
Abstract
As neoadjuvant and adjuvant chemotherapy schedules often consist of multiple treatment cycles over relatively long periods of time, it is important to know what effects protracted drug administration can have on the immune system. Here, we studied the long-term effects of doxorubicin on the capacity of dendritic cell (DC) precursors to differentiate into a particular DC subset, the Langerhans cells (LC). In order to achieve high telomerase activity as detected in hematological stem cells, precursor cells from the acute-myeloid leukemia (AML)-derived cell line MUTZ3 were stably transduced with human telomerase reverse transcriptase (hTERT) to facilitate their growth potential, while preventing growth, and drug-induced senescence, and preserving their unique capacity for cytokine-dependent DC and LC differentiation. The hTERT-MUTZ3 cells were selected with increasing concentrations of the anthracyclin doxorubicin. After 1–2 months of selection with 30–90 nM doxorubicin, the cells completely lost their capacity to differentiate into LC. This inhibition turned out to be reversible, as the cells slowly regained their capacity to differentiate after a 3- to 4-month drug-free period and with this became capable again of priming allogeneic T cells. Of note, the loss and gain of this capacity to differentiate coincided with the loss and gain of a subpopulation within the CD34+ proliferative compartment with surface expression of the stem cell factor receptor (SCF-R/CD117/c-Kit). These data are in favor of cytostatic drug-free intervals before applying autologous DC-based vaccination protocols, as specific DC precursors may need time to recover from protracted chemotherapy treatment and re-emerge among the circulating CD34+ hematopoietic stem and precursor cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recent therapeutic advances are turning cancer into a more chronic disease. With patients being treated on and off with cytotoxic drugs in order to control metastasis, the effects of such treatment on the immune system in the long run should be considered. Safeguarding the immune competence of cancer patients may be vital to their quality of life as well as overall survival. In addition, successful application of novel immunotherapies also requires intact immune effector functions. Dendritic cells (DC) are the main orchestrators of the immune system and have a key function in linking the innate with the adaptive immune response [28, 33]. For autologous DC vaccination strategies, most often patient-derived material is used as a source of DC precursor cells. We therefore examined whether a long history of drug exposure could hamper DC differentiation.
The human acute-myeloid leukemia (AML)-derived DC cell line MUTZ3 [16] can develop either into interstitial DC (MUTZ3-IDC) by culturing the cells with granulocyte and macrophage-colony stimulating factor (GM-CSF), tumor necrosis factor alpha (TNFα), and interleukin-4 (IL-4) or into Langerhans cells (MUTZ3-LC) by culturing the cells with GM-CSF, TNFα, and transforming growth factor beta (TGFβ) [21, 22]. Of note, MUTZ-3 DC development accurately reflects all stages observed in its physiological CD34+ precursor-derived counterparts, and as such, this cell line presents a relevant and sustainable human DC/LC differentiation [23]. We recently found that short-term exposure of CD34+ cells, including MUTZ-3, to the cytotoxic drugs mitoxantrone or doxorubicin induced accelerated DC and LC differentiation with corresponding functionalities like migration, allogeneic T-cell stimulation, and cytotoxic T lymphocyte (CTL) priming (van de Ven et al. manuscript in preparation). These observations support the use of single-dose cytostatic drugs, as a differentiating agent to facilitate in vitro DC culture for therapeutic purposes. In this study, we examined the effect of long-term drug exposure on the capacity of DC precursor cells to differentiate into functional LC. For these long-term cultures, we made use of MUTZ3 cells that had been stably transduced with the gene encoding the catalytic subunit of the telomerase complex, the human telomerase reverse transcriptase (hTERT), in order to avoid replicative senescence and achieve high passage numbers with maintained cytokine-dependent differentiation ability. Human hematological precursor cells in vivo also express telomerase [13]. Derivation of this cell line for the first time allowed the in vitro study of the effects of truly long-term exposure of human precursor cells to cytostatic drugs on DC differentiation. Drug selection of these CD34+ DC precursors revealed that the selective loss of a SCF-R/c-KIT+ subpopulation resulted in an inability for LC differentiation. After removal of doxorubicin from the cultures, this subpopulation re-emerged as did the LC differentiation capacity. The regained capacity to differentiate after drug removal, as well as the capacity of high-passage hTERT-MUTZ3 cells to differentiate, rules out an hTERT-induced effect on LC differentiation. These data strongly suggest that a drug-free period of adequate length should be allowed for when autologous DC-based therapies are considered consecutive to chemotherapy, in order for the required precursors to recover.
Materials and methods
MUTZ3 and dendritic cell cultures
The AML-derived MUTZ3 cell line was cultured as described before [21]. In short, MUTZ3 progenitors were cultured in MEM-α (minimum essential medium, Lonza, Vervier, Belgium) containing 20% fetal calf serum (FCS), 100 IU/ml sodium-penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 50 μM β-mercaptoethanol (2ME), and 10% 5637 (renal cell carcinoma) conditioned medium (MUTZ3 routine medium) in 12-well plates (Costar) at a concentration of 0.2 million cells/ml and were passaged twice weekly. Langerhans cells (MUTZ3-LC) were cultured from MUTZ-3 progenitors with 10 ng/ml TGF-β1 (Biovision, Mountain View, CA), 1,000 IU/ml rhGM-CSF (Sagramostim, Berlex), and 120 IU/ml TNFα (Miltenyi Biotec, Bergisch Gladbach, Germany) for 10 days to obtain immature MUTZ3-LC as described previously [30]. Immature MUTZ3-LC were matured by adding 33% monocyte condition medium (MCM) and 2,400 IU/ml TNFα for 48 h.
Retroviral constructs
The retroviral vector LZRS-hTERT-IRES-ΔNGFR has been described previously [25]. The ΔNGFR is a truncated form of the receptor, with no signaling moiety. Retroviruses were propagated in Phoenix A cells by plasmid DNA transfection using Lipofectamin 2000 (Invitrogen, Breda, The Netherlands) in serum-free medium for 3 h. Medium was refreshed after 24 h, and viral supernatants were harvested 48 h post-transfection and were either directly used for transduction or stored at −80°C in 0.5 ml aliquots.
MUTZ3 retroviral transduction
For the generation of MUTZ3 cells expressing hTERT, MUTZ3 progenitor cells were transduced with the LZRS-hTERT-IRES-ΔNGFR retrovirus as described before [7, 25]. In short, 0.5 million MUTZ3 progenitor cells were transferred to fibronectin-coated (40 μg/ml RetroNectin, Takara, Japan), non-tissue culture–coated plates (BD Biosciences, Heidelberg, Germany) in 0.5 ml viral supernatant supplemented with 10% 5637 conditioned medium. Plates were centrifuged for 90 min at 2,000 rpm at 25°C. Cells were re-transduced with 0.5 ml fresh viral supernatant after 24 h. The retrovirus infected and transduced the CD34+ proliferative progenitor population of the MUTZ3 cell line. hTERT-MUTZ3 cells were obtained by selecting the NGFR-positive cells by flow sorting, and cells were maintained in MUTZ3 routine medium as described above. Doxorubicin selection was started by culturing the hTERT-MUTZ3 cells with 5 nM doxorubicin (Amersham Pharmacia, Roosendaal, The Netherlands). Cells were passaged twice weekly as described above. Drug concentrations were gradually increased. Differentiation analysis was performed at different time points during selection with 30 nM (dox30+) and 90 nM (dox90+) cells. Control, non-exposed hTERT-MUTZ3 cells were kept in culture alongside the drug-selected cells and were tested for their differentiating capacity using cells with the same passage numbers.
hTERT PCR
RNA was isolated using RNA-Bee (Bio-connect, Huissen, The Netherlands), following manufacturer’s guidelines. The RNA concentration was determined on a Nanodrop spectrophotometer. cDNA synthesis and PCR reaction were carried out essentially as described before and were performed with 200 ng RNA [5, 26]. Primers and probes for hTERT and the household control gene U1 small nuclear ribonucleoprotein-specific A protein (SnRNP U1A) were used as described previously [26]: hTERT forward 5′ → 3′ primer: GAAGGCACTGTTCAGCGTGCTCAAC; hTERT reverse 5′ → 3′ primer: GGTTTGATGATGCTGGCGATGACC; hTERT probe: GGCGCCTGAGCTGTACTTTGTCAAGGTGGA. SnRNP U1A forward 5′ → 3′ primer: CAGTATGCCAAGACCGACTCAGA; snRNP U1A reverse 5′ → 3′ primer: GGCCCGGCATGTGGTGCATAA; snRNP U1A probe: AGAAGAGGAAGCCCAAGAGCCA. The RT-PCR reaction was performed at 35 cycles.
XTT cytotoxicity assay
Twenty thousand cells were seeded per well in 96-well round-bottom plates in a volume of 50 μl per well in culture medium. Doxorubicin was diluted to a maximum concentration of 300 nM in culture medium and 1:3 diluted to 120 pM. Fifty microliters of diluted doxorubicin was added per well in triplicate for each concentration, giving rise to a doxorubicin range from 0.06 to 150 nM. Cells were exposed to the drug for 96 h. Cell viability was measured by adding XTT supplemented with phenazine methosulphate (PMS) for 2–4 h, after which optical density was measured at 450 nm. Cell viability was determined relative to the control cells to which no doxorubicin had been added.
Flow cytometry
LC were immunophenotyped using FITC- and/or PE-conjugated Mabs directed against CD1a (1:25), CD54 (1:25), CD80 (1:25), CD86 (1:25), CD40 (1:10) (PharMingen, San Diego, CA), CD14 (1:25), HLA-DR (1:25), DC-SIGN (1:10) (BD Biosciences, San Jose, CA), CD83 (1:10), CD34 (1:10), Langerin (1:10) (Immunotech, Marseille, France). hTERT MUTZ3 cells, dox90+ and dox90− cells were phenotyped for cytokine receptors using the following antibodies: CD34-APC, CD116-FITC, CD117-PE (Pharmingen, San Diego, CA), CD14-PerCP_Cy5, and CD123-PE (BD Biosciences, San Jose, CA). In short, 2.5–5 × 104 cells were washed in PBS supplemented with 0.1% BSA and 0.02% NaN3 and incubated with specific or corresponding isotype-matched control Mabs for 30 min at 4°C. Cells were washed and analyzed with a FACS-Calibur flow cytometer (Becton and Dickinson, San Jose, CA) equipped with CellQuest analysis software, and the results were expressed as mean or median fluorescence intensities, the percentage of positive cells, or the relative ratios of positive cells compared with the control cultures.
Mixed leukocyte reaction (MLR)
MLR was performed as described [30]. In short, 1 × 102–3 × 104 LC were co-cultured with 1 × 105 allogeneic peripheral blood lymphocytes for 4 days in 96-well plates in IMDM containing 10% human pooled serum (HPS) (Sanquin, Amsterdam, The Netherlands), 100 IU/ml sodium penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, and 50 μM β-mercaptoethanol. At day 4, 2.5 μCi/ml [3H]-thymidine (6.7 Ci/mmol, MP Biomedicals, Irvine, CA) was added per well for 16 h. Plates were harvested onto glass fiber filtermats (Packard Instruments, Groningen, The Netherlands) using a Skatron cell harvester (Skatron Instruments, Norway), and [3H]-thymidine incorporation was quantified using a Topcount NXT Microbetacounter (Packard, Meriden, CT).
Statistical analysis
Statistical analysis of the data was performed using the paired or unpaired two-tailed Student’s t test. Differences were considered statistically significant when P < 0.05.
Results
Doxorubicin selection of MUTZ3 cells
The anthracyclin doxorubicin was chosen as a selection drug to study the effects of long-term drug exposure on DC precursor cells, because of its high clinical relevance. Doxorubicin is widely used to treat a variety of cancer types, mostly in combination with other cytotoxic drugs, as in CHOP (cyclophosphamide, doxorubicin (=hydroxydaunorubicin), vincristine (=Oncovin), and prednisone) or M-VAC (methotrexate, vinblastine, doxorubicin (=Adriamycin), and cisplatin) regimens [14, 19, 20, 31, 32]. As a source of DC precursor cells, we made use of the MUTZ3 cell line [16] that can be differentiated into either interstitial DC or LC [21, 22]. The IC50 value of doxorubicin in MUTZ3 cells was determined by means of a XTT cytotoxicity assay [IC50: 29 ± 18.3 nM (n = 3)]. Although the cells could be cultured in the presence of doxorubicin up to a concentration of 30 nM, the selected cells displayed marginal resistance to the drug compared with the parent MUTZ3 cells when analyzed in a cytotoxicity test (data not shown) and the 30 nM-selected cells already lost their proliferative capacity after 9 passages. As a consequence, increasing the concentration of doxorubicin, in order to generate a drug-resistant, long-term exposed cell line, did not prove feasible.
The telomerase complex plays a vital role in preventing replicative senescence. Of note, wtMUTZ3 cells were found to lack the expression of hTERT mRNA (Fig. 1a), which might have contributed to the failure to generate a continuously proliferating, doxorubicin-resistant cell population. We therefore generated MUTZ3 progenitor cells stably expressing hTERT via transduction with a retroviral construct encoding hTERT (designated hTERT-MUTZ3). Expression of hTERT (Fig. 1a) allowed for prolonged culture (and hence longer drug exposure time) while maintaining dependence on exogenous cytokines for differentiation and growth, which is a typical feature of the MUTZ3 cell line [16, 23]. Moreover, since hTERT is expressed by hematopoietic stem cells and by hematological tumors [13], drug exposure of hTERT-positive MUTZ3 cells more closely resembles physiological in vivo conditions.

Telomerase expression and doxorubicin sensitivity of MUTZ3 and hTERT-MUTZ3 cells. a MUTZ3 progenitors and hTERT-MUTZ3 progenitors were analyzed for telomerase expression by RT-PCR. b MUTZ3 and hTERT-MUTZ3 progenitors were analyzed for their sensitivity to the anthracyclin doxorubicin by means of a XTT assay over 96 h (experiment representative of 3)
Introduction of hTERT into MUTZ3 precursor cells did not influence the sensitivity of the cells for doxorubicin (Fig. 1b) but did enhance their proliferation rate compared with non-transduced MUTZ3 cells (average expansion factor over 3 days 7.8 ± 1.3 and 5.7 ± 1.7 for hTERT-MUTZ3 and MUTZ3, respectively (P < 0.02), averaged over 8 consecutive passages). hTERT-MUTZ3 cells retained their ability to differentiate into CD1a+ Langerin+ LC (Fig. 2a, b). Figure 2c shows the proliferation rate (i.e. expansion factor over the course of 3 days) of MUTZ3 versus hTERT-MUTZ3 upon exposure to 30 nM doxorubicin. Whereas the MUTZ3 cells stopped proliferating after 9 passages, the hTERT-MUTZ3 cells maintained a relatively consistent proliferation rate on drug selection for over 160 passages (data shown for the first 50 passages). Of note, wtMUTZ3 cells generally display a considerable fluctuation in expansion factors between passages. The same fluctuating pattern was observed in the hTERT-MUTZ3 cells (Fig. 2c).

Effects of hTERT introduction on MUTZ3-LC differentiation and proliferation. a Dot plots of flow cytometric analysis for the LC-typifying markers CD1a and Langerin on immature LC (iLC) cultures from MUTZ3 (iLC; left) and hTERT-MUTZ3 (hT-iLC; right). b Average percentages (+standard deviation) of CD14+, CD34+, CD1a+ and Langerin+ cells within iLC cultures from MUTZ3 or hTERT-MUTZ3 cells (n = 3). c Progenitor cell proliferation in the presence of 30 nM doxorubicin. Shown are the passage numbers against the expansion of the progenitor cells. MUTZ3 progenitor cells stopped proliferating after 9 passages in the presence of 30 nM doxorubicin, whereas hTERT-MUTZ3 cells could be cultured for over 160 passages (shown are data up to passage 50)
Doxorubicin selection hampers LC differentiation
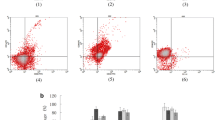
Doxorubicin concentrations used to condition the progenitor cell cultures were gradually increased, from 5 and 10 nM (Passage (P)22) up to 20 nM (P30), 30 nM (P55) (dox30+), and 90 nM (P67) (dox90+). The cells were analyzed for their sensitivity to doxorubicin and their LC differentiation capacity. Dox30+ cells were twofold and dox90+ cells were eightfold more resistant to doxorubicin compared with unselected hTERT-MUTZ3 cells (measured by XTT with P122 and P167). Culturing the cells under doxorubicin selection significantly affected their expansion factors, which were 7.3 ± 1.7 for unselected hTERT-MUTZ3, 5.0 ± 2.0 for dox30+, and 1.2 ± 2.0 for dox90+ (averaged over 13 passages) (P < 0.02 for both dox30+ and dox90+). LC differentiation from dox30+ to dox90+ cells was carried out in the absence of doxorubicin, and the non-exposed hTERT-MUTZ3 control cells were always differentiated from the same passage number as the drug-exposed dox30+ and dox90+ cells. Doxorubicin selection of the progenitor cells had no effect on the viability of the cells during LC culture (Fig. 3a). Cellular output after 10 days of differentiation (relative to input) was 1.06 ± 0.34 [range 0.58–1.35] for hTERT-MUTZ3 and 1.68 ± 0.88 [range 0.75–2.75] for dox90+ cells (n = 4). Selection did drastically reduce the capacity of the cells to differentiate into LC, reflected by significantly reduced levels of the LC-typifying markers CD1a and Langerin, as well as of the co-stimulatory markers CD40 and CD86 and of the antigen-presenting molecule HLA-DR (Fig. 3b) (n = 3; differentiation induced at P73, P83 and P189, data shown for dox 90+ cells; similar results were obtained with the dox30+ cells [data not shown]).

Doxorubicin selection of progenitor cells hampers LC differentiation. a Output of viable hTERT-MUTZ3 and dox90+ hTERT-MUTZ3 cells after 10 days of LC differentiation b hTERT-MUTZ3 and dox90+ hTERT-MUTZ3 cells were differentiated into immature LC following standard culture protocols and were analyzed for phenotypic marker expression by flow cytometry on day 10. Shown are the percentages of CD1a+ (top left), Langerin+ (top middle) and CD40+ (top right) cells and the mean fluorescence intensity levels of CD86 (bottom left), CD54 (bottom middle), and HLA-DR (bottom right) in both the hTERT-MUTZ3 and dox90+ LC cultures (n = 3, P < 0.05)
Inhibition of LC differentiation by doxorubicin selection is reversible
Dox90+ cells were next cultured in the absence of doxorubicin to study whether the doxorubicin-induced inability to differentiate into LC could be reversed. LC differentiation was analyzed after 1, 3, and 4 months of drug deprivation. Subsequent to differentiation, maturation was induced by addition of a cytokine cocktail, and after 2 days, the capacity of the matured cells to stimulate T-cell proliferation in an allogeneic (allo)MLR was determined. The differentiation capacity of the cells selected with 90 nM doxorubicin gradually returned after 3 months of drug depletion (dox90− LC), as evidenced by the expression of CD1a and Langerin (Fig. 4a). The differentiated drug-depleted cells also responded to maturation stimuli, reflected by the increase in the percentage of cells expressing the maturation marker CD83 (Fig. 4b). Accordingly, their ability to stimulate T-cell proliferation in an alloMLR also improved. After 3 and 4 months in the absence of drugs, dox90− mLC showed enhanced T-cell stimulatory abilities compared with their continuously drug-selected counterparts, and after 4 months of drug deprivation, their T-cell stimulatory capacity even matched that of the parental (passage-matched) hTERT-mLC (Fig. 4c).

Doxorubicin-induced suppression of LC differentiation is reversible. hTERT-MUTZ3 cells, dox90+ cells, and dox90− cells (drug-free) were differentiated into immature LC for 10 days after 1, 3, and 4 months of drug depletion of the dox90− cells. a dotplots showing CD1a and Langerin expression on hT-iLC, dox90+ iLC, and dox90− iLC. Upon a drug-free period of at least 3 months, the dox90− cells regained their differentiation potential. b Percentages of cells expressing the DC maturation marker CD83 in cultures started 1, 3, and 4 months after drug depletion of the dox90− cells. c MLR was performed using hT-mLC, dox90+ mLC, and dox90− mLC [depleted for drugs for 1 month (left), 3 months (middle), and 4 months (right)], to analyze the T-cell stimulatory capacities of the mLC
Capacity to differentiate coincides with the (re-)expression of the SCF-R
As hTERT-MUTZ3 cells remained dependent on exogenous cytokines for their growth and differentiation, we studied the expression of cytokine receptors involved in these processes on the drug-selected and subsequently drug-deprived cells, as well as on their unselected, parental counterparts. As the percentage of CD14+ cells within all cultures was less than five percent, we focused on the expression of cytokine receptors on the proliferative CD34+ cells. All CD34+ cells within all three cultures expressed high levels of the IL-3 receptor (CD123), low levels of the GM-CSF receptor (CD116, 5% positive), and TNF receptors I and II (5% positive) and were negative for the IL-6 receptor (CD126) (data not shown). There was, however, a striking difference in the expression of the stem cell factor receptor (SCF-R/CD117/c-kit) (Fig. 5a). While 36% of the parental hTERT-MUTZ3 CD34+ population expressed the SCF-R, expression of SCF-R was completely lost in the doxorubicin-selected cells. Of note, CD34+ dox90− cells again displayed normalized SCF-R expression levels, comparable to the parental hTERT-MUTZ3 cells (Fig. 5a).

Doxorubicin exposure down-regulates SCF-R/c-Kit expression on CD34+ progenitors. a hTERT-MUTZ3, dox90+, and dox90− (4 months drug-free) progenitor cells were analyzed for the expression of the SCF-R/c-Kit. Continuous exposure to doxorubicin abrogated the expression of SCF-R/c-Kit on a subset of CD34+ progenitors. SCF-R/c-Kit expression returned upon drug depletion. b wtMUTZ3 cells were cultured in the presence of 0, 30, 100, or 300 nM doxorubicin for 4 days. SCF-R+ and SCF-R− CD34+ cells were analyzed for their doxorubicin uptake. Left: percentages of SCF-R− (white squares) and SCF-R+ (black squares) cells positive for doxorubicin after 4 days with the indicated drug concentrations. Right: doxorubicin uptake in SCF-R− and SCF-R+ cells, determined by the mean fluorescence index (doxorubicin mean fluorescence intensity (FL-2 channel) of drug-exposed cells divided by the mean fluorescence intensity of non-exposed (0 nM) cells). *P < 0.05 (n = 3). c Percentages of SCF-R− and SCF-R+ cells that died upon doxorubicin treatment, as determined by flowcytometric analysis with propidium iodide (PI). *P < 0.05 (n = 3)
We next analyzed whether the absence of SCF-R-expressing CD34+ cells within the doxorubicin-selected cultures could be due to a higher sensitivity to the drug. When cultured in the presence of increasing concentrations of doxorubicin for 4 days, both the SCF-R− and SCF-R+ CD34+ MUTZ3 cells were found to take up doxorubicin (Fig. 5b). However, uptake levels for doxorubicin were higher in SCF-R+ cells compared with SCF-R− cells as determined by the mean fluorescence index (Fig. 5b). Moreover, whereas the percentages of cells taking up doxorubicin were comparable, significantly more SCF-R+ cells than SCF-R− cells died over the course of treatment at all indicated concentrations (Fig. 5c). Of note, sorting the SCF-R+ from the SCF-R− cells within untreated hTERT-MUTZ3 cultures showed that both populations were equally capable of differentiating into LC and that SCF-R expression was down-regulated in the isolated SCF-R+ fraction upon culture (two independent experiments: data not shown). These observations are consistent with the notion that SCF-R+ cells may constitute an early proliferative LC precursor pool that can quickly differentiate into SCF-R− cells with maintained LC differentiation ability but reduced proliferation capacity, resulting in an ultimate loss of differentiation capacity over time.
Discussion
This study describes a possible consequence of long-term exposure of DC precursors to cytostatic drugs, interfering with their capacity to develop into DC. By making use of hTERT-transduced MUTZ3 cells, we have demonstrated that prolonged doxorubicin exposure renders CD34+ precursor cells irresponsive to LC-differentiating cytokines. Fortunately, this detrimental effect of doxorubicin on the differentiation capacity of DC precursors proved to be a reversible phenomenon. The cells regained the capacity to differentiate and consequently their capacity to stimulate T-cell proliferation, after a 3- to 4-month drug-free period. Although this report focused on LC differentiation, we have made similar observations for differentiation of interstitial DC from long-term doxorubicin-exposed hTERT-MUTZ3 cells (data not shown). Analysis of cytokine receptor expression patterns on unexposed hTERT-MUTZ3 cells, doxorubicin-exposed dox90+ hTERT-MUTZ3 cells, and the dox90− hTERT-MUTZ3 cells that had been drug-free for 4 months revealed altered expression of the SCF-R (CD117/c-kit). Whereas a subpopulation of CD34+ hTERT-MUTZ3 cells expressed this receptor, a SCF-R-expressing population was absent in the doxorubicin-selected cells. Interestingly, when deprived of doxorubicin, in conjunction with the regained ability to differentiate, the SCF-R+ CD34+ precursor population re-emerged. Sensitivity analysis showed that SCF-R+ CD34+ precursors were more susceptible to doxorubicin-induced cell death compared to SCF-R− CD34+ precursors.
SCF-R+ CD34+ cells have been reported to be present both in bone marrow and in peripheral blood of healthy donors and AML patients [4, 6, 8, 12, 15, 17, 27]. In healthy donors, around 50% of the CD34+ cells have been reported to express SCF-R (range 19–85%), whereas in AML patients SCF-R+ cells were found in the majority of the patients and expression ranged between 3 and 96% of CD34+ cells co-expressing SCF-R [4, 6, 8, 12, 15, 17, 27]. It has been reported that SCF, the ligand for the SCF-R, in combination with FLT3-ligand, is crucial for the maintenance of a long-term proliferative pool of CD34+ DC precursor cells [10]. While blockade of the SCF-R did not directly inhibit DC differentiation from CD34+ precursors, it did interfere with continued proliferation of a subset of CD34+ precursors with the ability to differentiate into DC. Hence, the observation that the dox90+ cells lack this SCF-R+ subpopulation and simultaneously have lost their ability to differentiate suggests that due to chronic doxorubicin exposure, this DC precursor subset with long-term proliferative potential is selectively depleted from the general precursor population. A similar effect could be expected on the DC precursors among the circulating hematopoietic stem and precursor cells (HSPC) in patients treated with this drug and perhaps with related drugs. Research in patient groups treated with doxorubicin, or related drugs, is required to gain more insight into the effects of repetitive chemotherapy treatment on the presence and quality of DC precursor cells in vivo, in order to determine whether longer drug-free intervals should be considered when applying chemotherapy regimens prior to DC-based immunotherapies. In this regard, the use of allogeneic rather than autologous DC vaccines should be explored for patients with a long history of chemotherapy treatment [11].
One could argue that the altered differentiation capacity could be caused by the presence of hTERT in the MUTZ3 cells, which could lead to chromosomal instability and hence an altered differentiation potential [24]. However, it has been shown that hTERT-transduced T cells can be kept in culture for many months, without loosing their antigen specificity or their cytotoxic abilities [1]. Furthermore, in our studies, non-drug-exposed hTERT-MUTZ3 cells were kept in culture alongside the drug-selected cells and were used as passage-matched controls in the differentiation studies. Despite the continuous expression of hTERT, these cells maintained their cytokine dependence for growth and were still capable of LC differentiation at high passage numbers. Also, the observation that the drug-induced differentiation defect, along with SCF-R/c-kit expression on CD34+ cells, could be reversed upon withdrawal of the drug is additional proof that the observed effects are hTERT independent.
The negative effects of long-term exposure of DC precursor cells to cytostatic drugs are in sharp contrast with our own observation that a single low dose of the anthracyclin doxorubicin, or the related anthracenedione mitoxantrone, at the start of in vitro DC differentiation from CD34+ precursor cells strongly promotes and even accelerates differentiation (van de Ven et al. manuscript in preparation). Also, work by Zitvogel et al. has shown that systemic doxorubicin treatment can promote anti-tumor responses due to the release of danger signals like the high mobility group box 1 protein (HMGB1) released from dying tumor cells. Release of HMGB1 acts as a maturation signal for resident DC and promotes tumor antigen presentation to CD8+ T cells [3, 9, 29]. This notwithstanding, our data imply that in the long run, repetitive systemic treatment with cytostatic drugs could be disadvantageous due to its negative effect on the expansion of CD34+c-kit + DC precursor cells.
It is known that cytostatic drugs can induce the expression of ATP-binding cassette (ABC) transporters like P-glycoprotein (P-gp; ABCB1), multidrug resistance protein 1 (MRP1; ABCC1), or the breast cancer resistance protein (BCRP; ABCG2), thereby inducing multidrug resistance in tumor cells [2, 18]. As the protein expression levels of these ABC transporters were unaltered in doxorubicin-selected cells (data not shown), the effect of progenitor cell selection with doxorubicin seems ABC transporter independent, and to be caused mainly by the selective loss of SCF-R/c-kit expressing, CD34+ DC precursor cells with a high proliferative capacity and hence with an increased susceptibility to the DNA damaging effects of cytostatic drugs such as doxorubicin. Altogether our data suggest that, if feasible, sufficiently long drug-free intervals should be included in chemotherapy regimens in order to allow the immune system to recuperate. This is of particular relevance in view of accumulating evidence for a role of immune functions in clinical responsiveness to chemotherapy [33]. In addition, when DC vaccination is being considered, patients with a long history of chemotherapy treatment should be tested for their suitability for autologous DC vaccination strategies (assessment of the presence SCF-R+ CD34+ cells), as their DC precursor cells might give rise to DC of poor quality. These patients might actually benefit more from allogeneic DC vaccination approaches as recently reviewed by us [11].
References
Andersen H, Barsov EV, Trivett MT et al (2007) Transduction with human telomerase reverse transcriptase immortalizes a rhesus macaque CD8+ T cell clone with maintenance of surface marker phenotype and function. AIDS Res Hum Retroviruses 23:456–465
Annereau JP, Szakacs G, Tucker CJ et al (2004) Analysis of ATP-binding cassette transporter expression in drug-selected cell lines by a microarray dedicated to multidrug resistance. Mol Pharmacol 66:1397–1405
Apetoh L, Ghiringhelli F, Tesniere A et al (2007) Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13:1050–1059
Bene MC, Bernier M, Casasnovas RO et al (1998) The reliability and specificity of c-kit for the diagnosis of acute myeloid leukemias and undifferentiated leukemias. The European group for the Immunological classification of leukemias (EGIL). Blood 92:596–599
Bijl J, van Oostveen JW, Kreike M et al (1996) Expression of HOXC4, HOXC5, and HOXC6 in human lymphoid cell lines, leukemias, and benign and malignant lymphoid tissue. Blood 87:1737–1745
Blair A, Sutherland HJ (2000) Primitive acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo lack surface expression of c-kit (CD117). Exp Hematol 28:660–671
Bontkes HJ, Ruizendaal JJ, Kramer D et al (2006) Constitutively active STAT5b induces cytokine-independent growth of the acute myeloid leukemia-derived MUTZ-3 cell line and accelerates its differentiation into mature dendritic cells. J Immunother 29:188–200
Briddell RA, Broudy VC, Bruno E et al (1992) Further phenotypic characterization and isolation of human hematopoietic progenitor cells using a monoclonal antibody to the c-kit receptor. Blood 79:3159–3167
Casares N, Pequignot MO, Tesniere A et al (2005) Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 202:1691–1701
Curti A, Fogli M, Ratta M et al (2001) Stem cell factor and FLT3-ligand are strictly required to sustain the long-term expansion of primitive CD34+ DR- dendritic cell precursors. J Immunol 166:848–854
de Gruijl TD, van den Eertwegh AJ, Pinedo HM et al (2008) Whole-cell cancer vaccination: from autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol Immunother 57:1569–1577
De Waele M, Renmans W, Vander GK et al (2001) Growth factor receptor profile of CD34+ cells in AML and B-lineage ALL and in their normal bone marrow counterparts. Eur J Haematol 66:178–187
Deville L, Hillion J, Segal-Bendirdjian E (2009) Telomerase regulation in hematological cancers: a matter of stemness? Biochim Biophys Acta 1792:229–239
Dowdy SC, Boardman CH, Wilson TO et al (2002) Multimodal therapy including neoadjuvant methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC) for stage IIB to IV cervical cancer. Am J Obstet Gynecol 186:1167–1173
Gunji Y, Nakamura M, Osawa H et al (1993) Human primitive hematopoietic progenitor cells are more enriched in KITlow cells than in KIThigh cells. Blood 82:3283–3289
Hu ZB, Ma W, Zaborski M et al (1996) Establishment and characterization of two novel cytokine-responsive acute myeloid and monocytic leukemia cell lines, MUTZ-2 and MUTZ-3. Leukemia 10:1025–1040
Khoury E, Andre C, Pontvert-Delucq S et al (1994) Tumor necrosis factor alpha (TNF alpha) downregulates c-kit proto-oncogene product expression in normal and acute myeloid leukemia CD34+ cells via p55 TNF alpha receptors. Blood 84:2506–2514
Leonard GD, Fojo T, Bates SE (2003) The role of ABC transporters in clinical practice. Oncologist 8:411–424
Long HJ III, Monk BJ, Huang HQ et al (2006) Clinical results and quality of life analysis for the MVAC combination (methotrexate, vinblastine, doxorubicin, and cisplatin) in carcinoma of the uterine cervix: a gynecologic oncology group study. Gynecol Oncol 100:537–543
Long HJ III, Rayson S, Podratz KC et al (2002) Long-term survival of patients with advanced/recurrent carcinoma of cervix and vagina after neoadjuvant treatment with methotrexate, vinblastine, doxorubicin, and cisplatin with or without the addition of molgramostim, and review of the literature. Am J Clin Oncol 25:547–551
Masterson AJ, Sombroek CC, de Gruijl TD et al (2002) MUTZ-3, a human cell line model for the cytokine-induced differentiation of dendritic cells from CD34+ precursors. Blood 100:701–703
Santegoets SJ, Masterson AJ, van der Sluis PC et al (2006) A CD34+ human cell line model of myeloid dendritic cell differentiation: evidence for a CD14+ CD11b+ Langerhans cell precursor. J Leukoc Biol 80:1337–1344
Santegoets SJ, van den Eertwegh AJ, van de Loosdrecht AA et al (2008) Human dendritic cell line models for DC differentiation and clinical DC vaccination studies. J Leukoc Biol 84:1364–1373
Schreurs MW, Hermsen MA, Geltink RI et al (2005) Genomic stability and functional activity may be lost in telomerase-transduced human CD8+ T lymphocytes. Blood 106:2663–2670
Schreurs MW, Scholten KB, Kueter EW et al (2003) In vitro generation and life span extension of human papillomavirus type 16-specific, healthy donor-derived CTL clones. J Immunol 171:2912–2921
Snijders PJ, van Duin M, Walboomers JM et al (1998) Telomerase activity exclusively in cervical carcinomas and a subset of cervical intraepithelial neoplasia grade III lesions: strong association with elevated messenger RNA levels of its catalytic subunit and high-risk human papillomavirus DNA. Cancer Res 58:3812–3818
Strobl H, Takimoto M, Majdic O et al (1992) Antigenic analysis of human haemopoietic progenitor cells expressing the growth factor receptor c-kit. Br J Haematol 82:287–294
Ueno H, Klechevsky E, Morita R et al (2007) Dendritic cell subsets in health and disease. Immunol Rev 219:118–142
Ullrich E, Menard C, Flament C et al (2008) Dendritic cells and innate defense against tumor cells. Cytokine Growth Factor Rev 19:79–92
van de Ven R, de Jong MC, Reurs AW et al (2006) Dendritic cells require multidrug resistance protein 1 (ABCC1) transporter activity for differentiation. J Immunol 176:5191–5198
von Minckwitz G, Jonat W, Fasching P et al (2005) A multicentre phase II study on gefitinib in taxane- and anthracycline-pretreated metastatic breast cancer. Breast Cancer Res Treat 89:165–172
von Minckwitz G, Raab G, Caputo A et al (2005) Doxorubicin with cyclophosphamide followed by docetaxel every 21 days compared with doxorubicin and docetaxel every 14 days as preoperative treatment in operable breast cancer: the GEPARDUO study of the German breast group. J Clin Oncol 23:2676–2685
Zitvogel L, Kepp O, Aymeric L et al (2010) Integration of host-related signatures with cancer cell-derived predictors for the optimal management of anticancer chemotherapy. Cancer Res 70(23):9538–9543
Acknowledgments
The authors would like to thank Dr. J. de Wilde (VU University medical center, Amsterdam) for her help with the hTERT RT-PCR. This work was supported by a grant from the Dutch Cancer Society (KWF) to R.J.S., G.L.S. and T.D.G. (KWF2003-2830).
Conflict of interest
The authors have no financial conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van de Ven, R., Verbrugge, S.E., Reurs, A.W. et al. High susceptibility of c-KIT+CD34+ precursors to prolonged doxorubicin exposure interferes with Langerhans cell differentiation in a human cell line model. Cancer Immunol Immunother 60, 943–951 (2011). https://doi.org/10.1007/s00262-011-1003-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-011-1003-9