
Abstract
Based on gene expression changes measured in the peripheral blood within the first 2 days after irradiation, we predicted a pancytopenia in a baboon model. Eighteen baboons were irradiated with 2.5 or 5 Gy. According to changes in blood cell counts, the surviving baboons (n = 17) exhibited a hematological acute radiation syndrome (HARS) either with or without a pancytopenia. We used a two stage study design where stage I was a whole genome screen (microarrays) for mRNA combined with a qRT-PCR platform for simultaneous detection of 667 miRNAs using a part of the samples. Candidate mRNAs and miRNAs differentially upregulated or downregulated (>2-fold, p < 0.05) during the first 2 days after irradiation were chosen for validation in stage II using the remaining samples and using throughout more sensitive qRT-PCR. We detected about twice as many upregulated (mean 2128) than downregulated genes (mean 789) in baboons developing an HARS either with or without a pancytopenia. From 51 candidate mRNAs altogether, 11 mRNAs were validated using qRT-PCR. These mRNAs showed only significant differences between HARS groups and H0, but not between HARS groups with and without pancytopenia. Six miRNA species (e.g., miR-574-3p, p = 0.009, ROC = 0.94) revealed significant gene expression differences between HARS groups with and without pancytopenia and are known to sensitize irradiated cells. Hence, in particular, the newly identified miRNA species for prediction of pancytopenia will support the medical management decision making.
Similar content being viewed by others


Introduction
In a large-scale radiological emergency, early detection of exposed individuals would be required in order to evaluate the extent of radiation injuries and, when needed, decide in favor of a hospitalization and assign an appropriate treatment [1–3]. In particular, after high-dose exposure (≥2 Gy single whole body dose), severe acute health effects (acute radiation syndrome, ARS) will occur and early diagnosis within 1–3 days after exposure is pivotal to hospitalize exposed individuals in specialized clinics and to start the appropriate treatment as soon as possible.
In approaches like MEdical TREatment ProtocOLs (METREPOL), early detected clinical signs and symptoms are used for prediction of the late occurring hematologic ARS (HARS, [4]). METREPOL categorizes HARS into four severity degrees (H1–4) based on blood cell count (BCC) changes in the weeks that follow the exposure: no HARS (H0), low (H1), medium (H2), severe (H3), and fatal (H4) HARS. With the decrease in neutrophils and platelets in the peripheral blood, the hematological syndrome of the ARS is characterized mainly by immune suppression and hemorrhage over time. We hypothesized that the depletion of BCC would be preceded by changes in gene expression causally or timely related to their later decline and, therefore, could serve as an early indication of late occurring HARS severity score.
In previous studies, we successfully identified certain messenger RNAs (mRNAs) and microRNAs (miRNAs) predicting the late occurring HARS severity [5, 6]. However, when using METREPOL, we often experienced difficulties in the categorization, since, e.g., neutrophil counts during follow-up could reflect an H2 while platelets appeared more representative of an H1 degree HARS. However, medical management decisions for H1 (e.g., no hospitalization required) differ considerably from H2 (hospitalization and active supportive care required). As a result, we ultimately merged categories and came up with, e.g., H1–2 or H2–3 degree HARS, which adds additional categories to the four HARS severity categories according to METREPOL. Communications with clinicians confirmed the view that the prediction of patients developing a clinical relevant HARS degree either without or with a pancytopenia would be the most relevant categories regarding medical management decision making. Hence, we simplified the current study and searched for gene expression changes, namely mRNA and miRNAs, within the first 2 days after exposure in order to predict a clinical relevant HARS associated with or without a pancytopenia.
In collaboration with the French Army Biomedical Research Institute, we assessed blood samples obtained from irradiated baboons taken before (day 0) and 1 and 2 days after partial/total body exposure. BCC were measured in these baboons during the entire follow-up period in order to detect a clinical significant HARS associated either with or without pancytopenia. Pancytopenia was defined as a reduced number of neutrophils <500/μl over ≥10 days combined with platelets ≤10,000/μl measured at least once during the follow-up and a reduced number of red blood cells corresponding to an Hb ≤ 8 g/dl measured at least once during the follow-up [7, 8]. On the blood samples taken before and 1–2 days after exposure, we performed a whole genome screening and identified protein-coding mRNA genes associated with late occurring clinical relevant HARS with and without pancytopenia. These mRNAs were then validated using qRT-PCR. We also screened for 667 miRNAs using a qRT-PCR platform. The selected candidate miRNAs were also validated on the remaining samples in stage II using the same qRT-PCR platform but restricting the analysis on the candidate miRNAs from stage I.
Materials and methods
Animals
Eighteen baboons were bred by the Centre National de la Recherche Scientifique (Rousset sur Arc, France) for the purpose of biomedical research. In the nonhuman primate facility of the French Army Biomedical Research Institute, the baboons were placed in individual cages at 21 °C, with a relative humidity of 55% and a 12-/12-h light-dark schedule. The animals received fresh fruit and solid food twice a day and had access to water ad libitum. The male baboons had an average age of 8.1 years (±3.3 years) and weighed 23.7 (±5.2 kg). The experiment was approved by the French Army Animal Ethics Committee (no. 2010/12.0). All baboons were treated in compliance with the European legislation related to animal care and protection in order to minimize pain and damage. The total number of baboons evaluated in this study decreased to 17, for reasons described below.
Irradiation
The animals were anesthetized with a combination of tiletamine and zolazepam (6 mg kg−1 intramuscularly, Zoletil 100; Virbac, Carros, France) before irradiation. Then, the baboons were placed in restraint chairs, sitting orthogonally, front to a horizontal and homogeneous field of gamma rays delivered by a 60Co source (IRDI 4000; Alsthom, Levallois, France) to perform either total body irradiation (TBI) or partial body irradiation (PBI). In order to attain different patterns of PBI, a 20-cm thick lead screen was used to shield different parts of the body as detailed in Table 1. Two baboons were exposed to 5 Gy TBI and two others to 2.5 Gy TBI. Eight different exposure patterns were simulated and two baboons were exposed per pattern which summed up to 16 baboons receiving PBI (for details, see ref. [9]; Table 1) corresponding to an equivalent TBI dose of 2.5 or 5 Gy. Two dose rates were used (8 cGy/min for 5 Gy TBI and 5 Gy 50% PBI and 32 cGy/min for all other situations) because the Cobalt 60 source was changed during this study. Moreover, to achieve the same homogeneous radiation field whatever the dose rate, all baboons were irradiated at the same distance from the source. Consequently, radiation exposures lasted between 8 and 62 min. The midline tissue (right anterior iliac crest) dose in air was measured with an ionization chamber. Delivered doses were controlled by alumina powder thermoluminescent dosimeters placed on different cutaneous areas (thorax, thoracic and lumbar vertebrae, head, tibia, femur, femoral head; for details, see ref. [9]).
Blood collection, determination of HARS severity scores, and pancytopenia
Using changes in BCC observed days to weeks after irradiation, the severity scores (0, unexposed, 1–4, low-severe degree) of the HARS could be determined following METREPOL [4]. Often, changes in lymphocyte counts or platelets over time indicated HARS degrees differing from each other so that intermediates between, e.g., HARS 2 and 3 had to be defined. Pancytopenia was identified based on a reduced number of neutrophils (<500/μl over >10 days), platelets (<10,000/μl), and red blood cells (Hb < 8 g/dl) [7, 8]. Reduced numbers of platelets and red blood cells had to be measured at least once during the follow-up. The HARS score as well as pancytopenia was based on changes in differential blood counts taken at up to 22 time points over the course of 7–203 days after exposure. Of note, whole blood samples for gene expression measurements were taken only before irradiation (0 h) and at 1 and 2 days after irradiation in order to predict late occurring HARS using radiation induced changes in gene expression preceding the development of HARS.
RNA extraction and quality control
Whole blood samples (2.5 ml) were processed following the PAXgene Blood RNA system (BD Diagnostics, PreAnalytiX GmbH, Hombrechtikon, Switzerland). In brief, blood was drawn into a PAXgene Blood RNA tube at the French Army Biomedical Research Institute. The tube was gently inverted (10 times) and stored at room temperature overnight then at −20°. After all samples were collected, the PaxGene tubes were sent to Germany for further processing. After thawing, washing, and centrifugation, cells in the supernatant were lysed (proteinase K) followed by addition of lysis/binding solution taken from the mirVana Kit (Life Technologies, Darmstadt, Germany). With the mirVana kit, total RNA, including small RNA species, was isolated by combining a phenol-chloroform RNA precipitation with further processing using a silica membrane. After several washing procedures, DNA residuals became digested on the membrane (RNAse free DNAse Set, Qiagen, Hilden, Germany). RNA was eluted in a collection tube and frozen at −20 °C. Quality and quantity of isolated total RNA were measured spectrophotometrically (NanoDrop, PeqLab Biotechnology, Erlangen, Germany). RNA integrity was assessed by the 2100 Agilent Bioanalyzer (Life Science Group, Penzberg, Germany), and DNA contamination was controlled by conventional PCR using an actin primer. We used only RNA specimens with a ratio of A260/A280 ≥ 2.0 (Nanodrop) and RNA integrity number (RIN) ≥ 7.5 for whole genome microarray (IMGM Laboratories, Martinsried, Germany) or RIN ≥ 7.3 for qRT-PCR analyses.
Stage I screening: whole genome microarray
The whole genome screening for differentially expressed genes (DEG) (protein-coding mRNAs) was performed on 25 RNA samples with a subsequent range of HARS scores (H0 n = 5; HARS without pancytopenia n = 2 × 5, on days 1 and 2 after exposure; HARS with pancytopenia n = 2 × 5, on days 1 and 2 after exposure; online resource 1). We used the Agilent oligo microarray GE 8x60K (Agilent Technologies, Waldbronn, Germany) combined with a one-color-based hybridization protocol of GeneSpring GX12 software for data analysis as described in detail elsewhere [10]. We analyzed gene expression by quantile normalized log2-transformed probe signals as an outcome. We used the nonparametric Mann-Whitney (MW) test to compare gene expression across HARS with and without pancytopenia groups using the unexposed group (H0) as the reference (control). Only those gene transcripts that had a call “present” in at least 60% of RNA specimens were included in the analysis of gene expression, and only genes with MW p values ≤0.05 and a >2-fold gene expression difference among compared groups were considered to represent a candidate gene for validation in stage II. Due to the explorative nature of this study, the low sample size and the nonparametric statistics employed, we did not correct for multiple comparisons on the screening stage I of the study but considered this within our bioinformatic approach as well as the validation stage II of our study where the numbers of hypotheses tested in parallel becomes reduced from about 20,000 (stage I) to 51 mRNAs and 23 miRNAs in stage II (see below). Gene expression data presented in this publication have been deposited at the NCBI’s Gene Expression Omnibus (GEO accession number GSE77254).
Bioinformatics
All genes associated with p values ≤0.05 and a >2-fold gene expression difference (up or down) relative to the reference underwent gene set enrichment analyses using PANTHER pathway software (http://www.pantherdb.org/, version 10.0). PANTHER groups genes with similar biological function based on their annotation (reference list was the current Homo sapiens GO database). For these p values, we corrected for multiple testing by employing the Bonferroni algorithm.
Stage II: validation of stage I candidate genes via qRT-PCR
For validating the mRNA candidate genes from stage I (screening) using remaining RNA samples (online resource 1), we used a custom low density array (LDA; high-throughput qRT-PCR platform) and TaqMan chemistry. A 1-μg RNA aliquot of each RNA sample was reverse transcribed using a two-step PCR protocol (High Capacity Kit). Four hundred microliters of cDNA (1 μg RNA equivalent) was mixed with 400 μl 2× RT-PCR master mix and pipetted into the eight fill ports of the LDA. Cards were centrifuged twice (1200 rpm, 1 min, Multifuge 3S-R, Heraeus, Germany), sealed, and transferred into the 7900 qRT-PCR instrument. The qRT-PCR was run for 2 h following the qRT-PCR protocol for 384-well LDA format. All measurements were run in duplicate.
A commercially available 384-well LDA was used that provided the simultaneous detection of 380 different miRNAs. Two different LDAs (type A and B) were combined so that the detection of 667 miRNA species (partly spotted in duplicate to completely fill the LDA) was possible. Aliquots from each RNA sample (in general 2 μg total RNA/LDA type A/B) were reversely transcribed without preamplification over 3 h using “Megaplex pools without preamplification l for microRNA expression analysis protocol.” Using different sets of primers, two kinds of cDNAs suitable for each of both LDAs were created. In a second step, the whole template cDNA and 450 μl 2× RT-PCR master mix were adjusted to a total volume of 900 μl by adding nuclease free water, and aliquots of 100 μl were pipetted into each fill port of a 384-well human LDA. Cards were centrifuged twice (see above), sealed, and transferred into the 7900 RTQ-PCR instrument and again, the 384-well LDA RTQ-PCR protocol was run over 2 h.
All technical procedures for qRT-PCR were performed in accordance with standard operating procedures implemented in our laboratory in 2008 when the Bundeswehr Institute of Radiobiology became certified according to DIN EN ISO 9001/2008. All chemicals for qRT-PCR using TaqMan chemistry were provided by Life Technologies, Darmstadt, Germany.
For the custom LDA, CT values were normalized relative to the 18S ribosomal RNA (rRNA) measured in an aliquot of the RNA samples using a 96-well format TaqMan qRT-PCR platform. We have found that this approach to normalization was more robust compared to the use of the internal control (GAPDH and 18S rRNA) spotted on the LDA. For the commercial LDA, we used the median miRNA expression on each LDA for normalization purposes, because this proved to be the more robust and slightly more precise method compared to a normalization approach using a housekeeping miRNA species provided on the LDA (data not shown). The CT values of the housekeeping gene was subtracted from the CT value of each of the spotted genes, following the ∆CT—quantitative approach for normalization purposes.
Statistical analysis
Using the quantitative gene expression results from stage II, we examined none (H0) vs HARS groups with and without pancytopenia and we compared HARS groups with each other. Descriptive statistics (n, mean, standard deviation, min, max) and p values (t test and the nonparametric Kruskal-Wallis test (KW), where applicable) were calculated for each of the variables (candidate mRNAs and miRNAs) and per time point. Logistic regression analysis was performed on binary outcome variable for each of the variables (genes) of interest separately (univariate). Binary outcome variables comprised comparisons of either HARS groups relative to the unexposed H0 group or between HARS groups with and without pancytopenia. Odds ratios (OR), 95% confidence intervals (95% CI), and corresponding p values (Wald chi-square) were calculated. We also determined the area under a receiver-operator characteristic (ROC) curve providing a reasonable indication of overall diagnostic accuracy. ROC areas of 1.0 indicate complete agreement between the predictive model and the known HARS group and thus a clear distinction between healthy (H0) animals and baboons’ subsequently showing clinically relevant HARS with or without pancytopenia. All calculations were performed using SAS (release 9.2, Cary, NC, USA).
Results
Material available for the two-stage study design
Due to unusual blood cell counts before irradiation and a sudden death after irradiation, one out of the 18 baboons had to be excluded, leaving 17 baboons eligible for analysis.
During the screening approach at stage I, we assessed 25 whole genome microarrays for 25 blood samples (Table 1, online resource 1). Blood samples collected before irradiation from five baboons were selected randomly and represented H0 degree HARS (n = 5). Five baboons developed an HARS with pancytopenia and five blood samples were selected on day 1 and day 2 after irradiation for screening purposes (n = 10). Ten blood samples from another five baboons developing a clinically relevant HARS without pancytopenia were chosen randomly on the first 2 days after irradiation (n = 10). The same blood samples were used for screening of 667 miRNAs employing a commercially available LDA.
For the validation of mRNA and miRNAs at stage II, we used all available blood samples irrespective of whether they were already used for screening purposes. For examinations of mRNAs, the sample numbers were 17, 5, and 13 for H0, HARS with pancytopenia (3 samples on day 1 and 2 samples on day 2 after irradiation), and clinically relevant HARS without pancytopenia (7 samples on day 1 and 6 samples on day 2 after irradiation), respectively (Online resource 1; Table 1). For examinations of miRNAs altogether, 50 samples were utilized comprising H0 (n = 16), HARS with pancytopenia (5 samples on days 1 and 2 after irradiation, total n = 10), and clinically relevant HARS without pancytopenia (12 samples on days 1 and 2 after irradiation, n = 24).
Identification of HARS with and without pancytopenia
Changes in blood cell counts were observed over up to 202 days after irradiation. A decline in neutrophils, platelets and red blood cells was observed (Fig. 1). Based on the METREPOL definition for HARS and our criteria for pancytopenia (see above) we identified HARS with pancytopenia (red lines) and clinically relevant HARS without pancytopenia (green lines).

Changes in blood cell counts of neutrophils (upper graph), platelets (middle graph), and red blood cells (hemoglobin, lower graph) are shown for all 18 baboons up to 203 days after exposure. HARS severity was determined separately for count changes in neutrophils, lymphocytes, and platelets during the whole follow-up starting at day 7. Gray dashed lines indicate limits (neutrophils: 0.5 × 1000/μl; platelets, 10 × 1000/μl; red blood cells/hemoglobin, 8 g/dl) for the definition of a pancytopenia
Stage I: RNA isolation and whole genome microarray results
From 2.5 ml whole blood, we isolated 10, 8.5, and 6.3 μg total RNA on average before irradiation and 1 and 2 days after irradiation, respectively. RNA integrity (RIN) with a mean value of 8.6 (stdev ±0.6, min 7.3, max 9.5) suggested high-quality RNA sufficient for running both methods.
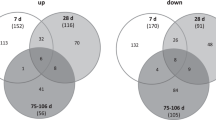
From about 20,000 protein-coding mRNAs, 46% on average (range: 34–54%) appeared expressed. An about equal number of 2000–2800 upregulated and downregulated DEG was observed on both days after irradiation in HARS either with or without pancytopenia (Fig. 2). As an exception, only 1379 DEG were observed at day 2 for HARS without pancytopenia. The overlapping number of DEG over both days was in the range of 71–86% for the upregulated genes and lower (22–29% for HARS with pancytopenia and 46–72% for HARS without pancytopenia) for the downregulated genes.

Venn diagrams showing the number of upregulated (left side) and downregulated (right side) protein coding genes (mRNA transcripts) observed for HARS with pancytopenia and HARS without pancytopenia. Differentially expressed genes (DEG) observed on both days after exposure are shown in the overlapping circle. Numbers outside the overlapping region represent the total number of differentially expressed genes that were not in common over day 1 to day 2. Percentages in parenthesis refer to the number of overlapping genes relative to the DEG of day 1 (first entry in parenthesis) and day 2 (second entry in parenthesis)
For the bioinformatic approach using PANTHER, at least 100 protein-coding genes (mRNAs) as input data are required. Therefore, PANTHER could be performed for the overlapping number of upregulated/downregulated mRNAs over both days and separately for HARS groups with and without pancytopenia (Table 2). Messenger RNAs coding for many biological processes (e.g., immune response or cell communication), protein classes (e.g., cytokine receptors), molecular functions (e.g., protein, RNA, or nucleic acid binding), and pathways (e.g., inflammation mediated by chemokine/cytokines or Toll receptor signaling) appeared overrepresented in HARS irrespective of whether a pancytopenia was developed or not. However, additional overrepresented numbers of mRNAs were observed for HARS with pancytopenia regarding biological processes (e.g., macrophage activation or protein phosphorylation), molecular functions (ion channel activity), and pathways namely the integrin and the apoptosis signaling pathways (bold entries, Table 2). For HARS without pancytopenia, additional overrepresented numbers of mRNAs were coding for biological processes or protein classes and were involved in mRNA processing or ribosomal proteins.
Based on the fold difference, the p value, and a preferable sustained changed mRNA expression over the 2 days after irradiation, we aimed to select candidate mRNAs for validation at stage II. Despite the high overlap in DEG over time (Fig. 2), we found no satisfying DEG being similarly expressed at both days. Also, all DEG of interest were differentially expressed in HARS with or without pancytopenia relative to H0. However, we experienced up to 6-fold differences in DEG in blood samples from baboons suffering from HARS with pancytopenia relative to HARS without pancytopenia. Using these prerequisites, we selected 51 candidate mRNAs (36 mRNAs for day 1 and 15 mRNAs for day 2) and forwarded them for validation in stage II using qRT-PCR.
During the screening of 667 miRNAs, we identified 23 miRNAs showing significant DEG of HARS with pancytopenia vs clinically relevant HARS without pancytopenia on days 1 (n = 17) and day 2 (n = 6) with two miRNAs (miR-584, miR-1290) overlapping on both days.
Stage II: validation using qRT-PCR measurements
During stage II validation of the 51 candidate mRNAs from stage I, 28 mRNAs showed either no amplification plot or amplification plots in a minority of all samples (≤3). Those were excluded from further analysis. Twelve genes revealed no significant changes in gene expression in baboons developing an HARS relative to H0 using qRT-PCR. There remained nine genes for identification of HARS with or without pancytopenia (Table 3) relative to H0 for the first day after exposure and two genes for the second day after exposure. Most of the genes from day 1 appeared 2-fold downregulated (e.g., CDCA7L or GBP2), but three were 3–5-fold upregulated (C11orf96, GLUL, TM4SF19) relative to H0 (Table 3) when developing a HARS without pancytopenia. For the second day, two genes appeared 2–3-fold downregulated when developing a HARS without pancytopenia. These fold differences increased up to 2-fold when developing an HARS with pancytopenia but did not become statistically significant (Table 3).
Examinations on miRNAs on day 1 after exposure showed five miRNA species (miR-124, miR-29c, miR-378, miR-574-3p, and rno-miR-7#) with significantly 1.7–2.6-fold higher mean DEG in baboons developing a HARS with pancytopenia vs those developing a clinically relevant HARS without pancytopenia (Table 3). For the second day, in particular, miR-133a appeared promising due to a 4.1-fold increased mean DEG for HARS with pancytopenia in comparison to HARS without pancytopenia (Table 3). The discrimination of HARS groups during screening using, e.g., miR-29c or miR-133 became reduced during the validation step (Fig. 3): Either the gene expression values of the unexposed group (miR-29C) or the HARS group without pancytopenia (miR-133) reached gene expression values overlapping with the HARS group comprising a pancytopenia (Fig. 3). During validation, in particular, miR-574-3p expression values (day 1 after exposure) of the HARS group with pancytopenia still discriminated from the HARS group without pancytopenia (ttest, p = 0.009; ROC = 0.96) or the unexposed control (ttest, p = 0.003; ROC = 0.94; Fig. 3). Also, HARS group without pancytopenia often revealed gene expression values comparable to H0 (e.g., validation of miR-574-3p, p = 0.48; Fig. 3).

Gene expression changes in the peripheral blood were examined in baboon groups being either unexposed (H0) or belonging to HARS groups with and without pancytopenia. We plotted the fold change (FC) of candidate genes from the screening stage relative to the mean H0 values on day 1 (miR-29c and miR-574-3p) and day 2 (miR-133a) after exposure. FCs of the genes from the screening stage are shown at the left side, and corresponding data from the validation stage are shown on the right side of the graphs. The validation stage includes additional samples not used during the screening stage I (see also Table 1). Symbols represent mean values and error bars reflect the SEM. The number of measurements per group is shown in Table 3
Discussion
We examined the possible clinical diagnostic utility of early radiation-induced gene expression changes on protein-coding mRNA species and noncoding miRNA species in the peripheral blood for the prediction of the late occurring hematological acute radiation syndrome (HARS) comprising a pancytopenia. We aimed to discriminate HARS with pancytopenia from baboons developing a clinically relevant HARS without suffering from pancytopenia. Regarding medical management decision making, it is desirable to know about a developing pancytopenia. During the screening approach, we identified 51 mRNAs and 23 miRNAs. Nine mRNA species and nine miRNA species showed significant differences of HARS groups with and without pancytopenia in comparison to the unexposed controls, but only six miRNA species revealed significant gene expression differences between HARS with pancytopenia and HARS without pancytopenia during the validation step. In particular, miR-574-3p appeared promising and the ROC (0.94) suggested a promising separation of both groups.
The nine mRNAs are coding for proteins involved in cell cycle regulation (CDCA7L), modification of the T cell and B cell immune response (GBP2, GLUL, HERC5, PPP3CC), erythropoiesis (GBP2), and cell migration of cancer and hematopoietic cells (HERC5, HMHA1). The synopsis of the literature search indicated a general link to hematopoiesis. Three genes were either of unknown function or unrelated. Interestingly, we did not find a reported association to radiation for all the mentioned mRNA species, in opposite to six out of nine identified miRNAs. Unexpectedly, none of the miRNAs were described in the context of hematopoiesis, but miR-146a and miR-378 showed an upregulation after high dose irradiation in vitro (for details, see online resource 2 (a/b)). Five miRNAs (miR-124#, rno-miR-7#, miR-133a, miR-29c, and miR-574-5p) were identified as being involved in radiosensitivity in cancer cell lines/stem cells. Since our miRNA candidates for pancytopenia prediction are not annotated for hematopoiesis but in part to radiosensitivity, it could be hypothesized that alterations in radiosensitivity controlled on a post-transcriptional level might be involved in the development of radiation induced pancytopenia. Noteworthy, since we examined gene expression in the whole blood, we can only speculate about their origin. This could be lymphocytes, granulocytes, and thrombocytes, an interaction of these cells with, e.g., the endothelium or other exposed body areas releasing miRNAs into the blood.
As an alternative to this approach, we already successfully predicted HARS severities defined according to METREPOL [5, 6]. Within these previous studies, we experienced difficulties with the categorization of the four HARS severity scores following the METREPOL protocol [4]. In these previous studies, we simplified the analysis and even merged HARS into new categories, namely developing a HARS (grades 1–3) or developing a more severe HARS (grades 2–3). With this approach, 29 mRNAs could be identified and discrimination was achieved independently with every single of 22 (H1–3) and 7 (H2–3) radiation-induced genes. Clearly, these two HARS categories seem to be more closely correlated to changes in gene expression. Hence, the newly identified candidate genes (in particular, miR-574-3p) for prediction of pancytopenia will add to the already identified gene set for the prediction of different HARS categories and support the medical management decision making.
Some limitations of our study should be kept in mind. We performed gene expression measurements on microarrays and qRT-PCR using human genomic sequences because the baboon transcriptome was not publically available. Given the high homology of both genomes (93%) and previously reports by other groups [11–13], we proceeded as described above. Since we used TaqMan chemistry with human primer and probe sequences (high sensitivity and specificity), it is more likely that we lost some of the detectable human RNA species due to a mismatch with the baboon genome, rather than producing false positives. Also, in preliminary validation studies using human samples from irradiated patients, we could already successfully reproduce most of our selected nine candidate genes from the previous baboon studies implying that differences in genomic sequences represent a minor problem.
The small sample size of our study represents a weakness and surely reduced the number of successfully validated candidate genes. Future work will consider larger sample sizes, and results will be validated on an additional species. As already mentioned, first preliminary validation studies of our previous baboon candidate genes in cancer patients undergoing bone marrow transplantation and TBI regimens are already looking promising and the new gene candidates for prediction of HARS with pancytopenia have to be validated accordingly.
In summary, we identified additional candidate genes (in particular miR-574-3p) for prediction of baboons developing a HARS with pancytopenia. These genes will add to the already identified gene set for the prediction of different HARS categories and, thus, support the medical management decision making.
References
Chaudhry MA (2008) Biomarkers for human radiation exposure. J Biomed Sci 15:557–563. doi:10.1007/s11373-008-9253-z
Weinstock DM, Case C, Bader JL et al (2008) Radiologic and nuclear events: contingency planning for hematologists/oncologists. Blood 111:5440–5445. doi:10.1182/blood-2008-01-134817
Wolbarst AB, Wiley AL, Nemhauser JB et al (2010) Medical response to a major radiologic emergency: a primer for medical and public health practitioners. Radiology 254:660–677. doi:10.1148/radiol.09090330
Friesecke I, Beyrer K, Fliedner TM (2001) How to cope with radiation accidents: the medical management. Br J Radiol 74:121–122. doi:10.1259/bjr.74.878.740121
Port M, Herodin F, Valente M, et al. (2016) First generation gene expression signature for early prediction of late occurring hematological acute radiation syndrome in baboons. 000:39–54. doi:10.1667/RR14318.1
Port M, Herodin F, Valente M, et al (2016) MicroRNA expression for early prediction of lateoccurring hematologic acute radiation syndrome in baboons. PLoS One 11:e0165307. doi:10.1371/journal.pone.0165307
(2009) Cross-sectional guidelines for therapy with blood components and plasma derivatives. doi:10.1159/isbn.978-3-8055-9393-9
Flowers CR, Seidenfeld J, Bow EJ et al (2013) Antimicrobial prophylaxis and outpatient management of fever and neutropenia in adults treated for malignancy: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 31:794–810. doi:10.1200/JCO.2012.45.8661
Valente M, Denis J, Grenier N et al (2015) Revisiting biomarkers of total-body and partial-body exposure in a baboon model of irradiation. PLoS One 10:e0132194. doi:10.1371/journal.pone.0132194
Abend M, Pfeiffer RM, Ruf C et al (2012) Iodine-131 dose dependent gene expression in thyroid cancers and corresponding normal tissues following the Chernobyl accident. PLoS One 7:e39103. doi:10.1371/journal.pone.0039103
Sherwin JRA, Hastings JM, Jackson KS et al (2010) The endometrial response to chorionic gonadotropin is blunted in a baboon model of endometriosis. Endocrinology 151:4982–4993. doi:10.1210/en.2010-0275
Shi Q, Cox LA, Glenn J et al (2010) Molecular pathways mediating differential responses to lipopolysaccharide between human and baboon arterial endothelial cells. Clin Exp Pharmacol Physiol 37:178–184. doi:10.1111/j.1440-1681.2009.05260.x
Aavik E, Mahapatra A, Boldrick J et al (2005) Correlation between gene expression and morphological alterations in baboon carotid after balloon dilatation injury. FASEB J 19:130–132. doi:10.1096/fj.04-2225fje
Acknowledgement
The sophisticated and carefully performed technical assistance of Sven Doucha-Senf and Eva Grumpelt is very much appreciated. We especially want to thank Dr. Michael Stadler and PD Dr. Christian Könecke from the Department of Hematology/Hanover Medical School for their advice in regard to clinical endpoints definitions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The experiment was approved by the French Army Animal Ethics Committee (no. 2010/12.0). All baboons were treated in compliance with the European legislation related to animal care and protection in order to minimize pain and damage.
Funding
This work was supported by both the French and the German Ministry of Defense.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Electronic supplementary material
Online resource 1.
The figure depicts a flow diagram of included samples, split study design, gene expression measurements and bioinformatics. Numbers in superscript: 1samples for stage II: a total of 35 samples for mRNA and another 50 samples for miRNA analysis were used for a validation including samples used in stage I as well. 2candidate genes were selected based on the p-value, the height and sustained differential gene expression over time. (PPT 102 kb)
Online resource 2 (a/b).
Gene description and annotation (Gene ID, gene name and chromosome location) of protein coding mRNAs and miRNAs (from Table 3) are provided in online resource Table 2 (a/b). A literature search (PubMed database/central) examined the association of these mRNA and miRNA species with the hematopoietic system (entries only found for mRNAs) and radiation exposure (entries only found for miRNA species). We employed either NCBI Entrez Gene database (mRNA) or mirBase (miRNA) for gene annotations. The online Resource 2b provides the miRNA ID of the utilized qRT-PCR platform (LDA) and the actual miRNA ID according to the mirBase (mirbase.org, November 2016) which was identified based on the deposited miRNA sequence of the LDA. 1miR-720 probably represents a fragment of a tRNA. (XLS 123 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Port, M., Hérodin, F., Valente, M. et al. Gene expression signature for early prediction of late occurring pancytopenia in irradiated baboons. Ann Hematol 96, 859–870 (2017). https://doi.org/10.1007/s00277-017-2952-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-017-2952-7