
Abstract
The neurotoxicity of lead has been well established through numerous studies. However, the cellular processes of lead neurotoxicity, as well as techniques to prevent or reverse cellular damage after lead exposure, remain unknown. If oxidative stress plays a primary role in lead-induced neurotoxicity, antioxidants should assist in reviving lead-exposed cells. The present study explores N-acetylcysteine (NAC) as an antioxidant agent in PC-12 cells after lead exposure. Selective oxidative stress parameters, including glutathione (GSH), glutathione disulfide (GSSG), and malondialdehyde (MDA), were measured in PC-12 cells exposed to various concentrations of lead acetate. Administering NAC after lead exposure improved cell survival as measured by Trypan Blue exclusion. NAC treatment also increased the GSH/GSSG ratio compared to the lead-only group, and reduced MDA to near control levels. These results imply that NAC protects cells from lead-induced oxidative damage by boosting the PC-12 cells’ antioxidant defense mechanisms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Lead poisoning in children is an entirely preventable disease; however, it still persists as a pervasive health problem (Hilliard et al. 1999). Lead adversely affects the nervous system, resulting in lower IQ, impaired cognitive functions, abnormal development, and behavioral irregularities (Kim et al. 1997). Children with lead poisoning are more prone to be impulsive and aggressive, and have greater difficulty paying attention and following directions (Wakefield 2002). Unfortunately, most of these symptoms are irreversible, and it is believed that incomplete development of the blood–brain barrier in children is the major reason for the differences seen in neurological symptoms (Goyer 1993).
Lead has been shown to interfere with the acquisition and recall of learned relationships in mollusks and mammals. Kuzirian demonstrated the impairing effects of lead on conditioned phototactic behavior in Hermissenda at the physiologically relevant concentration of 1.2 ppm (Kuzirian et al. 2001). In one study, 90 days of 250 ppm lead administration in drinking water increased behavioral suppression of contextual and auditory cues that predicted shock, suggesting impairment of the extinction of fear conditioning in Fischer 344 rats (Salinas and Huff 2002). In another study, chronic exposure to low levels (50 ppm) of lead for 1 week (from egg to adult) reduced the locomotor activity significantly in Drosophila melanogaster (Hirsch et al. 2003). Furthermore, exposure of pregnant Wistar rats to 300 ppm lead decreased the neurotransmitter levels, activity of alkaline phosphatase, and ATPase in the brains of the pups at day 12 after their birth, suggesting that gestational exposure of lead can cause neurochemical changes in the brain and further be responsible for behavioral and neurophysiological effects in lead-exposed animals (Antonio and Leret 2000). Lead exposure may cause low IQ levels, poor classroom performance, greater absenteeism, reading disabilities, and deficits in vocabulary, fine motor skills, and reaction time. It can also affect hand–eye coordination in young adults more than 10 years after childhood exposure (Needleman et al. 1990). These findings offer compelling reasons to seek therapies that might reduce or reverse the impact of lead exposure.
The biochemical and molecular elements of lead neurotoxicity have not been clearly elucidated, although many theories have been proposed. Lead is reported to alter Ca2+-mediated cellular processes in several biological systems; it may mimic Ca2+ in binding to regulatory proteins (Loikkanen et al. 1998; Ramesh et al. 1999). Additionally, lead can influence the release and reuptake of neurotransmitters normally controlled by voltage-gated Ca2+ channels (Bressler et al. 1996; Suszkiw 2004). Lead is also shown to activate protein kinase C at concentrations as low as 1 μM (Hilliard et al. 1999), nuclear transcription factor NF–κB (Ramesh et al. 1999), and the mRNA gene c-fos (Kim et al. 1997). Neuronal cells exhibit a greater sensitivity to lead-induced damage, perhaps due to a weakened oxidative stress-response (Opanashuk and Finkelstein 1995). Certain cell types have shown greater resistance to lead toxicity through isolating intercellular lead in nonmitochondrial granules (Holtzman et al. 1987).
Recently, oxidative stress has been the focus of research on lead-induced problems. Studies of neurons show that they respond to oxidative stress with increased catalase release, increased lipid peroxidation, and release of excitatory signal molecules (Loikkanen et al. 1998). Lead induces oxidative stress both directly by generation of reactive oxygen species (ROS), and indirectly by depletion of sulfhydryl-containing antioxidants (Gurer and Ercal 2000). Multiple studies with various species and cell types have explored the physiological and biochemical effects of lead exposure. In these previous studies, astroglial cells (Opanashuk and Finkelstein 1995), rats and mice (Gurer et al. 1999, Neal et al. 1999), fibroblast cells (Shelton et al. 1986), and Chinese hamster ovary (CHO) cells (Ercal et al. 1996b) were exposed to lead and several oxidative stress parameters were measured (Gurer et al. 1999, Hegg and Miletic 1998, Oravecz et al. 2001). In mice and rats, lead has been shown to induce the formation of reactive oxygen species and a well-known antioxidant, NAC, has been shown to protect cells from lead-induced oxidative stress.
The pheochromocytoma cell (PC-12) culture line is well characterized and established as a mammalian preneuronal preparation (Gurer et al. 1999; Hegg and Miletic 1998; Oravecz et al. 2001). The cells have been used as a neuronal model for studying the potential targets of neurotoxin action in particular (Hossain et al. 1997). Another study showed that in the presence of a nerve growth factor (NGF), PC-12 cells produce neurite outgrowths through the activation of the ERK/MAPK pathway (Williams et al. 2000). In the present study, rat PC-12 cells were used to investigate how lead induces oxidative stress, and the efficacy of N-acetylcysteine (NAC) as a postexposure antioxidant. There are many parameters for measuring oxidative stress. We have developed sensitive techniques for detecting and quantifying glutathione (GSH) and glutathione disulfide (GSSG) utilizing reverse-phase high-performance liquid chromatography (HPLC) (Ridnour et al. 1999). In this study, various oxidative stress parameters were measured including GSH, GSSG, malondialdehyde (MDA), and cell death (visualized with Trypan Blue).
Methods and Materials
Two media were used in culturing the PC-12 cells. The complete media was RPMI 1640 medium supplemented with 10% heat-inactivated horse serum, 5% FBS, 1% L-glutamine, and 1% Pen/Strep. The differentiation media was DMEM supplemented with 0.5% FBS, 1% L-glutamine, and 1% Pen/Strep.
Seven million (7 × 106) PC-12 cells were seeded into 12 flasks previously treated with lysine. After 24 hours, the flasks were randomly divided into four groups (control, Pb only, NAC only, Pb + NAC). The groups to receive lead were treated with a 500 μM lead acetate solution in DMEM. The cultures were incubated at 37°C for 72 hours. At the end of this period, the Pb-containing media was removed. The cultures were rinsed with straight DMEM, and collected by centrifugation. All the cells were removed from their original flasks, and reseeded in identical flasks. Differentiation media containing 1 mM NAC was added to the appropriate flasks. The cells were incubated for 24 hours, and then were removed from the culture and stored at –70°C until analyzed.
GSH Analysis
Reduced GSH levels were quantified using reverse-phase HPLC. The HPLC system (Shimadzu) consisted of a model LC-10A pump, an autoinjector with a Rheodyne injection valve with a 20 μL filling loop, and a Model Rf-535 fluorescence spectrophotometer operating at an excitation wave length of 330 nm and an emission wave length of 375 nm. The HPLC column was 250 × 4.6 mm and was packed with 5 μm particles of C18 packing material. Quantification of the peaks from the HPLC system was performed with a Chromatopac Model CR601 integrator (Shimadzu). The mobile phase was 70% acetonitrile and 30% water adjusted to a pH of approximately 2.5 through the addition of 1 mL/L of both acetic and o-phosphoric acids. The derivatives were eluted from the column isocratically at a flow rate of 1 mL/min.
Cells were homogenized in HPLC grade water. Twenty (20) μL of the diluted cell homogenate was added to 230 μL of HPLC grade water and 750 μL of NPM (1 mM in acetonitrile). The resulting solutions were incubated at room temperature for 5 minutes. The reaction was stopped by the addition of 5 μL of 2 N HCl. The samples were then filtered through a 0.2 mm Acrodisc filter and injected onto the HPLC system (Winters et al. 1995).
GSSG Analysis
Oxidized glutathione (GSSG) was measured by incubating 40 μL of straight cell homogenate, 44 μL of HPLC grade water, and 16 μL of 2-vinyl pyridine for 1 hour at room temperature in order to block any preexisting GSH. After 1 hour, 95 μL of a 2 mg/mL solution of NADPH and 5 μL of 2 units/mL glutathione reductase solution were mixed with the original solution. A 100 μL aliquot of this solution was then quickly removed and mixed with 150 μL of HPLC grade water and 750 μL of NPM (1 mM in acetonitrile). After a 5-minute incubation period, the reaction was stopped by adding 5 μL of 2 N HCl. The samples were filtered through a 0.2 mm Acrodisc filter and injected into the HPLC system (Winters et al. 1995).
MDA Analysis
To prepare the solution, 350 μL of straight cell homogenate, 100 μL of 500 ppm butylated hydroxytoluene, and 550 μL of 10% trichloroacetic acid were combined and boiled for 30 minutes. The tubes were cooled on ice and centrifuged for 10 minutes at 2500 rpm. Five hundred (500) μL of the supernatant was removed and 500 μL of thiobarbituric acid was added. The tubes were boiled again for 30 minutes, and then cooled on ice. From this solution, 500 μL were removed, added to 1.0 mL of n-butanol, vortexed, and centrifuged for 5 minutes at 1000 rpm to facilitate a phase separation. The top layer was then filtered through 0.45 μm filters and injected onto a 5 μm C18 column (250 × 4.6 mm) on a reverse-phase HPLC system. The mobile phase consisted of 69.4% 5 mM sodium phosphate buffer (pH = 7.0), 30% acetonitrile, and 0.6% tetrahydrofuran. The excitation wave length was 515 nm; the emission wave length was 550 nm (Draper et al. 1993).
Trypan Blue Cytotoxicity Assay
A 24-well, collagen-coated plate was seeded with 5 × 105 cells, and was treated with lead and NAC according to the procedure described above. The cells were then removed from the culture plates and treated with a dilute solution of Trypan Blue obtained from Sigma Chemical Company (St. Louis, MO). The blue and “clear” cells were counted in duplicate using a hemocytometer, and the survival fraction was calculated.
Protein Assay
The protein level was determined using the Bradford method with Coomassie Blue (Bio-Rad) (Bradford 1976).
Neurite Outgrowth Assay
PC-12 cells are plated at a density of 25 × 103 cell/well (24-well plates). The plate was divided into five groups in triplicate: 1) Control: no lead acetate, no NAC; 2) NGF control: NGF (100 ng/mL), no lead acetate, no NAC; 3) NAC only: NGF (100 ng/mL), no lead acetate, NAC (1 mM); 4) Lead only: NGF (100 ng/mL), lead acetate (100 μM), no NAC; 5) Lead + NAC: NGF (100 ng/mL), lead acetate (100 μM), NAC (1 mM). The media in the well was replaced with DMEM culture media supplemented with 0.5% FBS, 1% L-glutamine, and 1% penicillin/streptomycin. All wells received 100 ng/mL NGF every other day, except Group I. They also received 100 μM lead acetate (72 hours) and/or NAC (48 hours) in the DMEM media, according to their group number. Cells were fixed with 0.5 % glutaraldehyde in PBS and micropictures were taken.
Results
The concentrations of reduced glutathione (GSH), glutathione disulfide (GSSG), and the ratio of GSH/GSSG detected in the different groups are shown in Table 1. The highest concentrations were detected in the group treated with NAC, and the second highest concentrations were found in the control. The group treated with lead, then NAC, showed an increase in GSH when compared to the group that received only lead acetate, although the GSH level was not restored to the level of the control. The highest GSSG concentrations were detected in the lead acetate group. The Pb + NAC group showed an improved GSSG concentration when compared to the Pb-only group. Again, the NAC treatment did not return the GSSG levels to those of the control. The ratio of reduced to oxidized glutathione is a good indicator of the level of oxidative stress a cell is encountering. In a normal cell, the GSH level is kept high. In this study, the lead-treated group had a ratio of 0.81 ± 0.04, indicating a significant abundance of GSSG. The Pb + NAC group had a lower ratio than either the control or NAC only.
MDA is a byproduct of a free radical attack on lipids. A high concentration indicates the presence of oxidative stress. The highest levels of MDA were found in the lead-treated group (Table 2). Treatment with NAC, in the absence of lead, lowered the MDA level below the control level. NAC lowered the incidence of MDA in the lead- treated groups to a level almost as low as that of the control group.
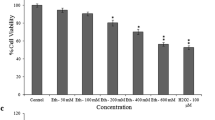
Cell viability is calculated by averaging the number of cells that survived in the control, and normalizing the treatment groups to the control. The cell survival fractions for all four concentrations of lead and lead + NAC are shown in Figure 1. The higher concentrations of lead were lethal to the cells, though administering NAC after the lead exposure improved the survival fraction significantly.

Survival curve of Pb only and Pb + NAC groups.
As shown in Figure 2, cells completely lost the normal morphology of their neurites in the presence of 100 μM lead acetate, as compared to the control cells. Neurites do not seem to be completely lost but look edematous. Treatment with NAC slightly decreased this bleb formation on neurites.

Effect of lead exposure on neurite formation in PC-12 cells in the presence or absence of NAC.
Discussion
Heavy metals, and lead in particular, interfere with cellular processes through many mechanisms, one of which is formation of reactive oxygen species. Previous reports from our laboratory and others have shown that lead exposure induces oxidative stress in CHO cells and animals (Gurer et al. 1999). We have shown that after 5 weeks of lead exposure, GSH levels in mice decreased and MDA levels increased in the brain (Ercal et al. 1996a). The results presented here support the idea that lead induces oxidative damage. It has been reported that low concentrations of lead increase neurite formation in PC-12 cells (Williams et al. 2000). It has also been shown that free radicals are required for this response to NGF, which strengthens the hypothesis that high lead levels induce oxidative stress (Oravecz et al. 2001, Suzukawa et al. 2000). Contrary to previous reports (Hilliard et al. 1999), our findings showed that lead caused swelling on neurites rather than affecting the number of neurites. In our observations, NAC improved the morphology of neurites in cells that were exposed to lead. In order to observe significant effect of lead on PC-12 cells, we used 100 μM lead acetate. At this concentration, the number of neurites was not affected. Thus, effects of lead on NGF-induced neurite outgrowth seem to be concentration dependent.
Oxidative stress is defined as an imbalance in the prooxidant/antioxidant ratio. Lead both directly and indirectly increases reactive oxygen species and decreases cells’ antioxidant defenses, thereby creating the oxidant imbalance. An in vivo imbalance is dangerous because it leads to signal cascades, activating the immune system to destroy the cell (Droge et al. 1994). Lead has a strong affinity for thiol groups and a majority of antioxidants, including GSH, have thiol groups at their active sites. Lead can bind to the –SH group, thereby deactivating the antioxidant. NAC was postulated to be a possible antioxidant capable of reversing lead-induced damage for two reasons. First, NAC possesses a cysteine with a free thiol group and thus can mimic the antioxidant action of GSH. Treatment with NAC may thereby increase the antioxidant available to sequester the damaging heavy metal, but it can also reduce other radical oxygen species generated by the metal. Second, NAC has been shown to be a biosynthetic precursor of GSH (Kelly 1998). Reduced glutathione (GSH) acts as a ROS scavenger within the cell. Increasing the synthesis of GSH would improve the antioxidant/prooxidant ratio within the cell.
In summary, lead causes toxicity and oxidative stress in PC-12 cells. NAC seems to reverse lead-induced damage in this cell line, particularly oxidative damage through two postulated routes—increasing the available GSH and by actively sequestering lead ions. These results indicate that further work should be done in vivo to quantify the usefulness of NAC as a potential therapy in the treatment of lead poisoning.
References
MT Antonio ML Leret (2000) ArticleTitleStudy of the neurochemical alterations produced in discrete brain areas by perinatal low-level lead exposure Life Sci 67 635–642
MM Bradford (1976) ArticleTitleA rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding Anal Biochem 72 248–254
JP Bressler L Belloni-Olivi S Forman GW Goldstein (1996) ArticleTitleDistinct mechanisms of neurotransmitter release from PC 12 cells exposed to lead J Neurosci Res 46 678–685
HH Draper EJ Squires H Mahmoodi J Wu S Agarwal M Hadley (1993) ArticleTitleA comparative evaluation of thiobarbituric acid methods for the determination of malondialdehyde in biological materials Free Radic Biol Med 15 353–363
W Droge K Schulze-Osthoff S Mihm D Galter H Schenk HP Eck S Roth H Gmunder (1994) ArticleTitleFunctions of glutathione and glutathione disulfide in immunology and immunopathology FASEB J 8 1131–1138
N Ercal P Treeratphan TC Hammond RH Matthews NH Grannemann DR Spitz (1996a) ArticleTitleIn vivo indices of oxidative stress in lead-exposed C57BL/6 mice are reduced by treatment with meso-2,3-dimercaptosuccinic acid or N-acetylcysteine Free Radic Biol Med 21 157–161
N Ercal P Treeratphan P Lutz TC Hammond RH Matthews (1996b) ArticleTitleN-acetylcysteine protects Chinese hamster ovary (CHO) cells from lead-induced oxidative stress Toxicology 108 57–64
RA Goyer (1993) ArticleTitleLead toxicity: current concerns Environ Health Perspect 100 177–187
H Gurer N Ercal (2000) ArticleTitleCan antioxidants be beneficial in the treatment of lead poisoning? Free Radic Biol Med 29 927–945
H Gurer H Ozgunes S Oztezcan N Ercal (1999) ArticleTitleAntioxidant role of alpha-lipoic acid in lead toxicity Free Radic Biol Med 27 75–81
CC Hegg V Miletic (1998) ArticleTitleDiminished blocking effect of acute lead exposure on high-threshold voltage-gated calcium currents in PC12 cells chronically exposed to the heavy metal Neurotoxicology 19 413–420
A Hilliard A Ramesh NH Zawia (1999) ArticleTitleCorrelation between lead-induced changes in cerebral ornithine decarboxylase and protein kinase C activities during development and in cultured PC 12 cells Int J Dev Neurosci 17 777–785
HV Hirsch J Mercer H Sambaziotis M Huber DT Stark T Torno-Morley K Hollocher H Ghiradella DM Ruden (2003) ArticleTitleBehavioral effects of chronic exposure to low levels of lead in Drosophila melanogaster Neurotoxicology 24 435–442
D Holtzman JE Olson C DeVries K Bensch (1987) ArticleTitleLead toxicity in primary cultured cerebral astrocytes and cerebellar granular neurons Toxicol Appl Pharmacol 89 211–225
MM Hossain A Takashima H Nakayama K Doi (1997) ArticleTitle5-Azacytidine induces toxicity in PC12 cells by apoptosis Exp Toxicol Pathol 49 201–206
GS Kelly (1998) ArticleTitleClinical applications of N-acetylcysteine Altern Med Rev 3 114–127
K Kim M Annadata GW Goldstein JP Bressler (1997) ArticleTitleInduction of c-fos mRNA by lead in PC 12 cells Int J Dev Neurosci 15 175–182
AM Kuzirian HT Epstein D Buck FM Child T Nelson DL Alkon (2001) ArticleTitlePavlovian conditioning-specific increases of the Ca2+- and GTP-binding protein, calexcitin in identified Hermissenda visual cells J Neurocytol 30 993–1008
JJ Loikkanen J Naarala KM Savolainen (1998) ArticleTitleModification of glutamate-induced oxidative stress by lead: the role of extracellular calcium Free Radic Biol Med 24 377–384
R Neal K Cooper G Kellogg H Gurer N Ercal (1999) ArticleTitleEffects of some sulfur-containing antioxidants on lead-exposed lenses Free Radic Biol Med 26 239–243
HL Needleman A Schell D Bellinger A Leviton EN Allred (1990) ArticleTitleThe long-term effects of exposure to low doses of lead in childhood An 11-year follow-up report. N Engl J Med 322 83–88
LA Opanashuk JN Finkelstein (1995) ArticleTitleRelationship of lead-induced proteins to stress response proteins in astroglial cells J Neurosci Res 42 623–632
K Oravecz E Bazso-Dombi F Jeney K Nagy M Gecse I Zs-Nagy (2001) ArticleTitleThe involvement of hydroxyl free radicals in differentiation of the PC-12 rat pheochromocytoma cell line Arch Gerontol Geriatr 33 61–69
GT Ramesh SK Manna BB Aggarwal AL Jadhav (1999) ArticleTitleLead activates nuclear transcription factor-kappaB, activator protein-1, and amino-terminal c-Jun kinase in pheochromocytoma cells Toxicol Appl Pharmacol 155 280–286
LA Ridnour RA Winters N Ercal DR Spitz (1999) ArticleTitleMeasurement of glutathione, glutathione disulfide, and other thiols in mammalian cell and tissue homogenates using high-performance liquid chromatography separation of N-(1-pyrenyl)maleimide derivatives Methods Enzymol 299 258–267
JA Salinas NC Huff (2002) ArticleTitleLead and conditioned fear to contextual and discrete cues Neurotoxicol Teratol 24 541–550
KR Shelton JM Todd PM Egle (1986) ArticleTitleThe induction of stress-related proteins by lead J Biol Chem 261 1935–1940
JB Suszkiw (2004) ArticleTitlePresynaptic disruption of transmitter release by lead Neurotoxicology 25 599–604
K Suzukawa K Miura J Mitsushita J Resau K Hirose R Crystal T Kamata (2000) ArticleTitleNerve growth factor-induced neuronal differentiation requires generation of Rac1-regulated reactive oxygen species J Biol Chem 275 13175–13178
J Wakefield (2002) ArticleTitleThe lead effect? Environ Health Perspect 110 A574–A580
TM Williams AM Ndifor JT Near RR Reams-Brown (2000) ArticleTitleLead enhances NGF-induced neurite outgrowth in PC12 cells by potentiating ERK/MAPK activation Neurotoxicology 21 1081–1089
RA Winters J Zukowski N Ercal RH Matthews DR Spitz (1995) ArticleTitleAnalysis of glutathione, glutathione disulfide, cysteine, homocysteine, and other biological thiols by high-performance liquid chromatography following derivatization by n-(1-pyrenyl)maleimide Anal Biochem 227 14–21
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aykin-Burns, N., Franklin, E.A. & Ercal, N. Effects of N-Acetylcysteine on Lead-Exposed PC-12 Cells. Arch Environ Contam Toxicol 49, 119–123 (2005). https://doi.org/10.1007/s00244-004-0025-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00244-004-0025-0