitive functions. Another potential immune-related mechanism for impaired cognition in RA involves the production of autoantibodies targeting brain antigens. During healthy aging, there is an increase in the levels of circulating autoantibodies, and this can be prematurely observed in RA. Of note, we have shown RA patients in remission had increased levels of autoantibodies specific for brain antigens, and this was inversely correlated with cognitive performance [44]. Therefore, the cognitive impairment in RA could also be explained by the detrimental action of CNS autoantibodies, as suggested by CNS demyelination observed in some RA patients [45]. More studies are necessary to investigate to what extent these autoantibodies are related to premature senescence in RA.
In summary, the immunosenescence may contribute to the aggravation of both articular and extra-articular manifestations in RA. Chronic inflammation may be fueling the generation of senescent T cells with pathogenic features, and novel therapeutic strategies aimed to limit senescent cells may be of great value in RA.
Cancer
Age is a major risk factor for solid cancers until the 9th decade of life. The incidence and mortality of cancer decrease dramatically after age 90 and is nearly absent in centenarians [46]. Several factors are involved with age-related increase in carcinogenesis, including the accumulation of senescent cells, inflammaging, and immunosenescence change associated with poor immune surveillance.
The concept of immunosurveillance was first proposed by Sir Frank MacFarlane Burnet in the 1950s, suggesting that immune efficacy may decrease during aging. Immunosurveillance includes the notion that cells of the immune system constantly supervises and control defective and senescent cells, avoiding development of cancerous cells. To date, it is widely known that the immune system can recognize and destroy the precursors of cancer [47], and this is largely mediated by cytotoxic CD8+ T and NK cells. However, recent advances have added an extension of this initial concept, in which the immune system not only acts to eliminate malignant cells but also in the selection of less immunogenic subclones. Immune cells may thus interact with the tumor cell not only in its formation but also in its progression, mainly through evasion mechanisms acquired by evading subclones—this process is known as immunoediting [48]. Cancer immunoediting includes three phases (elimination, equilibrium, and tumor escape) and are all affected by immunosenescence.
Deficits in cell-mediated immunity are largely involved with poor cancer immunosurveillance during aging. An efficient immunosurveillance is reached when cancer cells are fully eliminated before tumor formation. The cell-mediated mechanisms involved with efficient tumor control are complex but include effector actions of CD8+ and CD4+ T cells, NK, NK T, macrophages, dendritic cells (DCs), and secreted cytokines (such as IFNγ) [49]. Aging compromises both innate and adaptative immune responses, contributing to tumorigenesis and cancer progression. These immunosenescent changes include impaired cellular signaling (e.g., TLR and TCR signal transduction), blunted functional aspects (e.g., impaired cell proliferation, phagocytosis, and cytotoxic function), and various numerical changes involving T cell subsets [50]. There is an accumulation of highly differentiated T cells in cancer [51]. We have shown recently that women recently diagnosed with breast cancer had increased numbers of senescent T cells (CD27−CD28−) in parallel with a reduction of recent differentiated T cells (CD27+CD28+) [52]. The expansion of CD8+CD28− T cells has been observed in women with breast cancer during chemotherapy [53] and in lung cancer [54]. Also, the expansion of CD8+CD28− T cells in cancer patients was associated with the staging of cancer and low treatment response, probably because some immunotherapeutic interventions require the presence of these co-stimulatory molecules (i.e., CD28). The expansion of senescent cells may thus lead to an inefficient antitumor response [53, 55]. The T cell receptor (TCR) repertoire is significantly reduced during aging, particularly following 65 years of age and may limit the number of specific T cells targeting cancer antigens. Aging also leads to accumulation of cells with suppressor actions, characterized by the increased frequencies of regulatory T cells (Treg: CD4+CD25+FoxP3+) and myeloid-derived suppressor cells (MDSCs) [56]. These cells may inhibit the generation of antitumor responses by several mechanisms, including the secretion of cytokines (e.g., IL-10, TGFβ).
Inflammaging may also be implicated with higher incidence of cancer during aging. The inflammatory environment seems to favor the development of cancer through initiation, promotion, and progression of tumors. In addition, the aged microenvironment, of note within the tumor microenvironment, plays a key role in reprogramming tumor cells towards a SASP [57]. Although several chemotherapy therapies induce senescence in cancer, SASP can contribute negatively to cancer therapy. SASP has tumorigenic effects such as the increase in malignant phenotypes and tumor induction [5]. The chronic inflammation may facilitate carcinogenesis by inducing genetic mutations or via epigenetic mechanisms. It may also promote cancer progression and metastasis, by impairing T cell activation and expanding Tregs and MDSCs [56]. In addition, persistent chronic infections, such as the cytomegalovirus (CMV), have been associated with accelerated immunosenescence (of note in T cells and inflammaging) and could be another driving factor of poor cancer immunity [58]. Thus, immune dysfunction in elderly people could be a concerning factor for anti-tumor responses. Also, the ineffective response to the presence of new antigens, like reduced repertoire of T cells available and reactivation of chronic latent infections, such as herpesvirus are related to immune aging on the neoplastic process [59].
During progression of cancer, the tumor frequently induces a state of immunosuppression that favors the tumor escape. It is known that immunosuppressed individuals (e.g., therapeutic immunosuppression or HIV+ patients) have increased incidence of cancer, even though the malignant neoplasms observed in this population are different from those present in the older population [60]. Escape from immunosurveillance may occur due to several reasons, including the generation of exhausted and senescent CD8+ T cells exhibiting more marked upregulation of PD-1, CTLA-4, LAG-3, and TIM-3, which are all known to suppress the activation of T cells [61]. For example, PD-1 and its ligand (PD-L1) are targets for immunotherapy given the effectiveness of an antitumor response.
Cancer patients may have characteristics of premature cell aging. Premature cellular aging has been associated with poor clinical outcomes. Indeed, it was shown that cancer patients have shortened leukocyte telomeres [62], particularly patients with more advanced tumors. Patients with colorectal cancer with reduced telomeric size in circulating leukocytes had shorter survival than those who do not [63]. However, it is not clear whether these characteristics are present before the development of cancer or may appear as consequences of tumorigenesis.
Cardiovascular diseases
Aging is the most important determinant for cardiovascular diseases (CVD) and the principal cause of death worldwide. In the last decades, there has been a significant improvement in CVD treatment that contributed to increased lifespan, and the number of older people is likely to double by 2050. In this regard, in a fast-changing demographics, the cardiovascular risk factors (tobacco use, unhealthy diet, obesity, physical inactivity, excessive alcohol consumption, hypertension, diabetes, and hyperlipidemia) are highly prevalent; it is urgent to fully understand molecular mechanisms involved with age-related conditions for better prevention and therapeutic treatment.
Cardiovascular diseases are classified as a group of heart and blood vessel disorders, associated with features of organismal aging, loss of homeostasis, with increased morbidity and mortality rates [64]. Extensive evidence suggests that immunosenescence is associated with detrimental clinical outcomes (e.g., mortality, frailty, poor response to vaccination, etc.) [65]. Of note, inflammaging is a major risk factor for all chronic diseases and geriatric syndromes including CVD [66]. Elevated levels of pro-inflammatory cytokines contribute to the inflammaging process, augments the probability of endothelial damage, vascular remodeling impairment, atherosclerosis, and insulin resistance [67, 68]. The adverse effects of inflammaging are observed in frailty and hypertension [69], suggesting that the inability to fine control systemic inflammation could be a marker of tissue dysfunction associated with pathological aging.
During cardiac stress, ischemic injury, hypertension, and metabolic syndrome, necrotic cells release high amounts of high-mobility group box 1 (HMGB1) and heat shock protein 60 (HSP 60). These damage-associated molecular patterns (DAMPs) are recognized by pattern recognition receptors (PRRs) expressed mainly by innate immune cells. As a result, tissue cells (mainly M1 macrophages) and non-immune cells secrete large amounts of pro-inflammatory cytokines for recruitment of phagocytic immune cells, clearance of apoptotic and pyroptotic cells and tissue renewal. Classical biomarkers of inflammation of the innate immune response, including CRP, IL-1β, IL-6, TNFα, and several cell adhesion molecules are similarly linked to the occurrence of myocardial infarction and stroke, in healthy and individuals with known coronary disease [70]. Elevated secretion of TNFα, IL-6, and IL-1β from cardiomyocytes and peripheral tissues have been shown to play an important role in the pathogenesis and progression of myocardial dysfunction and plasma levels of these pro-inflammatory cytokines can predict the short- and long-term survival in patients with cardiac heart failure. Yet, persistent macromolecular damage is a hallmark of aging and a trigger of cellular senescence.
Considering that senescence is a physiological program also occurring during embryonic development and wound healing, the presence of senescent cells may be useful in some pathological conditions. For example, the presence of senescent cells in myocardial infarction, cardiac fibrosis, and atherosclerosis is beneficial as they may limit tissue fibrosis [71]. In this sense, the clearance of TP53-positive senescent myofibroblasts through recruitment of inflammatory phagocytic cells, driven by SASP factors, reduces fibrosis and collagen matrix deposition. In a similar way, the atherosclerotic plaque formation is inhibited by senescent cells. Initially, endothelial dysfunction is associated with reduced nitric oxide (NO) production and augmented vascular permeability. Increased activity from macrophage-derived release of pro-inflammatory cytokines induce proliferation of vascular smooth muscle cells (VSMC) and accumulation of oxidized cholesterol-containing low-density lipoprotein (LDL) particles, trapped by foam cells in the vessel walls. Inflammation induces the accumulation of VSMC and endothelial senescent cells characterized by expression of P21, TP53, SA-βGAL. However, pre-clinical models with gain or loss of function of TP53 suggested that senescent cells may limit proliferation of VCMS and protect the organism from the development of atherosclerotic plaques [72].
Anti-inflammatory M2 macrophages play an important role in tissue repair and remodeling for inflammation resolution. Recent evidence demonstrates yet a prevalence of pro-inflammatory M1 macrophages in the plaque atherosclerotic responsible for the acute coronary syndrome (ACS). Pro-atherosclerotic inflammatory cytokines (such as IL-6 and IL-12), as well as reactive oxygen (ROS) and nitrogen (RNS) species amplifies the oxidative stress present in the plaque. Increased levels of Th1 and Th17 and its cytokines, such as IL-17, IL-21, and IL-23, have been described in atherosclerotic carotid artery plaques. These findings are found to be associated with the progression of disease and plaque vulnerability [17]. Intermediate senescent CD14+CD16+ monocytes are also present in the atherosclerotic plaques and express high levels of vascular adhesion molecules necessary for adhesion and diapedesis through endothelial cells [73].
The involvement of macrophages in aging and chronic inflammatory diseases has been described as “macroph-aging” [74]. This definition highlights a key role for the innate immune system, specially pro-inflammatory macrophages as contributors and intensifiers of premature aging and are present in chronic conditions such as CVD. However, accumulating evidence suggests that senescent T cells, mainly cytotoxic CD8+ T cells are also implicated in CVD pathogenesis.
In atherosclerosis and ACS, the CD8+CD28− T cells were found to be expanded and constituted a risk factor for vascular dysfunction in a cohort of CMV-infected individuals [75]. Many studies have reported the presence of senescent T cells (with shorter telomeres) in peripheral mononuclear cells (PBMCs) from patients with atherosclerosis [76] and myocardial infarction [77]. Although it is not possible to establish a causal effect, its accumulation suggests that the release of active mediators contribute to the pathophysiology of different age-related diseases.
Peripheral late-differentiated CD4+CD28− T cells are present in acute coronary events [39], and CD4+ effector memory T cells (CD3+CD4+CD45RA−CD45RO+CCR7−) were associated with atherosclerosis. The anti-inflammatory IL-10 might also be involved in the aging of the cardiovascular system, and high levels were found in hearts from healthy old mice and reduced atherosclerosis after Treg transfer. The involvement of IL-10 has been described in humans where low levels of IL-10 were associated with hard recovery after coronary artery bypass grafting [78].
Metabolic diseases
During the last decades, the prevalence of metabolic diseases such as type 2 diabetes (T2D), obesity, dyslipidemia, hepatic steatosis, and metabolic syndrome, has dramatically increased worldwide. This plethora of pathologies shows overlapping features with CVD and mortality. The systemic low-grade chronic inflammatory process presented in elderly individuals is a critical etiological component of physiological decline and risk factor for age-related diseases. Metabolic-driven inflammation, termed as metainflammation, is a major aspect of metabolic disorders which parallel resembles the same critical inflammatory process of aging [79]. The metabolic imbalance associated with the expansion of adipose tissue, metabolic dysfunction, systemic inflammation, insulin resistance, atherosclerosis, genomic instability, and dysregulated cellular metabolism is closely associated with cellular processes of premature aging and naturally present in elderly individuals. This suggests that both aging and metabolic diseases are interconnected processes.
This scenario has gained considerable attention in the last decades, especially when considering metabolic disorders as models of accelerated aging [66]. Inflammaging and metainflammation are present in T2D. One of the first mechanisms described to promote insulin resistance was the interference of insulin signaling driven by TNFα, establishing the first connection between metabolism and immune response [80]. Since then, several studies have been performed to clarify the link between T2D and immunity. Since T2D is commonly presented in the elderly, it is expected that its immunological profile overlaps immunosenescence but not necessarily in a causal relation. However, since inflammation-driven T2D is observed at early ages, this provides evidence that T2D is a model of premature immunosenescence [81].
A classical feature of immunosenescence presented in T2D is the decreased pool of CD4+ naïve T cells, concomitant with an increased pool of memory CD4+ T cells and effector CD4+ and CD8+ T cells. The heightened population of effector T cells is identified as the major producer of IFNγ and TNFα, enhancing the systemic pro-inflammatory status. Late-differentiated or senescent T cells (CD8+CD57+ and CD8+CD28−) have recently been demonstrated to predict the development of hyperglycemia in humans [82]. Similar to CVD, T2D patients had a reduced T cell repertoire, related to the increased naïve/effector T cell ratio. However, both adaptive and innate immune compartment shows impaired function and activation in T2D. Komura et al. have demonstrated diminished phagocytic activity and TLR responsiveness in peripheral blood monocytes of diabetic patients, as well as higher susceptibility to apoptosis in culture [83]. Those features were all correlated to increased markers of endoplasmic reticulum stress and related to poor glycemic control, a characteristic feature of this condition [83].
Hyperglycemia has already been described as a trigger of pro-inflammatory cytokines in innate immune cells. Elevated levels of glycated hemoglobin (HbA1c) are correlated with decreased phagocytic activity of circulatory monocytes and neutrophils, which is restored in patients treated with metformin, a drug that ameliorates insulin sensitivity and controls glycemic levels [84]. One of the proposed mechanisms of hyperglycemia-driven innate immune impairment is through over-activation of the Advanced Glycation End Products and Receptor for AGE (AGE-RAGE) pathway [81].
Obesity presents itself as a serious health condition strongly associated with inflammaging and constitutes a model of accelerated aging [85]. Adipose tissue dysfunction is implied as the central etiological component of obesity due to its role in metabolic homeostasis and high content of immune cells [86]. In this regard, impaired function of the immune system has been demonstrated in murine models of obesity. Experimental models confirm decreased migration rates, super anion production, and phagocytic activity in peritoneal macrophages that predispose organisms to increased susceptibility to pathogens [87]. Decreased chemotaxis of peritoneal lymphocytes and impaired proliferative and cytokine production in vitro stimulation, as well as increased oxidative stress markers in these cells has been observed [87].
Recent data provides evidence that mediators in the plasma of obese individuals are likely to promote premature immunosenescence. Healthy PBMCs exposed to plasma from obese individuals showed an increase in the pool of late-differentiated CD8+CD28− T cells with downregulation of CD28, a major aspect of T cell aging. Also, PBMC from obese individuals had shortened telomeres, indicating replicative senescence [88, 89].
The immunosenescent profile in obesity onset can also be demonstrated in early ages. Obese children (12 years) also have lower proportion of early CD4+ and CD8+ T cells and a higher proportion of effector memory and intermediate CD8+ T cells, as well as late and senescent CD4+ T cells. Attenuated alterations were found in overweight children (12 years), pointing out a correlation between increased body fat mass and the development of an immunosenescent profile [90].
As described for CVD, obesity shows hallmarks of immunosenescence, diminished responsiveness to vaccines, and heightened susceptibility to infections [91]. Paich et al. demonstrated impaired CD4+ and CD8+ T cell activation and function upon pH1N1 challenge in PBMC from post-vaccinated obese individuals, as a consequence of augmented frequencies of CD28− T cell associated with diminished in vitro CD8+ T cell activation after H1N1 challenge 12 months post-vaccination and reduced serum antibody titer [92].
It should be noted that adipose tissue has also been considered an endocrine organ. In addition to adipocytes, the vascular stromal fraction (VSF) is composed of a significant number of immune cells, mainly M2 macrophages, lymphocytes, and innate lymphoid cells (ILC2) that secrete Th2 cytokines, such as IL-4, IL-5, IL-10, IL-13, and IL-33, which are classically associated with an anti-inflammatory profile [65, 66]. Importantly, the adipose tissue is also made up of cells that do not belong to the immune system, such as mesenchymal stem cells (MSC) named for their location, stem cells derived from adipocytes (ADSC), endothelial cells, and fibroblasts, among others, in charge of producing extracellular matrix and spontaneous or induced cell regeneration under cell stress [93]. In this context, adipose tissue produces and releases several bioactive molecules that impact the individual in a systemic way, such as the secretion of several hormones, cytokines, chemokines, adipokines, enzymes, and lipids and more recently discovered the secretion of microRNAs, which are associated with the regulation of the immune system and immunometabolism.
Obesity alters the composition, structure and function of adipose tissue cells. There is recruitment of up to 60% of macrophages (M1) and lymphocytes (Th1) from the periphery with a pro-inflammatory profile (TNFα, IL-1β, IL-6), extracellular matrix remodeling associated with fibrosis, insulin resistance, and metabolic dysfunction, characteristics of the obesogenic state. The ADCS isolated from adipose tissue of individuals with obesity have different functions from those present in eutrophic individuals, secretes large amounts of monocyte chemo-attracting protein 1 (MCP-1) responsible for the greater recruitment of macrophages [94].
Obesity may also drive important immunosenescent changes in B cells. Frasca et al. have shown decreased in vitro antibody response to influenza on serum from obese individuals, associated with increased percentage of late/exhausted memory B cells—with high pro-inflammatory phenotype. Additionally, obese patients presented higher unstimulated B cell production of TNFα, resembling those presented in elderly individuals. These cells were negatively correlated with anti-influenza antibody after in vitro stimulation and were associated with higher basal TLR4 expression and increased pro-inflammatory cytokine production [95]. The heightened pro-inflammatory B cell pool is implied as the main driver of T cell inflammatory profile in obesity and T2D. Inflammatory B cells are characterized by increased basal secretion of IL-6 and IFNγ concomitant with accentuated decreased secretion of IL-10, sustaining chronic low-grade inflammation. B cell-null (μMT) obese mice show improved insulin sensitivity and reduced pro-inflammatory T cell Th17/Th1 function, supporting the role of B cells on the inflammatory obesity state [96]. Sustained elevated leptin levels due to adipose tissue endocrine dysregulation have recently been implied as a key factor driving B cell immunosenescence [97]. Briefly, leptin is secreted by adipose tissue and presents a positive immunomodulatory function that became deleterious at high chronical levels. Comparable obesity leptin levels downregulate class switch and IgG production in vitro of B cells from eutrophic individuals to the same extent as observed in non-leptin-treated B cells from obese individuals. Such impairment is attributed to the upregulation of inflammatory markers in leptin-treated B cells [97, 98].
Conclusions
In the last decades, lifespan has increased rapidly (of note in developing countries) in parallel with increased incidence of age-related pathological conditions. Changes in innate and adaptive immune responses observed in healthy older adults are found to be anticipated in chronic inflammatory conditions (Table 1). Classical alterations of immunosenescence include thymic involution, expansion of late-differentiated effector T cells, and increased levels of peripheral pro-inflammatory cytokines, known as inflammaging.
Not surprisingly, immunosenescence and SASP have been observed in older adults and during the developmental course of many immune-mediated conditions. Age-related diseases such as neurodegenerative diseases, RA, CVD, metabolic disorders, and cancer share common features of immunosenescence. Adverse effects of chronic low-grade inflammation increase the risk for the early appearance of diseases associated with age, suggesting that both aging and chronic (immune-mediated) diseases are interconnected states with common characteristics. A causal association between regulation of the immune response, induction of immunosenescence, and lifespan in humans remains elusive. Defining disease-specific signaling pathways that regulate the immune response towards immunosenescence is an important milestone for defining the role of immunosenescence in chronic diseases. In this way, it is important to intervene more quickly and multidimensionally with novel preventive and therapeutic approaches. The study of immunosenescence can bring viable solutions for the prevention and treatment of age-related diseases as well as to increase the healthspan of elderly populations.
References
Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, Ferrucci L, Gilroy DW, Fasano A, Miller GW, Miller AH, Mantovani A, Weyand CM, Barzilai N, Goronzy JJ, Rando TA, Effros RB, Lucia A, Kleinstreuer N, Slavich GM (2019) Chronic inflammation in the etiology of disease across the life span. Nat Med 25:1822–1832. https://doi.org/10.1038/s41591-019-0675-0
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
Franceschi C, Bonafè M, Valensin S, Olivieri F, de Luca M, Ottaviani E, de Benedictis G (2000) Inflamm-aging: an evolutionary perspective on Immunosenescence. Ann N Y Acad Sci 908:244–254. https://doi.org/10.1111/j.1749-6632.2000.tb06651.x
Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S (2017) Inflammaging and ‘Garb-aging’. Trends Endocrinol Metab 28:199–212. https://doi.org/10.1016/j.tem.2016.09.005
Sun Y, Coppé JP, Lam EWF (2018) Cellular senescence: the sought or the unwanted? Trends Mol Med 24:871–885. https://doi.org/10.1016/j.molmed.2018.08.002
Chen JM, Cui GH, Jiang GX, Xu RF, Tang HD, Wang G, Chen SD, Cheng Q (2014) Cognitive impairment among elderly individuals in Shanghai suburb, China: association of C-reactive protein and its interactions with other relevant factors. Am J Alzheimers Dis Other Dement 29:712–717. https://doi.org/10.1177/1533317514534758
Komulainen P, Lakka TA, Kivipelto M, Hassinen M, Penttila IM, Helkala EL, Gylling H, Nissinen A, Rauramaa R (2007) Serum high sensitivity C-reactive protein and cognitive function in elderly women. Age Ageing 36:443–448. https://doi.org/10.1093/ageing/afm051
Lindqvist D, Hall S, Surova Y, Nielsen HM, Janelidze S, Brundin L, Hansson O (2013) Cerebrospinal fluid inflammatory markers in Parkinson’s disease - associations with depression, fatigue, and cognitive impairment. Brain Behav Immun 33:183–189. https://doi.org/10.1016/j.bbi.2013.07.007
Trollor JN, Smith E, Agars E, Kuan SA, Baune BT, Campbell L, Samaras K, Crawford J, Lux O, Kochan NA, Brodaty H, Sachdev P (2012) The association between systemic inflammation and cognitive performance in the elderly: the Sydney memory and ageing study. Age (Omaha) 34:1295–1308. https://doi.org/10.1007/s11357-011-9301-x
Yaffe K, Kanaya A, Lindquist K, Simonsick EM, Harris T, Shorr RI, Tylavsky FA, Newman AB (2004) The metabolic syndrome, inflammation, and risk of cognitive decline. J Am Med Assoc 292:2237–2242. https://doi.org/10.1001/jama.292.18.2237
Miller AH, Maletic V, Raison CL (2009) Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry 65:732–741. https://doi.org/10.1016/j.biopsych.2008.11.029
Di Benedetto S, Müller L, Wenger E et al (2017) Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci Biobehav Rev 75:114–128. https://doi.org/10.1016/j.neubiorev.2017.01.044
Goshen I, Kreisel T, Ounallah-Saad H, Renbaum P, Zalzstein Y, Ben-Hur T, Levy-Lahad E, Yirmiya R (2007) A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology 32:1106–1115. https://doi.org/10.1016/j.psyneuen.2007.09.004
Strioga M, Pasukoniene V, Characiejus D (2011) CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology 134:17–32. https://doi.org/10.1111/j.1365-2567.2011.03470.x
Larbi A, Pawelec G, Witkowski JM, Schipper HM, Derhovanessian E, Goldeck D, Fulop T (2009) Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis 17:91–103. https://doi.org/10.3233/JAD-2009-1015
Serre-Miranda C, Roque S, Santos NC et al (2015) Effector memory CD4+ T cells are associated with cognitive performance in a senior population. Neurol Neuroimmunol NeuroInflammation 2:e54. https://doi.org/10.1212/NXI.0000000000000054
Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, Chen K, Lehallier B, Channappa D, de Los Santos MB, McBride A, Pluvinage J, Elahi F, Tam GKY, Kim Y, Greicius M, Wagner AD, Aigner L, Galasko DR, Davis MM, Wyss-Coray T (2020) Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577:399–404. https://doi.org/10.1038/s41586-019-1895-7
Ritzel RM, Crapser J, Patel AR, Verma R, Grenier JM, Chauhan A, Jellison ER, McCullough LD (2016) Age-associated resident memory CD8 T cells in the central nervous system are primed to potentiate inflammation after ischemic brain injury. J Immunol 196:3318–3330. https://doi.org/10.4049/jimmunol.1502021
Katsel P, Haroutunian V (2019) Is Alzheimer disease a failure of mobilizing immune defense? Lessons from cognitively fit oldest-old. Dialogues Clin Neurosci 21:7–19
Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M (2004) T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci U S A 101:8180–8185. https://doi.org/10.1073/pnas.0402268101
Syrjälä H, Surcel H-M, Ilonen J (1991) Low CD4/CD8 T lymphocyte ratio in acute myocardial infarction. Clin Exp Immunol 83:326–328. https://doi.org/10.1111/j.1365-2249.1991.tb05636.x
Wikby A, Ferguson FG, Forsey RJ, Thompson J, Strindhall J, Lofgren S, Nilsson BO, Ernerudh J, Pawelec G, Johansson B (2005) An immune risk phenotype, cognitive impairment, and survival in very late life: impact of allostatic load in Swedish octogenarian and nonagenarian humans. J Gerontol Ser A 60:556–565
Luz Correa B, Ornaghi AP, Cerutti Muller G, Engroff P, Pestana Lopes R, Gomes da Silva Filho I, Bosch JA, Bonorino C, Bauer ME (2014) The inverted CD4:CD8 ratio is associated with cytomegalovirus, poor cognitive and functional states in older adults. Neuroimmunomodulation 21:206–212. https://doi.org/10.1159/000356827
Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, Kavanaugh A, McInnes IB, Solomon DH, Strand V, Yamamoto K (2018) Rheumatoid arthritis. Nat Rev Dis Prim 4:1–23. https://doi.org/10.1038/nrdp.2018.1
Doran MF, Pond GR, Crowson CS, O'Fallon WM, Gabriel SE (2002) Trends in incidence and mortality in rheumatoid arthritis in Rochester, Minnesota, over a forty-year period. Arthritis Rheum 46:625–631. https://doi.org/10.1002/art.509
van Onna M, Boonen A (2016) The challenging interplay between rheumatoid arthritis, ageing and comorbidities. BMC Musculoskelet Disord 17:184. https://doi.org/10.1186/s12891-016-1038-3
Bauer ME (2020) Accelerated immunosenescence in rheumatoid arthritis: impact on clinical progression. Immun Ageing 17:6. https://doi.org/10.1186/s12979-020-00178-w
Pawlik A, Ostanek L, Brzosko I, Brzosko M, Masiuk M, Machalinski B, Gawronska-Szklarz B (2003) The expansion of CD4+CD28- T cells in patients with rheumatoid arthritis. Arthritis Res Ther 5:R210–R213. https://doi.org/10.1186/ar766
Petersen LE, Grassi-Oliveira R, Siara T, Ribeiro dos Santos SG, Ilha M, de Nardi T, Keisermann M, Bauer ME (2015) Premature immunosenescence is associated with memory dysfunction in rheumatoid arthritis. Neuroimmunomodulation 22:130–137. https://doi.org/10.1159/000358437
Effros RB, Dagarag M, Spaulding C, Man J (2005) The role of CD8+ T-cell replicative senescence in human aging. Immunol Rev 205:147–157. https://doi.org/10.1111/j.0105-2896.2005.00259.x
Martens PB, Goronzy JJ, Schaid D, Weyand CM (1997) Expansion of unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum 40:1106–1114. https://doi.org/10.1002/art.1780400615
Sallusto F, Lenig D, Forster R et al (1999) Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401:708–712. https://doi.org/10.1038/44385
Del Rey MJ, Valín Á, Usategui A et al (2019) Senescent synovial fibroblasts accumulate prematurely in rheumatoid arthritis tissues and display an enhanced inflammatory phenotype. Immun Ageing 16:1–9. https://doi.org/10.1186/s12979-019-0169-4
Goronzy JJ, Matteson EL, Fulbright JW et al (2004) Prognostic markers of radiographic progression in early rheumatoid arthritis. Arthritis Rheum 50:43–54. https://doi.org/10.1002/art.11445
Bryl E, Vallejo AN, Matteson EL, Witkowski JM, Weyand CM, Goronzy JJ (2005) Modulation of CD28 expression with anti-tumor necrosis factor α therapy in rheumatoid arthritis. Arthritis Rheum 52:2996–3003. https://doi.org/10.1002/art.21353
Scarsi M, Ziglioli T, Airo P (2010) Decreased circulating CD28-negative T cells in patients with rheumatoid arthritis treated with abatacept are correlated with clinical response. J Rheumatol 37:911–916. https://doi.org/10.3899/jrheum.091176
Solomon DH, Reed GW, Kremer JM, Curtis JR, Farkouh ME, Harrold LR, Hochberg MC, Tsao P, Greenberg JD (2015) Disease activity in rheumatoid arthritis and the risk of cardiovascular events. Arthritis Rheum 67:1449–1455. https://doi.org/10.1002/art.39098
Gerli R, Schillaci G, Giordano A, Bocci EB, Bistoni O, Vaudo G, Marchesi S, Pirro M, Ragni F, Shoenfeld Y, Mannarino E (2004) CD4+CD28- T lymphocytes contribute to early atherosclerotic damage in rheumatoid arthritis patients. Circulation 109:2744–2748. https://doi.org/10.1161/01.CIR.0000131450.66017.B3
Dumitriu IE, Araguás ET, Baboonian C, Kaski JC (2009) CD4+CD28null T cells in coronary artery disease: when helpers become killers. Cardiovasc Res 81:11–19. https://doi.org/10.1093/cvr/cvn248
Koller L, Richter B, Goliasch G, Blum S, Korpak M, Zorn G, Brekalo M, Maurer G, Wojta J, Pacher R, Hülsmann M, Niessner A (2013) CD4+CD28null cells are an independent predictor of mortality in patients with heart failure. Atherosclerosis 230:414–416. https://doi.org/10.1016/j.atherosclerosis.2013.08.008
Ormseth MJ, Solus JF, Oeser AM, Bian A, Gebretsadik T, Shintani A, Raggi P, Stein CM (2016) Telomere length and coronary atherosclerosis in rheumatoid arthritis. J Rheumatol 43:1469–1474. https://doi.org/10.3899/jrheum.151115
Prelog M (2006) Aging of the immune system: a risk factor for autoimmunity? Autoimmun Rev 5:136–139. https://doi.org/10.1016/j.autrev.2005.09.008
Petersen LE, Baptista TSA, Molina JK, Motta JG, do Prado A, Piovesan DM, de Nardi T, Viola TW, Vieira ÉLM, Teixeira AL, Grassi-Oliveira R, Bauer ME (2018) Cognitive impairment in rheumatoid arthritis: role of lymphocyte subsets, cytokines and neurotrophic factors. Clin Rheumatol 37:1171–1181. https://doi.org/10.1007/s10067-018-3990-9
Baptista TSA, Petersen LE, Molina JK, de Nardi T, Wieck A, do Prado A, Piovesan DM, Keisermann M, Grassi-Oliveira R, Bauer ME (2017) Autoantibodies against myelin sheath and S100β are associated with cognitive dysfunction in patients with rheumatoid arthritis. Clin Rheumatol 36:1959–1968. https://doi.org/10.1007/s10067-017-3724-4
Hamed SA, Selim ZI, Elattar AM, Elserogy YM, Ahmed EA, Mohamed HO (2012) Assessment of biocorrelates for brain involvement in female patients with rheumatoid arthritis. Clin Rheumatol 31:123–132. https://doi.org/10.1007/s10067-011-1795-1
Pavlidis N, Stanta G, Audisio RA (2012) Cancer prevalence and mortality in centenarians: a systematic review. Crit Rev Oncol Hematol 83:145–152. https://doi.org/10.1016/j.critrevonc.2011.09.007
Zitvogel L, Tesniere A, Kroemer G (2006) Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol 6:715–727. https://doi.org/10.1038/nri1936
Teng MWL, Kershaw MH, Smyth MJ (2013) Cancer immunoediting: from surveillance to escape. In: Cancer immunotherapy, 2nd edn. Immune suppression and tumor growth 3:85–99. https://doi.org/10.1016/B978-0-12-394296-8.00007-5
Swann JB, Smyth MJ (2007) Review series immune surveillance of tumors. J Clin Invest 117:1137–1146. https://doi.org/10.1172/JCI31405.antigens
Fulop T, Larbi A, Kotb R, Pawelec G (2013) Immunology of aging and cancer development. Interdiscip Top Gerontol 38:38–48. https://doi.org/10.1159/000343599
Hotta K, Sho M, Fujimoto K, Shimada K, Yamato I, Anai S, Konishi N, Hirao Y, Nonomura K, Nakajima Y (2011) Prognostic significance of CD45RO memory T cells in renal cell carcinoma. Br J Cancer 105:1191–1196. https://doi.org/10.1038/bjc.2011.368
Trintinaglia L, Bandinelli LP, Grassi-Oliveira R, Petersen LE, Anzolin M, Correa BL, Schuch JB, Bauer ME (2018) Features of immunosenescence in women newly diagnosed with breast cancer. Front Immunol 9:1651. https://doi.org/10.3389/fimmu.2018.01651
Song G, Wang X, Jia J, Yuan Y, Wan F, Zhou X, Yang H, Ren J, Gu J, Lyerly HK (2013) Elevated level of peripheral CD8+CD28- T lymphocytes are an independent predictor of progression-free survival in patients with metastatic breast cancer during the course of chemotherapy. Cancer Immunol Immunother 62:1123–1130. https://doi.org/10.1007/s00262-013-1424-8
Meloni F, Morosini M, Solari N, Passadore I, Nascimbene C, Novo M, Ferrari M, Cosentino M, Marino F, Pozzi E, Fietta AM (2006) Foxp3 expressing CD4+ CD25+ and CD8+CD28- T regulatory cells in the peripheral blood of patients with lung Cancer and pleural mesothelioma. Hum Immunol 67:1–12. https://doi.org/10.1016/j.humimm.2005.11.005
Chen IH, Lai YL, Wu CL, Chang YF, Chu CC, Tsai IF, Sun FJ, Lu YT (2010) Immune impairment in patients with terminal cancers: influence of cancer treatments and cytomegalovirus infection. Cancer Immunol Immunother 59:323–334. https://doi.org/10.1007/s00262-009-0753-0
Verschoor CP, Johnstone J, Millar J, Dorrington MG, Habibagahi M, Lelic A, Loeb M, Bramson JL, Bowdish DME (2013) Blood CD33(+)HLA-DR(−) myeloid-derived suppressor cells are increased with age and a history of cancer. J Leukoc Biol 93:633–637. https://doi.org/10.1189/jlb.0912461
Fane M, Weeraratna AT (2020) How the ageing microenvironment influences tumour progression. Nat Rev Cancer 20:89–106. https://doi.org/10.1038/s41568-019-0222-9
Bauer ME, De la Fuente M (2016) The role of oxidative and inflammatory stress and persistent viral infections in immunosenescence. Mech Ageing Dev 158:27–37. https://doi.org/10.1016/j.mad.2016.01.001
Fülöp T, Larbi A, Pawelec G (2013) Human T cell aging and the impact of persistent viral infections. Front Immunol 4:1–9. https://doi.org/10.3389/fimmu.2013.00271
Bonafè M, Valensin S, Gianni W, Marigliano V, Franceschi C (2001) The unexpected contribution of immunosenescence to the leveling off of cancer incidence and mortality in the oldest old. Crit Rev Oncol Hematol 39:227–233. https://doi.org/10.1016/S1040-8428(01)00168-8
Kouidhi S, Elgaaied AB, Chouaib S (2017) Impact of metabolism on T-cell differentiation and function and cross talk with tumor microenvironment. Front Immunol 8:270. https://doi.org/10.3389/fimmu.2017.00270
Pan W, Du J, Shi M et al (2017) Short leukocyte telomere length, alone and in combination with smoking, contributes to increased risk of gastric cancer or esophageal squamous cell carcinoma. Carcinogenesis 38:12–18. https://doi.org/10.1093/carcin/bgw111
Jia H, Wang Z (2016) Telomere length as a prognostic factor for overall survival in colorectal cancer patients. Cell Physiol Biochem 38:122–128. https://doi.org/10.1159/000438614
Fulop T, Witkowski JM, Pawelec G et al (2014) On the immunological theory of aging. Aging Facts Theor 39:163–176. https://doi.org/10.1159/000358904
Pawelec G (2018) Age and immunity: what is “immunosenescence”? Exp Gerontol 105:4–9. https://doi.org/10.1016/j.exger.2017.10.024
Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A (2018) Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 14:576–590. https://doi.org/10.1038/s41574-018-0059-4
Ferrucci L, Fabbri E (2018) Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol 15:505–522. https://doi.org/10.1038/s41569-018-0064-2
Soysal P, Arik F, Smith L et al (2020) Inflammation, frailty and cardiovascular disease. Adv Exp Med Biol 1216:55–64. https://doi.org/10.1007/978-3-030-33330-0_7
Barton M, Husmann M, Meyer MR (2016) Accelerated vascular aging as a paradigm for hypertensive vascular disease: prevention and therapy. Can J Cardiol 32:680–686.e4. https://doi.org/10.1016/j.cjca.2016.02.062
Biasucci LM, La Rosa G, Pedicino D et al (2017) Where does inflammation fit? Curr Cardiol Rep 19:1–10. https://doi.org/10.1007/s11886-017-0896-0
Zhu F, Li Y, Zhang J, Piao C, Liu T, Li HH, du J (2016) Functional plasticity of adipose-derived stromal cells during development of obesity. Stem Cells Transl Med 5:893–900. https://doi.org/10.1371/journal.pone.0074535
Sanz-González SM, Barquín L, García-Cao I et al (2007) Increased p53 gene dosage reduces neointimal thickening induced by mechanical injury but has no effect on native atherosclerosis. Cardiovasc Res 75:803–812. https://doi.org/10.1016/j.cardiores.2007.05.002
Merino A, Buendia P, Martin-Malo A, Aljama P, Ramirez R, Carracedo J (2011) Senescent CD14 + CD16 + monocytes exhibit proinflammatory and proatherosclerotic activity. J Immunol 186:1809–1815. https://doi.org/10.4049/jimmunol.1001866
Franceschi C, Campisi J (2014) Chronic inflammation (Inflammaging) and its potential contribution to age-associated diseases. J Gerontol - Ser A Biol Sci Med Sci 69:S4–S9. https://doi.org/10.1093/gerona/glu057
Grahame-Clarke C, Chan NN, Andrew D, Ridgway GL, Betteridge DJ, Emery V, Colhoun HM, Vallance P (2003) Human cytomegalovirus seropositivity is associated with impaired vascular function. Circulation 108:678–683. https://doi.org/10.1161/01.CIR.0000084505.54603.C7
Grahame-Clarke C (2005) Human cytomegalovirus, endothelial function and atherosclerosis. Herpes 12:42–45
Tae Yu H, Youn JC, Lee J, Park S, Chi HS, Lee J, Choi C, Park S, Choi D, Ha JW, Shin EC (2015) Characterization of CD8+ CD57+ T cells in patients with acute myocardial infarction. Cell Mol Immunol 12:466–473. https://doi.org/10.1038/cmi.2014.74
Fulop T, Franceschi C, Hirokawa K, Pawelec G (2019) Handbook of Immunosenescence, 2a. Springer International Publishing, Cham
Hotamisligil GS (2017) Inflammation, metaflammation and immunometabolic disorders. Nature 542:177–185. https://doi.org/10.1038/nature21363
Hotamisligil GS, Peraldi P, Budavari A et al (1996) IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science (80-) 271:665–670. https://doi.org/10.1126/science.271.5249.665
Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, Schmidt AM, Orchard TJ, Fisher EA, Tall AR, Goldberg IJ (2013) Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab 17:695–708. https://doi.org/10.1016/j.cmet.2013.04.001
Ho LY, Kim SR, Han DH et al (2019) Senescent T cells predict the development of hyperglycemia in humans. Diabetes 68:156–162. https://doi.org/10.2337/db17-1218
Komura T, Sakai Y, Honda M, Takamura T, Matsushima K, Kaneko S (2010) CD14+ monocytes are vulnerable and functionally impaired under endoplasmic reticulum stress in patients with type 2 diabetes. Diabetes 59:634–643. https://doi.org/10.2337/db09-0659
Lecube A, Pachón G, Petriz J, Hernández C, Simó R (2011) Phagocytic activity is impaired in type 2 diabetes mellitus and increases after metabolic improvement. PLoS One 6:e23366. https://doi.org/10.1371/journal.pone.0023366
Salvestrini V, Sell C, Lorenzini A (2019) Obesity may accelerate the aging process. Front Endocrinol (Lausanne) 10:266. https://doi.org/10.3389/fendo.2019.00266
Trim W, Turner JE, Thompson D (2018) Parallels in immunometabolic adipose tissue dysfunction with ageing and obesity. Front Immunol 9:169. https://doi.org/10.3389/fimmu.2018.00169
Hunsche C, Hernandez O, De La Fuente M (2016) Impaired immune response in old mice suffering from obesity and premature immunosenescence in adulthood. J Gerontol - Ser A Biol Sci Med Sci 71:983–991. https://doi.org/10.1093/gerona/glv082
Parisi MM, Grun LK, Lavandoski P, Alves LB, Bristot IJ, Mattiello R, Mottin CC, Klamt F, Jones MH, Padoin AV, Guma FCR, Barbé-Tuana FM (2017) Immunosenescence induced by plasma from individuals with obesity caused cell signaling dysfunction and inflammation. Obesity 25:1523–1531. https://doi.org/10.1002/oby.21888
Grun LK, da Rosa Teixeira N, von Mengden L et al (2018) TRF1 as a major contributor for telomeres’ shortening in the context of obesity. Free Radic Biol Med 129:286–295. https://doi.org/10.1016/j.freeradbiomed.2018.09.039
Spielmann G, Johnston CA, O’Connor DP et al (2014) Excess body mass is associated with T cell differentiation indicative of immune ageing in children. Clin Exp Immunol 176:246–254. https://doi.org/10.1111/cei.12267
Tagliabue C, Principi N, Giavoli C, Esposito S (2016) Obesity: impact of infections and response to vaccines. Eur J Clin Microbiol Infect Dis 35:325–331. https://doi.org/10.1007/s10096-015-2558-8
Paich HA, Sheridan PA, Handy J, Karlsson EA, Schultz-Cherry S, Hudgens MG, Noah TL, Weir SS, Beck MA (2013) Overweight and obese adult humans have a defective cellular immune response to pandemic H1N1 influenza a virus. Obesity 21:2377–2386. https://doi.org/10.1002/oby.20383
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa JI, Ohtani M, Fujii H, Koyasu S (2010) Innate production of T H 2 cytokines by adipose tissue-associated c-Kit + Sca-1 + lymphoid cells. Nature 463:540–544. https://doi.org/10.1038/nature08636
Hui RZ, Kim EK, Kim H, Claycombe KJ (2007) Obesity-associated mouse adipose stem cell secretion of monocyte chemotactic protein-1. Am J Physiol Endocrinol Metab 293:E1153–E1158. https://doi.org/10.1152/ajpendo.00186.2007
Frasca D, Ferracci F, Diaz A, Romero M, Lechner S, Blomberg BB (2016) Obesity decreases B cell responses in young and elderly individuals. Obesity 24:615–625. https://doi.org/10.1002/oby.21383
DeFuria J, Belkina AC, Jagannathan-Bogdan M, Snyder-Cappione J, Carr JD, Nersesova YR, Markham D, Strissel KJ, Watkins AA, Zhu M, Allen J, Bouchard J, Toraldo G, Jasuja R, Obin MS, McDonnell ME, Apovian C, Denis GV, Nikolajczyk BS (2013) B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci U S A 110:5133–5138. https://doi.org/10.1073/pnas.1215840110
Frasca D, Diaz A, Romero M, Blomberg BB (2020) Leptin induces immunosenescence in human B cells. Cell Immunol 348:103994. https://doi.org/10.1016/j.cellimm.2019.103994
Frasca D, Diaz A, Romero M, Landin AM, Blomberg BB (2014) High TNF-α levels in resting B cells negatively correlate with their response. Exp Gerontol 54:116–122. https://doi.org/10.1016/j.exger.2014.01.004
Taipa R, das Neves SP, Sousa AL et al (2019) Proinflammatory and anti-inflammatory cytokines in the CSF of patients with Alzheimer’s disease and their correlation with cognitive decline. Neurobiol Aging 76:125–132. https://doi.org/10.1016/j.neurobiolaging.2018.12.019
Scheiblich H, Trombly M, Ramirez A, Heneka MT (2020) Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol 41:300–312. https://doi.org/10.1016/j.it.2020.02.002
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S, Gao Y, Chen J (2012) Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43:3063–3070. https://doi.org/10.1161/STROKEAHA.112.659656
Rossol M, Kraus S, Pierer M, Baerwald C, Wagner U (2012) The CD14 brightCD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the Th17 cell population. Arthritis Rheum 64:671–677. https://doi.org/10.1002/art.33418
Schauer D, Starlinger P, Reiter C, Jahn N, Zajc P, Buchberger E, Bachleitner-Hofmann T, Bergmann M, Stift A, Gruenberger T, Brostjan C (2012) Intermediate monocytes but not TIE2-expressing monocytes are a sensitive diagnostic Indicator for colorectal Cancer. PLoS One 7:e44450. https://doi.org/10.1371/journal.pone.0044450
Yang M, Gan H, Shen Q, Tang W, du X, Chen D (2012) Proinflammatory CD14 +CD16 + monocytes are associated with microinflammation in patients with type 2 diabetes mellitus and diabetic nephropathy uremia. Inflammation 35:388–396. https://doi.org/10.1007/s10753-011-9374-9
Anani W, Shurin MR (2017) Targeting myeloid-derived suppressor cells in cancer. In: Advances in Experimental Medicine and Biology. Springer New York LLC, New York, pp 105–128
Veglia F, Perego M, Gabrilovich D (2018) Myeloid-derived suppressor cells coming of age review-article. Nat Immunol 19:108–119. https://doi.org/10.1038/s41590-017-0022-x
Koetz K, Bryl E, Spickschen K, O'Fallon WM, Goronzy JJ, Weyand CM (2000) T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci U S A 97:9203–9208. https://doi.org/10.1073/pnas.97.16.9203
Wagner UG, Koetz K, Weyand CM, Goronzy JJ (1998) Perturbation of the T cell repertoire in rheumatoid arthritis. Proc Natl Acad Sci U S A 95:14447–14452. https://doi.org/10.1073/pnas.95.24.14447
Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, Chen W, Zhang C, Zhang Y (2016) Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol 13:167–179
Lurain NS, Hanson BA, Martinson J, Leurgans SE, Landay AL, Bennett DA, Schneider JA (2013) Virological and immunological characteristics of human cytomegalovirus infection associated with Alzheimer disease. J Infect Dis 15:564–572. https://doi.org/10.1093/infdis/jit210
Pierer M, Rothe K, Quandt D, Schulz A, Rossol M, Scholz R, Baerwald C, Wagner U (2012) Association of anticytomegalovirus seropositivity with more severe joint destruction and more frequent joint surgery in rheumatoid arthritis. Arthritis Rheum 64:1740–1749. https://doi.org/10.1002/art.34346
Moro-García MA, López-Iglesias F, Marcos-Fernández R, Bueno-García E, Díaz-Molina B, Lambert JL, Suárez-García FM, Morís de la Tassa C, Alonso-Arias R (2018) More intensive CMV-infection in chronic heart failure patients contributes to higher T-lymphocyte differentiation degree. Clin Immunol 192:20–29. https://doi.org/10.1016/j.clim.2018.03.015
Blum A, Peleg A, Weinberg M (2003) Anti-cytomegalovirus (CMV) IgG antibody titer in patients with risk factors to atherosclerosis. Clin Exp Med 3:157–160. https://doi.org/10.1007/s10238-003-0019-7
Wu J, Li L (2016) Autoantibodies in Alzheimer’s disease: potential biomarkers, pathogenic roles, and therapeutic implications. J Biomed Res 30:361–372
Zerche M, Weissenborn K, Ott C et al (2015) Preexisting serum autoantibodies against the NMDAR subunit NR1 modulate evolution of lesion size in acute ischemic stroke. Stroke 46:1180–1186. https://doi.org/10.1161/STROKEAHA.114.008323
Awadallah SM, Hamad M, Jbarah I, Salem NM, Mubarak MS (2006) Autoantibodies against oxidized LDL correlate with serum concentrations of ceruloplasmin in patients with cardiovascular disease. Clin Chim Acta 365:330–336. https://doi.org/10.1016/j.cca.2005.09.021
Funding
This article was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Rio Grande do Sul (FAPERGS), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES—Finance Code 001).
Author information
Authors and Affiliations
Contributions
MEB conceived the manuscript. FBT and MEB drafted the manuscript. All authors have revised and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no conflict of interest.
Additional information
This article is a contribution to the special issue on: Immunosenescence: New Biomedical Perspectives - Guest Editors: Claudio Franceschi, Aurelia Santoro and Miriam Capri
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Barbé-Tuana, F., Funchal, G., Schmitz, C.R.R. et al. The interplay between immunosenescence and age-related diseases. Semin Immunopathol 42, 545–557 (2020). https://doi.org/10.1007/s00281-020-00806-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-020-00806-z