
Abstract
The genus Herbaspirillum gained the spotlight due to the several reports of diazotrophic strains and promising results in plant-growth field assays. However, as diversity exploration of Herbaspirillum species gained momentum, it became clearer that the plant beneficial lifestyle was not the only form of ecological interaction in this genus, due to reports of phytopathogenesis and nosocomial infections. Here we performed a deep search across all publicly available Herbaspirillum genomes. Using a robust core genome phylogeny, we have found that all described species are well delineated, being the only exception H. aquaticum and H. huttiense clade. We also uncovered that the nif genes are only highly prevalent in H. rubrisubalbicans; however, irrespective to the species, all nif genes share the same gene arrangement with high protein identity, and are present in only two main types, in inverted strands. By means of a NifHDKENB phylogenetic tree, we have further revealed that the Herbaspirillum nif sequences may have been acquired from the same last common ancestor belonging to the Nitrosomonadales order.
Similar content being viewed by others


Introduction
Biological nitrogen fixation (BNF) is a process mediated by a group of prokaryotes, called diazotrophs, capable of reducing atmospheric nitrogen to ammonium [1]. Some of these microorganisms are closely associated with plants, being able to transfer fixed nitrogen to the host, thus making them potential bioinoculants [2]. However, free-living prokaryotes may also contribute to the nitrogen budgets, particularly in soil lacking leguminous plants [3], deep soil [4], and canopy soil [5]. BNF is catalyzed by nitrogenase, a complex metalloenzyme that bears conserved structural and functional characteristics. The nitrogenase expression is regulated by, at least, 20 structural and regulatory genes [1, 6]. While some regulatory genes may vary according to the diazotrophic species and conditions in which BNF occurs, it has been shown that nifHDKENB are essential genes and therefore constitute a minimum criterion for the in silico prediction of diazotrophy [7]. This approach has been used to screen diazotrophic strains in different studies (reviewed in [8]).
The genus Herbaspirillum is known for its beneficial association with plants. Field assays have already shown that strains of this genus can promote plant growth when being used as bioinoculants [9,10,11,12]. These findings were further corroborated by several reports of isolates capable of fixing atmospheric nitrogen along with nif occurrence in many strains [6]. While the majority of data supporting nitrogen fixation is related to H. seropedicae [6, 13], the nif genes have already been reported in H. rubrisubalbicans and H. frisingense [14]. Further, the nitrogen-fixing potential of H. frisingense was validated using acetylene reduction assays, PCR assays, and genome sequencing [15,16,17].
Whole-genome sequencing studies have uncovered key molecular features of H. seropedicae interaction with plant host [18]. These findings were further extended by genomic comparisons among clinical and environmental isolates of H. seropedicae [13]. Moreover, striking similarities of amino acid identity and gene arrangement have been revealed between the nif-region of H. seropedicae and H. frisingense [15]. Despite these achievements, the major part of the Herbaspirillum species diversity was often underexplored in genomic comparison studies, including the mild pathogen H. rubrisubalbicans, which may cause disease in some susceptible sugarcane and sorghum varieties [19].
Genome comparisons can help to elucidate the evolutionary patterns that have led to the presence and conservation of protein families. To gain full insight into the nif distribution and gene arrangement conservation in the Herbaspirillum genus, we conducted a broad genomic study comparing all genome sequences publicly available. We explored the Herbaspirillum nif diversity and conservation by means of genomic comparisons, pangenome, and both core genes and Nif phylogenies.
Material and Methods
Bacterial Strains and Dataset Curation
All whole-genome sequences of the genus Herbaspirillum, publicly available in February of 2021, were downloaded (n = 62) from the RefSeq database [20]. The characteristics of all strains are summarized in the Supplementary Table 1. The BUSCO 3.0 tool [21] was employed to assess the assembly quality, using the database of single-copy orthologs of Betaproteobacteria. All genomes had at least 90% of single-copy orthologs present in the database; thus no genome was removed at this step.
Genome Filtering and Comparisons
The percentage of conserved proteins (POCP) was used to confirm that all the obtained genomes belonged to the Herbaspirillum genus. The total protein content of the genomes was predicted using PROKKA 1.14.6 software [22]. The orthologous proteins between genome pairs were estimated using the orthofinder 2.5.2 software [23]. The POCP values were calculated using the formula [(C1 + C2)/(T1 + T2)] × 100%, where C1 and C2 represent the conserved number of proteins in the two genomes being compared, respectively, and T1 and T2 represent the total number of proteins in the two genomes being compared, respectively [21]. The amino acid identity was calculated using EzAAI [24]. Genomes with all pairwise POCP values greater than 50% were considered of the Herbaspirillum genus. The POCP matrix was visualized as a heatmap using pheatmap 1.0.12 package in the R environment [25]. The average nucleotide identity (ANI) was calculated for genomes with POCP ≥ 50% using the pyani software 0.2.11 [26]. The ANI distance matrix was used to infer a phylogeny by neighbor-joining using the APE 5.5 package [27]. The resulting phylogenetic tree was visualized by iTOL v4 [28].
Herbaspirillum spp. pangenome
The protein sequences were used for pangenome determination, using a clustering threshold of 50% identity with Roary 3.13.0 [29]. Sequences shared by ≥ 99% of the genomes were considered part of the core genome of the genus, whereas the total gene content was considered the pangenome. The sequences belonging to the core genome were aligned using mafft 7.0 [30]. These alignments were concatenated and used to obtain a maximum likelihood phylogeny using the FastTree 2.1.10 software [31]. The pangenome presence/absence matrix visualization was obtained using roary_plots.py, provided by the Roary tool. Genes statistically associated to the presence of the nif cluster were assessed using Scoary 1.6.16 [32]. COG and KEGG categories were inferred for the pangenomes of nif-containing and nif-absent genomes using the BPGA 1.3 [33].
Distribution of Nitrogen Fixation Genes in Herbaspirillum spp
To determine the distribution of the nif cluster genes in Herbaspirillum genus, the sequences of 18 genes belonging to four regions of the nif cluster of H. seropedicae SmR1 were obtained. The sequences of nifA (WP_013234836.1), nifB (WP_006463072.1), nifH (WP_013234820.1), nifD (WP_013234819.1), nifK (WP_013234818.1), nifE (WP_013234817.1), nifN (WP_013234816.1), nifX (WP_006463095.1), modA (WP_013234812.1), modB (WP_013234811.1), modC (WP_013234810.1), fixX (WP_006463103.1), fixC (WP_013234809.1), fixB (WP_013234808.1), fixA (WP_013234807.1), nifW (WP_013234806.1), nifV (WP_013234805.1), and fdxN (WP_013234804.1) were used as queries for blastp searches [29], which searched these sequences against a database formed by all protein sequences from all genomes of Herbaspirillum spp., only hits with at least 50% coverage and 70% identity were considered. The presence or absence of genes related to nitrogen fixation was visualized by integrating the maximum likelihood phylogenetic tree of the core genome and a matrix of presence and absence of the genes described above, using the ITOL v4 software [28].
The nif Cluster Conservation in H. seropedicae and H. rubrisubalbicans
Gene sequences from the nif cluster of H. seropedicae SmR1 were used as queries in an online blastn using the SimpleSynteny platform [34]. Only blastn hits above 50% coverage and 70% identity were taken into account. The visualization of the nif cluster organization was finalized using Inkscape 1.0.2–2 [35].
Nif Protein Phylogeny
The NifHDK and NifENB protein sequences of all Herbaspirillum genomes were obtained as described above. The H. seropedicae SmR1 Nif sequences were used as queries in blastp against NCBI NR database restricted to the Betaproteobacteria class, excluding the genus Herbaspirillum, as subject. The first 500 hits for each one of the six Nif sequences were obtained. Using in-house scripts, 191 unique sequences occurring as hits for all six Nif sequences were filtered. At last, 14 Herbaspirillum Nif sequences, from genomes encoding the full nif cluster, were added to the filtered Nif hits. The software MUSCLE 3.8.31 [36] was used to perform the multiple-sequence alignment, which was further concatenated and used to reconstruct a maximum likelihood phylogeny using IQ-TREE 2 [37] with the inputs -nt 14 -m MFP -bb 1000. The visualization of the Nif phylogenetic tree was finalized using the ITOL v4 software [28] and Inkscape 1.0.2–2 software [35].
Results
Whole-Genome Comparisons
Overall protein conservation was assessed to infer whether the downloaded genomes belonged to the same genus. Pairwise comparisons revealed that the percentage of conserved proteins ranged from 37% to 99% (Fig. 1). Fifty-eight strains were classified as genus Herbaspirillum due to POCP values ranging from 51% to 99%. However, four strains showed POCP values lower than 50%, indicating that they do not belong to the Herbaspirillum genus. The following strains were removed: Herbaspirillum sp. K2R10-39, Herbaspirillum sp. K1R23-30, Herbaspirillum sp. SJZ107, and Herbaspirillum sp. ST 5–3. On the contrary, POCP values higher than 90% were observed among genomes of the same species, indicating the suitability of this approach to determine genome-to-genome relatedness. The removed genomes also harbored low Amino Acid Identity (AAI) and orthologous fraction, when compared to the other Herbaspirillum genomes (Supplementary Tables 2,3,4,5).

Heatmap of percentage of conserved proteins of 62 Herbaspirillum genomes. Warm colors depict higher percentage of shared proteins orthologs. Outliers genomes presented values < 50%
The average nucleotide identity (ANI) of the remaining 58 genomes belonging to the genus Herbaspirillum was used to build a distance-based dendrogram (Fig. 2). Thirty genomes formed species clusters, whereas 28 genomes did not group with any described species. Overall, the genus Herbaspirillum comprises 11 described species and five clusters without official nomenclature that form species-like clusters. Nine genomes deposited as Herbaspirillum sp. clustered together with known species. Herbaspirillum sp. WT00C (GCF_001929405.1) showed 95.6% ANI with H. seropedicae SmR1. Herbaspirillum sp. B65 (GCF_000333555.1) and B501 (GCF_000333575.1) had 98.4% and 99.8% ANI, respectively, with H. rubrisubalbicans Os45. Herbaspirillum sp. VT-16–41 (GCF_001994935.1), Herbaspirillum sp. BH1 (GCF_002870055.1), and Herbaspirillum sp. SG826 (GCF_011759625.1) grouped into the clade of H. frisingense (97%, 97.9%, and 98% ANI, respectively, with H. frisingense GSF30). Herbaspirillum sp. XFZ15_10_5 (GCF_003634405.1) and Herbaspirillum sp. B39 (GCF_000333495.1) showed 99% and 97.2% ANI, respectively, with H. huttiense NFYY53159. At last, Herbaspirillum sp. CAH-3 (GCF_009668275.1) was grouped into the H. aquaticum clade (98.5% ANI). The only case of misclassified strain was H. huttiense 1147, which should be reclassified to H. aquaticum (Supplementary Table 6). Moreover, five species-like clusters harboring only strains with no known species had an ANI value greater than 95% among themselves, thus indicating the presence of undescribed species within the Herbaspirillum genus.

Distance-based dendrogram of average nucleotide identity (ANI) of 58 Herbaspirillum genomes. Outer arcs delimit clades with more than 95% ANI. Black arcs delimit a described species; outer red arcs delimit an undescribed specie-like cluster
The Pangenome and nif Distribution of the Genus Herbaspirillum
Fifty-eight genomes were used to build the pangenome of Herbaspirillum, which comprises 32,954 gene families. The pangenome size increased along with the genome count, indicating high genome diversity yet to be uncovered (Supplementary Figure S1). The core genome constituted of 840 genes present in more than 99% of genomes. The pangenome presence and absence matrix revealed the occurrence of large gene regions shared by the majority of genomes, accounting for the 730 gene families comprising the soft core genome. As 4809 was the mean amount of protein coding genes, 32.65% of the total genomic content was highly conserved (present in more than 95% of genomes) across all the Herbaspirillum genus (Fig. 3; Supplementary Table 7). The low number of unique genes per species or cluster, with at least three genomes, also indicate that the Herbaspirillum species retain a high amount of shared sequences (Supplementary Table 8).

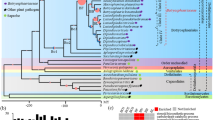
The Herbaspirillum pangenome presence/absence matrix mapped onto the core gene maximum likelihood phylogenetic tree. A Core genome phylogenetic tree. Inner black boxes delimit a described species; inner red boxes delimit an undescribed specie-like cluster. B Blue filled squares account for a gene presence, while whitespaces represent gene absence
The nif genes were found in 16 genomes of various Herbaspirillum species; we conducted a deeper exploration of the pangenome by searching for genes statistically associated to the nif occurrence. While the vast majority of strongly associated gene families belong to the nif cluster itself, other genes such as tolQ and cntO are related to the membrane integrity and zinc uptake (Supplementary Table 9). By exploring the pangenomes of nif-containing and nif-absent genomes, we could not uncover clear distinctions regarding the genes COG and KEGG categories (Supplementary Figures S2 and S3, respectively).
We further explored the maximum likelihood core genome phylogenetic tree along with the nif cluster gene distribution. The species H. rubrisubalbicans showed three peculiar features, an outgroup behavior, smallest number of nucleotide substitution events along with widespread presence of nif cluster genes. Also, only in H. rubrisubalbicans B65 and H. rubrisubalbicans 501 are found incomplete nif clusters (Fig. 4A and B). Overall, the nif genes are present in four of the 11 assessed species; the total prevalence of Herbaspirillum spp. containing nif was 27.5% (16 genomes). H. seropedicae SmR1, H. seropedicae Z67, H. seropedicae Z67_2, H. frisingense GSF30, H. huttiense 1147, Herbaspirillum sp. AP02, and all other H. rubrisubalbicans harbor the full nif cluster (Fig. 4B). The aforementioned similarity among H. aquaticum and H. huttiense can also be visualized in the maximum likelihood phylogenetic tree, as well as the high precision of the current taxonomic affiliations.

The Herbaspirillum nif cluster distribution. A Core gene maximum likelihood phylogenetic tree of 58 Herbaspirillum genomes. Bootstrap values are shown as inner branch values. B Presence or absence of the nif-related operons. Filled squares depict the presence of a full nif-related operon, half-filled black squares indicate that at least one nif-related gene was not found within the operon using the applied thresholds, and white squares represent the absence of all genes of a nif-related operon
All the 14 full nif-encoding genomes harbored homologs of nifA, nifB, nifH, nifD, nifK, nifE, nifN, nifX, nifW, nifV, nifQ, nifS, nifT, nifW, nifZ1, and nifZ, with exceptions within H. rubrisubalbicans. Other nitrogenase accessory genes, modABC, operon fixXCBA were also detected along with fdxN, fdxB, and unknown genes. Remarkably, only two main nif cluster structures were found, being all genes in the opposite strand in each structure (Fig. 5).

The two gene arrangements of the nif cluster in Herbaspirillum genomes. Genes sharing the same color shared identity higher than 70%
We next investigated the Nif phylogeny across the Betaproteobacteria class. The maximum likelihood phylogenetic tree confirmed the high conservation of the Herbaspirillum Nif protein sequences (Fig. 6). Interestingly, the branch closest to the Herbaspirillum genus harbors sequences taxonomically distinct, belonging to Azoarcus, Aromatoleum, and Rugosibacter, whereas the inner branch clusters sequences of the Sulfuricellaceae and Gallionellaceae families. Other Nif sequences show taxonomically coherent clustering, such as Azonexaceae, Burkholderiaceae, and Comamonadaceae.

Unrooted maximum likelihood phylogenetic tree of NifHDKENB concatenated protein sequences of 205 strains of the Betaproteobacteria class. Color legend depicts the taxonomic affiliations. Inner bootstrap values are shown
Discussion
Nitrogen fixation is one of the metabolic attributes most sought in potential bacterial bioinoculants, even serving as a parameter to differentiate among beneficial isolates in screening assays. Here we analyzed all publicly available genomes of the Herbaspirillum genus, well-known for its diazotrophic strains. However, our results deconstruct this view, showing, for the first time, that diazotrophism is a scarce metabolic competence even within Herbaspirillum-sequenced strains. In previous efforts to study the Herbaspirillum diversity, four species were reclassified to the novel genus Noviherbaspirillum using phenotypic, chemotaxonomic, and 16S rRNA gene phylogenetics [38]. Genomic studies also corrected misclassified genomes in H. seropedicae [13] and H. frisingense [39]. In spite of these achievements, Monteiro et al. [40] defined the Herbaspirillum genus as a poorly defined and studied genus. To gain a deeper understanding of Herbaspirillum diversity, we created the first multi-locus genus-wide phylogenetic tree. Our findings support the boundaries between the described species, even among species containing a single genome such as H. lusitanum, H. rhizosphaerae, and H. autotrophicum. We also confirm the closeness between H. huttiense and H. aquaticum, as reported by Regunath et al. [41], reinforcing that 16S rRNA gene sequencing is unable to differentiate between these two species.
So far, the only described pangenome of a Herbaspirillum species is of H. seropedicae [13], obtained using the genome sequences of five strains. Due to the small number of genomes, even within the largely represented species, we built a genus-wide pangenome to gain a deeper understanding regarding gene conservation across all Herbaspirillum-sequenced strains. We have found that 32.65% of the pangenome was conserved in more than 95% of the 58 genomes. This high conservation had already been reported for other soil- and plant-related bacteria [42]. Irrespective to the presence or absence of the nif cluster within the genomes, we could not detect clear genomic patterns that could be related to the plant-associated lifestyle. A reason for this could be the low number of available genomes for bacterial population approaches (e.g., population structure).
While H. lusitanum, H. rhizosphaerae, and H. autotrophicum have only one sequenced genome, we have found five species-like genome clusters; one of those comprises the isolates Herbaspirillum sp. AP02 and Herbaspirillum sp. AP21, which promote ryegrass growth [43]. Interestingly, the nif cluster is present in the first and absent in the latter, a similar pattern as observed in H. seropedicae, H. frisingense, and H. huttiense. As nif absence has been correlated to the transition to opportunistic lifestyle in H. seropedicae [13] and H. frisingense [39], an in-depth assessment of genomic distinctions between nif-encoding and nif-absent Herbaspirillum spp. genomes should be considered for biotechnological applications.
Comparisons between clinical and environmental isolates of H. seropedicae have revealed that the opportunistic strains AU13965 and AU14040 lack the nif cluster and had acquired gene regions related to lipopolysaccharide biosynthesis [13]. Similarly, H. frisingense GSF-30, isolated from C4-fiber plants [44], harbors the full set of nif genes, while the closely related uropathogenic strain Herbaspirillum sp. VT-16–41 [45] lacks this gene region. Oliveira et al. [39] employed in silico tools to reveal that environmental and clinical strains of H. frisingense share the same set of virulence and antibiotic resistance genes. H. huttiense has also been associated to nosocomial infections [46]; however Andreozzi et al. [47] have reported an augmented expression of nifH in H. huttiense RCA24 when treated with IAA. While we have found that H. huttiense 1147 nif cluster structure is similar to others in the genus, we also uncovered that this strain shares high ANI percentage with H. aquaticum IEH 4430, possibly belonging to H. aquaticum species. Thus, further research is needed to uncover whether the distinctions observed in H. seropedicae and H. frisingense also occur in H. huttiense/H. aquaticum clade.
A clear distinction of the aforementioned sparse nif occurrence could be observed in H. rubrisubalbicans; on the contrary, every strain of this species harbored the core nif genes along with the major part of nif accessory genes. H. rubrisubalbicans has been shown to associate with roots, stems, and leaves of sugarcane [48, 49], being capable to significantly increase the stalk yield of several sugarcane varieties [50, 51]. However, H. rubrisubalbicans may also develop a phytopathogenic lifestyle in some sugarcane varieties [52], as it has already been reported both in rice [53]and sorghum [54].
The nif genes are organized in an array of arrangements across the prokaryotes; an example of this diversity has been reported in Bradyrhizobium, in which seven distinct types of nif clusters were found, each with unique gene arrangements [55]. Here, we found only two types of nif gene arrangements, differing only between forward or complementary strands. Irrespective to the strand orientation, all genes were highly conserved; the high identity and conservation of nif genes had already been reported in comparisons between H. frisingense GSF-30 and H. seropedicae SmR1 [15]. Thus, our results expanded these observations for the whole Herbaspirillum genus.
Due to the high diversity of the nif cluster gene arrangements, researchers established a minimum set of six nif genes, coding for structural and biosynthetic components, as a criterion tool for prediction of nitrogen fixation [7]. This minimum nif gene set was used in a large-scale multi-locus Nif protein phylogeny, which categorized Betaproteobacteria Nif sequences, including three Herbaspirillum strains, to the IB cluster [8]. Our results of the Betaproteobacteria Nif diversity, using the same set of minimum protein sequences, revealed that all 14 Herbaspirillum spp. Nif sequences clustered together in the same tip, having sequences of Azoarcus, Aromatoleum, and Rugosibacter as closest neighbors. Using nifH and nifD gene sequences, it has been shown that Azoarcus and Aromatoleum nif clusters may have been acquired from Dechloromonas and Azospira, respectively [56]. This was further supported by employing RAFTS3G-32 cluster analysis on the nifHDKENB sequences [57]. Interestingly, our maximum likelihood phylogenetic tree showed no distinctions between the 14 Herbaspirillum NifHDKENB sequences, indicating that these sequences were not acquired from horizontal gene transfer events.
The inner tips of the same branch, where Herbaspirillum sequences were placed, hold several sequences from Sulfuricellaceae and Gallionellaceae families, which are currently taxonomically separated [58]. The Sulfuricellaceae and Gallionellaceae families belong to the order Nitrosomonadales, which harbor many strains related to the nitrogen biological cycle. It has been shown that nif genes were repeatedly horizontally acquired during lifestyle transitions of symbiotic rhizobial lineages [59]; however our results indicate that all Herbaspirillum nif genes may have been acquired from the same source, possibly the last common ancestor, as independent events of horizontal acquisitions from distinct donors would unlikely result in conserved nif patterns as observed.
Conclusion
The high conservation of the nif genes arrangement and identity of Nif protein sequences across all nif-encoding Herbaspirillum species indicate that this region may have not been horizontally acquired. In this case, non nif-encoding Herbaspirillum strains could have lost these genes along their evolution. Comparative genomic analyses between nif-encoding Herbaspirillum and nif-encoding neighbor taxa (e.g., Azoarcus) may be useful to test this hypothesis.
References
Dixon R, Kahn D (2004) Genetic regulation of biological nitrogen fixation. Nat Rev Microbiol 2:621–631. https://doi.org/10.1038/nrmicro954
Carvalho TLG, Balsemão-Pires E, Saraiva RM et al (2014) Nitrogen signalling in plant interactions with associative and endophytic diazotrophic bacteria. J Exp Bot 65:5631–5642. https://doi.org/10.1093/jxb/eru319
Smercina DN, Evans SE, Friesen ML, Tiemann LK (2021) Temporal dynamics of free-living nitrogen fixation in the switchgrass rhizosphere. GCB Bioenergy 13:1814–1830. https://doi.org/10.1111/gcbb.12893
Zhang, W., Hou, W., Zeng, X., et al.: High abundance of Thaumarchaeota found in deep metamorphic subsurface in eastern China. Microorganisms. 10, (2022). https://doi.org/10.3390/microorganisms10030542
Matson AL, Corre MD, Burneo JI, Veldkamp E (2015) Free-living nitrogen fixation responds to elevated nutrient inputs in tropical montane forest floor and canopy soils of southern Ecuador. Biogeochemistry 122:281–294. https://doi.org/10.1007/s10533-014-0041-8
Chubatsu LS, Monteiro RA, de Souza EM et al (2012) Nitrogen fixation control in Herbaspirillum seropedicae. Plant Soil 356:197–207. https://doi.org/10.1007/s11104-011-0819-6
Dos Santos PC, Fang Z, Mason SW et al (2012) Distribution of nitrogen fixation and nitrogenase-like sequences amongst microbial genomes. BMC Genomics 13:1–12. https://doi.org/10.1186/1471-2164-13-162
Koirala, A., Brözel, V.S.: Phylogeny of nitrogenase structural and assembly components reveals new insights into the origin and distribution of nitrogen fixation across bacteria and archaea. Microorganisms 9, (2021). https://doi.org/10.3390/microorganisms9081662
AE Araújo da S, Baldani VLD, P Galisa de S et al (2013) Response of traditional upland rice varieties to inoculation with selected diazotrophic bacteria isolated from rice cropped at the Northeast region of Brazil. Appl Soil Ecol 64:49–55. https://doi.org/10.1016/j.apsoil.2012.10.004
Alves GC, Dos Santos CLR, Zilli JE et al (2021) Agronomic evaluation of Herbaspirillum seropedicae strain ZAE94 as an inoculant to improve maize yield in Brazil. Pedosphere 31:583–595. https://doi.org/10.1016/S1002-0160(21)60004-8
Alves GC, Videira SS, Urquiaga S, Reis VM (2015) Differential plant growth promotion and nitrogen fixation in two genotypes of maize by several Herbaspirillum inoculants. Plant Soil 387:307–321. https://doi.org/10.1007/s11104-014-2295-2
Liu Y, Ludewig U (2020) Improved establishment of Miscanthus × giganteus stem propagation by Herbaspirillum inoculation. Ind Crops Prod 150:112339. https://doi.org/10.1016/j.indcrop.2020.112339
Faoro H, Oliveira WK, Weiss VA et al (2019) Genome comparison between clinical and environmental strains of Herbaspirillum seropedicae reveals a potential new emerging bacterium adapted to human hosts. BMC Genomics 20:1–15. https://doi.org/10.1186/s12864-019-5982-9
Baldani JI, Rouws L, Cruz LM et al (2014) The family Oxalobacteraceae. In: Schmid M, Hartmann A (eds) The Prokaryotes. Springer, Berlin Heidelberg, Berlin, Heidelberg, pp 919–974
Straub D, Rothballer M, Hartmann A, Ludewig U (2013) The genome of the endophytic bacterium H. frisingense GSF30T identifies diverse strategies in the Herbaspirillum genus to interact with plants. Front Microbiol 4:1–10. https://doi.org/10.3389/fmicb.2013.00168
Straub D, Yang H, Liu Y et al (2013) Root ethylene signalling is involved in Miscanthus sinensis growth promotion by the bacterial endophyte Herbaspirillum frisingense GSF30 T. J Exp Bot 64:4603–4615. https://doi.org/10.1093/jxb/ert276
Rothballer M, Eckert B, Schmid M et al (2008) Endophytic root colonization of gramineous plants by Herbaspirillum frisingense. FEMS Microbiol Ecol 66:85–95. https://doi.org/10.1111/j.1574-6941.2008.00582.x
Pedrosa FO, Monteiro RA, Wassem R et al (2011) Genome of Herbaspirillum seropedicae strain SmR1, a specialized diazotrophic endophyte of tropical grasses. PLoS Genet 7:e1002064. https://doi.org/10.1371/journal.pgen.1002064
Tuleski TR, Kimball J, Do Amaral FP et al (2020) Herbaspirillum rubrisubalbicans as a phytopathogenic model to study the immune system of Sorghum bicolor. Mol Plant-Microbe Interact 33:235–246. https://doi.org/10.1094/MPMI-06-19-0154-R
Li W, O’Neill KR, Haft DH et al (2021) RefSeq: Expanding the Prokaryotic Genome Annotation Pipeline reach with protein family model curation. Nucleic Acids Res 49:D1020–D1028. https://doi.org/10.1093/nar/gkaa1105
Manni M, Berkeley MR, Seppey M et al (2021) BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol 38:4647–4654. https://doi.org/10.1093/molbev/msab199
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. https://doi.org/10.1093/bioinformatics/btu153
Emms DM, Kelly S (2019) OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol 20:1–14. https://doi.org/10.1186/s13059-019-1832-y
Kim D, Park S, Chun J (2021) Introducing EzAAI: a pipeline for high throughput calculations of prokaryotic average amino acid identity. J Microbiol 59:476–480. https://doi.org/10.1007/s12275-021-1154-0
R Core Team: A Language and Environment for Statistical Computing R Foundation for Statistical Computing, Vienna, Austria (2020)
Pritchard L, Glover RH, Humphris S et al (2016) Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal Methods 8:12–24. https://doi.org/10.1039/c5ay02550h
Paradis E, Claude J, Strimmer K (2004) APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20:289–290. https://doi.org/10.1093/bioinformatics/btg412
Letunic I, Bork P (2019) Interactive Tree of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47:256–259. https://doi.org/10.1093/nar/gkz239
Page AJ, Cummins CA, Hunt M et al (2015) Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 31:3691–3693. https://doi.org/10.1093/bioinformatics/btv421
Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. https://doi.org/10.1093/molbev/mst010
Price, M.N., Dehal, P.S., Arkin, A.P.: FastTree 2 — approximately maximum-likelihood trees for large alignments. PLoS One 5, (2010). https://doi.org/10.1371/journal.pone.0009490
Brynildsrud O, Bohlin J, Scheffer L, Eldholm V (2016) Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol 17:1–9. https://doi.org/10.1186/s13059-016-1108-8
Chaudhari NM, Gupta VK, Dutta C (2016) BPGA—an ultra-fast pan-genome analysis pipeline. Sci Rep 6:1–10. https://doi.org/10.1038/srep24373
Veltri D, Wight MM, Crouch JA (2016) SimpleSynteny: a web-based tool for visualization of microsynteny across multiple species. Nucleic Acids Res 44:W41–W45. https://doi.org/10.1093/nar/gkw330
Software Inkscape Project (2020) Inkscape. https://inkscape.org. Accessed 10 Jan 2021
Edgar RC (2004) MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Minh BQ, Schmidt HA, Chernomor O et al (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:1530–1534. https://doi.org/10.1093/molbev/msaa015
Lin SY, Hameed A, Arun AB et al (2013) Description of Noviherbaspirillum malthae gen. nov., sp. nov., isolated from an oil-contaminated soil, and proposal to reclassify Herbaspirillum soli, Herbaspirillum aurantiacum, Herbaspirillum canariense and Herbaspirillum psychrotolerans as Noviherbaspirillum soli comb. nov., Noviherbaspirillum aurantiacum comb. nov., Noviherbaspirillum canariense comb.nov. and Noviherbaspirillum psychrotoleranscomb.nov. based on polyphasic analysis. Int J Syst Evol Microbiol 63:4100–4107. https://doi.org/10.1099/ijs.0.048231-0
Oliveira, W.K., Ávila, H.L., Tadra, M.Z. et al.: High genomic identity between clinical and environmental strains of Herbaspirillum frisingense suggests pre-adaptation to different hosts and intrinsic resistance to multiple drugs. Antibiotics 10, (2021). https://doi.org/10.3390/antibiotics10111409
Monteiro RA, Cruz LM, Wassem R et al (2014) Comparative genomics of Herbaspirillum species. In: Katsy EI (ed) Plasticity in Plant-Growth-Promoting and Phytopathogenic Bacteria. Springer, New York, New York, NY, pp 171–198
Regunath H, Kimball J, Smith LP, Salzer W (2015) Severe community-acquired pneumonia with bacteremia caused by Herbaspirillum aquaticum or Herbaspirillum huttiense in an immune-competent adult. J Clin Microbiol 53:3086–3088. https://doi.org/10.1128/JCM.01324-15
Maistrenko OM, Mende DR, Luetge M et al (2020) Disentangling the impact of environmental and phylogenetic constraints on prokaryotic within-species diversity. ISME J 14:1247–1259. https://doi.org/10.1038/s41396-020-0600-z
Cortés-Patiño S, Vargas C, Álvarez-Flórez F et al (2021) Potential of Herbaspirillum and Azospirillum consortium to promote growth of perennial ryegrass under water deficit. Microorganisms 9:1–16. https://doi.org/10.3390/microorganisms9010091
Kirchhof G, Eckert B, Stoffels M et al (2001) Herbaspirillum frisingense sp. nov., a new nitrogan-fixing bacterial species that occurs in C4-fibre plants. Int J Syst Evol Microbiol 51:157–168. https://doi.org/10.1099/00207713-51-1-157
Tetz V, Tetz G (2017) Draft genome sequence of the uropathogenic Herbaspirillum frisingense strain ureolyticus VT-16-41. Genome Announc 5:14–16. https://doi.org/10.1128/genomeA.00279-17
Liu C, Kwon MJ, Kim M et al (2020) Septicemia caused by Herbaspirillum huttiense secondary to pneumonia. Ann Lab Med 39:340–342. https://doi.org/10.3343/ALM.2019.39.3.340
Andreozzi A, Prieto P, Mercado-Blanco J et al (2019) Efficient colonization of the endophytes Herbaspirillum huttiense RCA24 and Enterobacter cloacae RCA25 influences the physiological parameters of Oryza sativa L. cv. Baldo rice Environ Microbiol 21:3489–3504. https://doi.org/10.1111/1462-2920.14688
Baldani JI, Pot B, Kirchhof G et al (1996) Emended description of Herbaspirillum; inclusion of [Pseudomonas] rubrisubalbicans, a mild plant pathogen, as Herbaspirillum rubrisubalbicans comb. nov.; and classification of a group of clinical isolates (EF group 1) as Herbaspirillum species 3. Int J Syst Bacteriol 46:802–810. https://doi.org/10.1099/00207713-46-3-802
Olivares FL, Baldani VLD, Reis VM et al (1996) Occurrence of the endophytic diazotrophs Herbaspirillum spp. in roots, stems, and leaves, predominantly of Gramineae. Biol Fertil Soils 21:197–200
De Oliveira ALM, De Canuto EL, Urquiaga S et al (2006) Yield of micropropagated sugarcane varieties in different soil types following inoculation with diazotrophic bacteria. Plant Soil 284:23–32. https://doi.org/10.1007/s11104-006-0025-0
Schultz N, de Morais RF, da Silva JA et al (2012) Avaliação agronômica de variedades de cana-de-açúcar inoculadas com bactérias diazotróficas e adubadas com nitrogênio. Pesqui Agropecuária Bras 47:261–268. https://doi.org/10.1590/s0100-204x2012000200015
Olivares FL, James EK, Baldani JI, Döbereiner J (1997) Infection of mottled stripe disease-susceptible and resistant sugar cane varieties by the endophytic diazotroph Herbaspirillum. New Phytol 135:723–737. https://doi.org/10.1046/j.1469-8137.1997.00684.x
Valdameri G, Alberton D, Moure VR et al (2017) Herbaspirillum rubrisubalbicans, a mild pathogen impairs growth of rice by augmenting ethylene levels. Plant Mol Biol 94:625–640. https://doi.org/10.1007/s11103-017-0629-1
Tuleski TR, de Baura VA, Donatti L et al (2019) Cellulose production increases sorghum colonization and the pathogenic potential of Herbaspirillum rubrisubalbicans M1. Sci Rep 9:1–10. https://doi.org/10.1038/s41598-019-40600-y
Tao J, Wang S, Liao T, Luo H (2021) Evolutionary origin and ecological implication of a unique nif island in free-living Bradyrhizobium lineages. ISME J 15:3195–3206. https://doi.org/10.1038/s41396-021-01002-z
Faoro H, Rene Menegazzo R, Battistoni F et al (2017) The oil-contaminated soil diazotroph Azoarcus olearius DQS-4T is genetically and phenotypically similar to the model grass endophyte Azoarcus sp. BH72. Environ Microbiol Rep 9:223–238. https://doi.org/10.1111/1758-2229.12502
Raittz RT, De Pierri CR, Maluk M et al (2021) Article comparative genomics provides insights into the taxonomy of Azoarcus and reveals separate origins of nif genes in the proposed Azoarcus and Aromatoleum genera. Genes (Basel) 12:1–21. https://doi.org/10.3390/genes12010071
Kojima H, Kanda M, Umezawa K, Fukui M (2021) Sulfurimicrobium lacus gen. nov., sp. nov., a sulfur oxidizer isolated from lake water, and review of the family Sulfuricellaceae to show that it is not a later synonym of Gallionellaceae. Arch Microbiol 203:317–323. https://doi.org/10.1007/s00203-020-02029-0
Remigi P, Zhu J, Young JPW, Masson-Boivin C (2016) Symbiosis within symbiosis: evolving nitrogen-fixing legume symbionts. Trends Microbiol 24:63–75. https://doi.org/10.1016/j.tim.2015.10.007
Author information
Authors and Affiliations
Contributions
A. M. P., conceptualization, writing, original draft, methodology, and investigation. F. P. M., writing — final draft, methodology, data curation, investigation, and visualization. A. C. M. A. and C. R. F. S. S., conceptualization, supervision, and writing — review and editing.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pedrolo, A.M., Matteoli, F.P., Soares, C.R.F.S. et al. Comparative Genomics Reveal the High Conservation and Scarce Distribution of Nitrogen Fixation nif Genes in the Plant-Associated Genus Herbaspirillum. Microb Ecol 86, 563–574 (2023). https://doi.org/10.1007/s00248-022-02084-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-022-02084-8