
Abstract
Purpose
Cerebellar ataxias are a large and heterogeneous group of disorders. The evaluation of brain parenchyma via MRI plays a central role in the diagnostic assessment of these conditions, being mandatory to exclude the presence of other underlying causes in determining the clinical phenotype. Once these possible causes are ruled out, the diagnosis is usually researched in the wide range of hereditary or sporadic ataxias.
Methods
We here propose a review of the main clinical and conventional imaging findings of the most common hereditary degenerative ataxias, to help neuroradiologists in the evaluation of these patients.
Results
Hereditary degenerative ataxias are all usually characterized from a neuroimaging standpoint by the presence, in almost all cases, of cerebellar atrophy. Nevertheless, a proper assessment of imaging data, extending beyond the mere evaluation of cerebellar atrophy, evaluating also the pattern of volume loss as well as concomitant MRI signs, is crucial to achieve a proper diagnosis.
Conclusion
The integration of typical neuroradiological characteristics, along with patient’s clinical history and laboratory data, could allow the neuroradiologist to identify some conditions and exclude others, addressing the neurologist to the more appropriate genetic testing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cerebellar ataxias are a large group of disorders characterized by various clinical presentations, ranging from the presence of a pure cerebellar phenotype to a heterogeneous combination of cerebellar signs along with extra-cerebellar symptoms [1]. Although still very challenging, different workflows have been proposed to help clinicians in identifying the cause of the clinical presentation and reach a proper diagnosis [2,3,4]. In all cases, the in vivo evaluation of brain parenchyma via MRI plays a pivotal role in the diagnostic assessment of cerebellar ataxias [5]. Indeed, it is mandatory to exclude that the observed cerebellar involvement could be due to structural damage secondary to non-degenerative conditions (e.g., stroke, neoplasm, metabolic or toxic disorders). Once these possible causes are ruled out in determining the cerebellar symptoms, the diagnosis is usually researched in the wide range of hereditary or sporadic ataxias, which are all usually characterized only by the presence of non-specific and sometimes overlapping imaging findings, with cerebellar atrophy being the least common denominator. Nevertheless, although challenging, a proper evaluation of imaging data and the integration of the patient’s clinical history could allow the neuroradiologist to identify some conditions and exclude others, addressing the neurologist to the more appropriate genetic testing.
Given this background, here, we propose a review of the main clinical and conventional imaging findings of the most common hereditary degenerative ataxias, to highlight the main features in these conditions. We will discuss degenerative ataxias according to their frequency and clinical relevance [6], while in this review, we will not cover the malformative conditions (e.g., ponto-cerebellar hypoplasias, some tubulinopathies, or certain dystroglycanopathies), given their different pathophysiology and imaging appearance.
Autosomal dominant ataxias
Spinocerebellar ataxia type 1
Spinocerebellar ataxia type 1 (SCA1) accounts for 6% of autosomal dominant cerebellar ataxias [7]. Affected individuals have 39 or more CAG trinucleotide repeats in the ATXN1 gene, which encodes for the Ataxin1 protein [8]. Onset is typically between the third and the fourth decades, even though childhood onset has been reported [9, 10]. The phenotype comprises a cerebellar syndrome with ataxia of gait, stance, and limbs, dysarthria, and oculomotor abnormalities. Pyramidal signs are common, but amyotrophy and sensory loss also occur [7]. The duration of disease from onset to death is 10 to 30 years in the adult-onset forms, whereas it is more rapid and severe in juvenile-onset forms [10].
Brain MRI typically shows olivo-ponto-cerebellar atrophy and white matter volume decrease, with a similar distribution but less severe than SCA2 [11, 12].
The presence of a midline T2-hyperintensity in the pontine base has been reported [13] as well as a cruciform pontine T2-hyperintensity, the “hot cross bun” sign, due to ponto-cerebellar fibers degeneration [14] (Fig. 1). Usually, the supratentorial compartment is relatively spared by the disease, with high-intensity areas on T2-weighted images in the frontal white matter only anecdotally reported [15]. Finally, spinal cord volume reduction might be present [12] and correlates with the SARA score, length of CAG expansion, and disease duration [16].

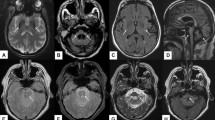
Brain MRI scan of a 69-year-old female SCA1 patient. Axial T2-weighted (a) and coronal FLAIR (b) images show a global cerebellar volume loss, along with brainstem atrophy (c–d) and the presence of the “hot cross bun” sign (arrows)
Spinocerebellar ataxia type 2
Spinocerebellar ataxia type 2 (SCA2) is caused by an abnormal expansion of CAG repetition (>33) in the ATXN2 gene, coding for Ataxin2 protein [17], with an estimated prevalence in certain areas of 6.57 cases per 100,000 individuals [18]. Showing a mean disease duration of around 10 years, the onset usually occurs in the fourth decade, but can vary from childhood to late adulthood with an inverse correlation between age of onset and CAG repeat length [17]. Patients with SCA2 present with a cerebellar syndrome associated more often than SCA1 with saccadic slowing, hyporeflexia, tremor, or titubation. In some cases (especially those with a smaller number of CAG repeat expansions), SCA2 can present as parkinsonism [7]. First symptoms are slowly progressive gait ataxia and leg cramps, whereas dysarthria, kinetic, or postural tremor, decreased muscle tone, and tendon reflexes appear later [19]. Dystonia, chorea, and dementia have also been described as relatively common (almost 40% of the patients) [20]. Rarely, it can be associated with L-dopa-responsive parkinsonism [21,22,23], while ocular findings (i.e., nystagmus, slow saccadic eye movements, and supranuclear ophthalmoplegia) are more common [24].
Brain MRI shows significant global atrophy of the cerebellum, with marked volume loss involving both hemispheres and the vermis. Furthermore, pontine atrophy (with flattening of the inferior part) (Fig. 2) and a variable degree of medulla oblongata and spinal cord volume loss are also usually depicted [18], with the severity of the olivo-ponto-cerebellar volume loss that correlates with clinical disability [25]. The “hot cross bun” sign may be present, resembling a sporadic multi-system atrophy pattern, while basal ganglia T2-hyperintensity are only rarely reported [25]. In advanced stages of the disease, a pattern of fronto-temporal atrophy with ventricular enlargement can also be depicted [26].

Imaging findings in a 38-year-old female SCA2 patient. Sagittal (a) and axial (b) multiplanar reconstructions of 3D T1-weighted volume show a global cerebellar volume loss, involving both the vermis and cerebellar hemispheres, along with significant brainstem atrophy, particularly affecting the pons which demonstrates a flattening of its inferior profile (arrows)
Spinocerebellar ataxia type 3
Spinocerebellar ataxia type 3 (SCA3), also known as Machado-Joseph disease, is the most common SCA subtype worldwide and is caused by abnormal CAG trinucleotide repeats (52–86) in ATXN3 gene encoding for the Ataxin3 protein [9, 27]. Its onset usually occurs between the second and fifth decade, being characterized by ataxia, dysarthria, hyperreflexia, diplopia, and nystagmus [28]. Ambulation difficulty progressively increases, with assistive devices usually required 10 to 15 years after onset [28], while upper motor neuron signs are often present and may mimic hereditary spastic paraplegia [29, 30]. Sleep disturbance and impaired executive and emotional functioning have also been reported [31, 32]. The disease duration is variable from few up to 30 years after onset, with exitus usually occurring due to pulmonary complication and cachexia [33].
Brain imaging reveals a variable degree of ponto-cerebellar atrophy, less severe compared to the one found in SCA1 and SCA2 patients, mainly involving the vermis and dentate nuclei with subsequent enlargement of the fourth ventricle [34] (Fig. 3). Also, similar to what is reported in SCA1 and SCA2, pontine T2-weighted hyperintensities may be present in these patients, resembling in some cases the “hot cross bun” sign morphology [12]. As the disease progresses, frontal and temporal lobe atrophy can be observed [35], while abnormal pallidal linear hyperintensities on T2-weighted and FLAIR sequences have been sporadically reported [36].

Axial (a, c) and coronal (b) T2-weighted images showing mild cerebellar atrophy, with enlargement of the 4th ventricle (arrow), in a 30-year-old female SCA3 patient
Spinocerebellar ataxia type 6
Spinocerebellar ataxia type 6 (SCA6), characterized by an abnormal CAG trinucleotide repeat expansion in CACNA1A gene [37], shows an estimated prevalence of about 0.02 on 100,000 individuals [38]. The onset can be extremely variable, ranging from 19 to 73 years (mean onset age = 43–52 years), with a relatively preserved lifespan [39, 40]. Clinically, SCA6 usually presents with a “pure” cerebellar ataxia, although a mild peripheral neuropathy (as well as bradykinesia, dystonia, and pyramidal signs) can also occur in some patients [41]. Ocular abnormalities are also quite common, as they have been described in 50% of patients, mainly consisting of diplopia and downbeat nystagmus [42,43,44].
At brain imaging, a variable degree of cerebellar atrophy has been described, involving the hemispheres but mostly the vermis (Fig. 4), with volume loss affecting the pons and middle cerebellar peduncles that has been only anecdotally reported in these patients [34]. No brain signal abnormalities have been reported in SCA6, nor supratentorial atrophy.

Brain MRI findings in a 56-year-old female SCA6. In the sagittal T1- (a) and coronal T2- (b) weighted images, it is appreciable a global cerebellar atrophy, with particular involvement of the vermis (arrow) and a relative sparing of the pons
Spinocerebellar ataxia type 7
Spinocerebellar ataxia type 7 (SCA7) shows a prevalence of less than 1:100,000, representing 2% of all SCAs [45, 46]. It is associated with the presence of more than 36 CAG trinucleotide repeat expansion in the ATXN7 gene, although the development of symptoms has been also reported in patients with a lower number of repeats [47]. Disease onset ranges from childhood up to the sixth decade, with early-onset forms being more aggressive and rapidly progressive [48, 49]. Typical symptoms include cerebellar syndrome and visual loss caused by retinal degeneration, ultimately leading to complete blindness [50, 51].
MRI findings include cerebellar atrophy, mainly involving the superior part of the vermis, along with marked pontine atrophy [52] (Fig. 5), with the latter preceding and showing some degree of independence from the presence of cerebellar degeneration [53]. The “hot cross bun” sign has been reported in only one SCA7 patient [54], while no further reports describing signal alterations in SCA7 are available in literature. Finally, a variable degree of supratentorial atrophy can be present in these patients [52] (Fig. 5).

Neuroradiological findings in a 79-year-old male SCA7 patient. Axial T2- (a) and sagittal T1- (b) weighted sequences demonstrate a global cerebellar, as well as pontine (arrow), atrophy. Along with the infratentorial involvement, a diffuse supratentorial gray matter volume loss is also present (c)
Spinocerebellar ataxia type 8
Spinocerebellar ataxia type 8 (SCA8) accounts for 2–5% of autosomal dominant forms of inherited ataxia and is more common in Finland [55, 56]. It is caused by the abnormal expansion of both an expanded CTG trinucleotide repeat in the ATXN8OS gene and the complementary CAG repeat in the ATXN8 gene [57]. The onset is extremely variable, ranging from 1 to 73 years, and the phenotype is characterized by ataxia, scanned dysarthria, and tremor, with reflex hyperactivity that may be present in severe cases [58]. Progression is usually independent of the age of onset and may take decades, though it does not significantly shorten lifespan [59, 60].
On brain MRI, cerebellar atrophy affecting both the hemispheres and the vermis is usually found, with preservation of the brainstem and the cerebral hemispheres [61] (Fig. 6). No signal abnormalities have been described, except for a case of “hot cross bun” sign [54], while mild spinal cord atrophy can sometimes be present [58].

Brain MRI scan of 43-year-old male SCA8 patient. Coronal T2- (a) and sagittal T1- (b) weighted images show global cerebellar atrophy, with a relative sparing of the pons
Spinocerebellar ataxia type 17
Spinocerebellar ataxia type 17 (SCA17) is caused by the expansion of a CAG trinucleotide (42 or more repeats) in the TATA-box-binding protein gene (TBP) [62, 63]. Similarly to SCA8, it is also characterized by wide variability in its age of onset (from 3 to 75 years, mean = 34.6 years) [64]. The clinical features include cerebellar ataxia, cognitive decline, psychiatric symptoms, parkinsonism, and hyperkinetic disorders (e.g., chorea or dystonia, hence SCA17 being also referred to as Huntington disease-like 4 [65]). Among the abovementioned symptoms, ataxia and psychiatric abnormalities usually represent the initial manifestations of the disease, being then followed by involuntary movement, parkinsonism, dementia, and pyramidal signs [62, 66,67,68].
Conventional brain MRI shows a variable degree of cerebellar atrophy, affecting both the vermis and the hemispheres [25]. No significant changes are usually reported in the supratentorial areas, both in terms of signal changes or atrophy, with the exception of a single case showing a T2-hyperintense putaminal rim [69].
Autosomal recessive ataxias
Friedreich’s ataxia
Friedreich’s ataxia (FRDA) is the most common autosomal recessive ataxia, with an estimated prevalence in Europe between 1 in 750,000 (Finland) and 1 in 20,000 (Northern Spain) [70]. It is caused by biallelic GAA trinucleotide repeat expansions in intron 1 of the FXN gene on chromosome 9q21, encoding Frataxin [71]. FRDA first symptoms typically present between the age of 10 and 15, with onset after 25 and 40 years considered as late and very late onset, respectively [70, 72]. FRDA patients experience a shortened lifespan (average 35–40 years), with the most common cause of exitus represented by cardiac dysfunction [73]. From a neurological standpoint, FRDA is a disorder affecting both the central and peripheral nervous systems, with gait and limb ataxia, dysarthria, and lower limb areflexia that are present in almost all cases [74]. Pyramidal weakness is a relatively late sign, much more prominent in the lower limbs, while hearing difficulties due to acoustic neuropathy and eye movement abnormalities (e.g., square-wave jerks) are common signs of FRDA [74]. Systemic involvement is present in this condition, as shown by the presence of musculoskeletal (i.e., scoliosis and equinovarus deformities), cardiac (i.e., cardiomyopathy), and endocrine (i.e., diabetes mellitus) abnormalities [70, 74,75,76].
Unlike most of the inherited ataxias, brain MRI in FRDA patients typically shows normal cerebellar volume (Fig. 7), or only mild atrophy of the upper portion of the vermis, while global cerebellar atrophy can be rarely found in very-late-onset patients [77, 78]. Indeed, the main imaging finding in this condition is a decrease of the antero-posterior diameter of the medulla oblongata and the cervical spinal cord, sometimes associated to signal abnormalities in the posterior or lateral columns [79], a finding consistent with degeneration of the ascending dorsal column system [80]. Furthermore, dentate nuclei atrophy, with increased iron accumulation, may be detected by susceptibility-weighted imaging [78, 81,82,83]. The supratentorial compartment is usually spared in this condition, both in terms of signal changes and volume loss.

Axial (a) and sagittal (b) multiplanar reconstructions of a 3D T1-weighted volume show the typical brain MRI appearance in FRDA patients, as depicted in this 20-year-old male subject where a preserved cerebellar volume is present
Autosomal recessive spastic ataxia of Charlevoix-Saguenay
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) shows a high prevalence in northeastern Quebec Canada [84], although cases have been reported outside Canada in recent decades [85,86,87]. It is caused by autosomal recessive mutations in the SACS gene (13q11), which encodes a large protein named Sacsin, that might have roles in mitochondrial function, protein chaperoning, and the ubiquitin-proteasome system [88, 89]. The mean age at onset is approximately 6 years (range: 0–40 years), but an increasing number of cases with disease onset in teenage or early adult ages have been reported [90]. Disease progression is slow, and patients become wheelchair bound by the third or fourth decade of life. Most ARSACS patients show a typical clinical triad characterized by early-onset cerebellar ataxia, lower limb spasticity, and peripheral neuropathy [91]. Other symptoms and signs include dysarthria, nystagmus, and hypermyelination of the retinal fibers [91].
Brain MRI features include early and progressive superior vermis atrophy and linear hypointensities on T2-weighted images in the pons near the pyramidal tracts [92] (Fig. 8). Recent studies also found T2-hyperintensities of the lateral pons when merging into the middle cerebellar peduncles that appear thickened, probably related to abnormally large transverse ponto-cerebellar fibers, along with a frequent association with posterior fossa arachnoid cysts [93]. Furthermore, bilateral parietal atrophy and short-stretched thinning of the posterior mid-body of the corpus callosum can be depicted in ARSACS patients [93], as well as a thinning of the cervical spinal cord [94] (Fig. 8).

Brain MRI findings in a 45-year-old female ARSACS patient. A predominant superior vermian atrophy is shown on a sagittal T1-weighted sequence (a, white arrow), along with as a mild T2-weighted hypointensity affecting the transverse pontine fibers (b, black arrows). Parietal (c) and cervical spinal cord (a, arrowheads) atrophy is also evident
Ataxia with oculomotor apraxia type 1
Ataxia with oculomotor apraxia type 1 (AOA1) is caused by biallelic mutations in the APTX gene at locus 9p13.3, which encodes for a nuclear histidine triad (HIT) protein, named aprataxin [95]. The first manifestation is usually represented by progressive gait imbalance (mean age of onset = 4.3 years; range = 2–10 years), with a mean disease duration of 29.8 ± 14.8 years [96]. The clinical phenotype of AOA1 is characterized by an early-onset cerebellar syndrome, nystagmus, dysarthria, oculomotor apraxia, areflexia, peripheral axonal neuropathy, muscle weakness, and a variable degree of intellectual disability [97]. Laboratory findings include hypoalbuminemia and hypercholesterolemia, while alpha-fetoprotein (AFP) value is usually relatively increased, although less than what was reported in AOA2 patients [98].
MRI findings include diffuse cerebellar atrophy, mainly involving the anterior vermis, and possible brainstem atrophy, without T2-weighted signal changes [96] (Fig. 9). Disappearance of the dentate nuclei hypointensity on susceptibility-weighted imaging (SWI) has been described in AOA1 patients, along with a preserved volume, findings suggestive of an iron content change in this structure [99].

Axial T2- (a) and sagittal T1- (b) weighted images of an 18-year-old male AOA1 patient showing a severe global cerebellar atrophy, with relative sparing of the brainstem
Ataxia with oculomotor apraxia type 2
Ataxia with oculomotor apraxia type 2 (AOA2) is caused by mutations in the senataxin (SETX) gene at locus 9q34, which encodes for a protein suspected to be a DNA/RNA helicase [95, 100, 101]. It is a progressive, disabling cerebellar ataxia occurring within the second decade [102]. The clinical phenotype is characterized by progressive cerebellar ataxia, sensorimotor peripheral neuropathy, occasional oculomotor apraxia (relatively less frequently than the frequency reported in AOA1 patients), strabismus, chorea, and/or dystonia [102]. Laboratory examination reveals elevated AFP serum levels (more than those reported in AOA1 patients) and less frequently elevated creatine kinase serum level [98].
From a neuroradiological standpoint, MRI findings are very similar to those found in AOA1 patients (Fig. 10), with cerebellar atrophy (with prominent involvement of the vermis and the anterior lobe) and loss of dentate SWI hypointensity and relative supratentorial sparing [99, 103].

Coronal T2- (a) and sagittal T1- (b) weighted images of a 25-year-old female AOA2 patient showing a moderate global cerebellar and a relative brainstem sparing
Spastic paraplegia 7
Spastic paraplegia 7 (SGP7) is the fourth cause of genetic ataxia in the UK, and the second most common cause of recessive ataxia (Kara2016). Indeed, recent studies demonstrated that SPG7 mutations are a frequent cause of undiagnosed cerebellar ataxias with adult-onset and pyramidal signs and provided the minimum prevalence of SPG7-related disease at 0.72/100,000 [104]. It appears to have a predilection for male patients (83%), with an average age of symptoms onset at 41.7 years [105]. Almost all patients show cerebellar ataxia at the diagnosis, usually along with mild spasticity and ocular findings [104]. Some patients present with a complicated phenotype of spastic paraplegia, associated with optic neuropathy, urinary urgency, scoliosis, pes cavus, neuropathy, and amyotrophy [106].
At the brain MRI examination, the most frequent feature is represented by mild cerebellar atrophy, mostly involving the vermis [105] (Fig. 11). An increased T2-weighted signal in dentate nuclei is reported in this condition, while the red nucleus signal is usually normal [105]. No supratentorial involvement has been reported so far in SPG7 patients.

Imaging finding in a 69-year-old male SPG7 patient. Brain MRI axial (a) and sagittal T2- (b) weighted images show the presence of a mild global cerebellar atrophy, with particular involvement of the vermis (black arrow)
Ataxia-telangiectasia
Ataxia-telangiectasia (AT) is a primary immunodeficiency disease caused by mutations in AT mutated (ATM) gene encoding a serine/threonine protein kinase [107, 108]. Disease onset is usually between 6 and 18 months, and the clinical phenotype can be highly variable, including progressive cerebellar ataxia, oculo-cutaneous telangiectasia, variable immunodeficiency, radiosensitivity, susceptibility to malignancies, and metabolic disorders [109]. Involuntary movements (e.g., chorea, dystonia, athetosis, myoclonic jerks, or various tremors) can be present, while cognitive impairment is usually observed in up to 30% of patients [109]. Disease duration is less than 25 years, with the two most common causes of exitus that are chronic pulmonary diseases and malignancy [110].
At brain MRI, AT patients typically demonstrates progressive cerebellar atrophy, with significant vermian involvement [111] (Fig. 12). In addition, in some cases, supratentorial white matter T2-weighted and SWI hypointensities can be detected, representing hemosiderin deposits and deep cerebral telangiectatic vessels [111, 112] (Fig. 12). In older patients, diffuse T2-weighted hyperintensity of the cerebral white matter can be found as an expression of vascular damage [112].

Neuroradiological findings in a 26-year-old female AT patient. A mild global cerebellar atrophy is shown in both axial (a) and coronal (b) T2-weighted images, while the T2*-weighted sequence allows for the depiction of small punctuate hypointense foci (arrows in c) representing both hemosiderin deposits and telangiectatic vessels
Ataxia with isolated vitamin E deficiency
Ataxia with isolated vitamin E deficiency (AVED) is caused by mutations in the alpha tocopherol transfer protein (TTPA) gene, mapped on chromosome 8q13 that codifies for a protein which binds alpha tocopherol and very-low-density lipoproteins (VLDLs) in the liver [113]. The mutated protein impairs the incorporation of vitamin E into plasma VLDL, with subsequent systemic oxidative stress damage [114].
The age of onset is variable, from early childhood to very late adult life [115]. The neurological phenotype is very similar to the one found in FRDA patients (progressive cerebellar ataxia with posterior column involvement, Romberg’s sign, and pyramidal spasticity) [116], although AVED patients are more frequently experiencing head titubation and dystonia, with less pronounced cardiovascular impairment and neuropathy [117]. In the presence of this phenotype, the evidence of very low serum vitamin E levels, in the absence of fat malabsorption, is highly suggestive of AVED [118].
Similarly to the neurological phenotype, also brain MRI findings in AVED patients resemble those found in FRDA, with the presence of preserved cerebellar volumes in almost all cases [119], although mild hemispheric atrophy has been reported in some subjects [120]. Finally, no cervical spine abnormalities are usually reported in AVED patients.
Cerebrotendinous xanthomatosis (CTX)
Cerebrotendinous xanthomatosis CTX is a neurometabolic storage disorder caused by mutations in the CYP27A1 gene, mapped on chromosome 2q33 and codifying for the 27-hydroxylase, with mutations that lead to reduced enzymatic activity and elevated levels of cholestanol, cholesterol, and bile alcohols [121]. The age of onset may range from infancy to adulthood, with a median age of clinical presentation that ranges between 9 and 19 years, although diagnosis usually occurs only later during adulthood [122].
Clinical symptoms and signs are both neurological and non-neurological. Among systemic manifestations, xanthomas present early in childhood and enlarge over time, with the Achilles tendon being the most common affected site, although they can also be found in other subcutaneous tissues, especially at the level of the elbow [123]. Other non-neurological manifestations include neonatal cholestatic jaundice, chronic diarrhea, and ocular manifestations (e.g., cataract, optic neuropathy, optic disk paleness, and premature retinal senescence) [124]. Hallmark neurological signs are represented by intellectual disability, pyramidal signs (i.e., spasticity, hyperreflexia, extensor plantar responses), cerebellar signs (ataxia, dysarthria, nystagmus), and peripheral neuropathy, while other symptoms include epileptic seizures and parkinsonism [122].
MRI studies show, along with a variable degree of cerebral and cerebellar atrophy, focal or diffuse subcortical and periventricular white matter T2-weighted hyperintensities, sometimes also found affecting the midbrain [125]. Areas of low T2-weighted signal can also be observed, referable to vacuolation and calcifications, and have been proved to be a possible biomarker of disease progression [125]. Furthermore, a non-homogeneous T2-weighted hyperintense signal in dentate nuclei and surrounding cerebellar white matter has been demonstrated in most of the CTX patients, apparently showing some degree of correlation with clinical severity [125] (Fig. 13).

Brain MRI findings in a 42-year-old male CTX patient. Along with a mild global cerebellar atrophy, it is present gliosis and calcifications of the deep cerebellar WM, extending to the peri-dentate region (arrows), as shown in the axial FLAIR (a), T1-weighted (b), and SWI (c) sequences
Autosomal recessive spinocerebellar ataxia type 10
Autosomal recessive spinocerebellar ataxia type 10 (SCAR10), also known as autosomal recessive cerebellar ataxia type 3 (ARCA3), is caused by homozygous or compound heterozygous mutations in the anoctamin 10 (ANO10) gene, mapped on 3p21.33 [126].
The onset is reported in the teenage or young adult years [127], usually presenting with gait and limb ataxia, dysarthria, nystagmus, and occasional involvement of lower motor neurons, while the cognitive status may be normal or impaired [128]. A characteristic finding of this condition is the presence of low levels of coenzyme Q10 (CoQ10) [129].
At the brain MRI scan, SCAR10 patients show moderate to marked cerebellar atrophy [129,130,131], in some cases coupled to a mild T2-weighted hyperintensity of the DN [130, 131] (Fig. 14). Diffuse supratentorial cortical atrophy, more pronounced in the fronto-parietal regions, can be also detected in older patients [129, 131].

A 36-year-old female SCAR10 patient showing a global cerebellar atrophy, along with a mild T2-weighted hyperintensity affecting both dentate nuclei (black arrows in b)
X-linked ataxias
Fragile X-associated tremor/ataxia syndrome
FXTAS constitutes a progressive neurodegenerative movement disorder caused by a fragile X “premutation,” defined as 55–200 CGG repeats in the 50-untranslated region of the FMR1 gene, while the presence of more than 200 repeats results in the development of the fragile X syndrome (a heritable form of cognitive impairment) [132].
FXTAS is more common in males than in females, the onset is usually over the age of 50 [133], with a life expectancy that ranges between 5 and 25 years [134]. Classical neurological manifestations include kinetic tremor and cerebellar ataxia, but cognitive decline, psychiatric disorders, peripheral neuropathy, and autonomic dysfunction are also frequently described [135, 136].
At brain MRI scan, FXTAS patients show characteristic features, useful for a correct diagnosis along with clinical signs and genetic tests. Indeed, the revised FXTAS diagnostic criteria [137] include the presence of two major radiological features, namely the presence of white matter lesions in middle cerebellar peduncles and in corpus callosum splenium. The first finding is considered a radiological hallmark of this condition (Fig. 15), although also being reported in cerebellar type multiple system atrophy (MSA-C) or acquired hepato-cerebral degeneration [86], while the second has been recently reported as a more reliable MRI sign [134]. Other common and minor findings include the presence of T2-weighted hyperintensities in the pons (i.e., the “hot cross bun” sign) and in the afferent projections of the middle and superior cerebellar peduncles, as well as supratentorial areas (e.g., insula or periventricular white matter), along with a generalized brain, brainstem, and cerebellar atrophy [138].

Imaging findings in a 55-year-old male patient with FXTAS. Axial T1- (a) and T2- (b) weighted sequences show the presence of a mild global cerebellar atrophy, associated with the typical T2-weighted hyperintensity of the middle cerebellar peduncles (black arrows in b)
Conclusion
Neuroradiological diagnosis of hereditary degenerative ataxias can be very challenging, given that usually brain MRI scans show, in most of these conditions, the presence of non-specific and sometimes overlapping imaging findings.
In this work, we have reviewed the main clinical and conventional imaging findings of the most common hereditary degenerative ataxias. A proper assessment of imaging and clinical data is crucial for the neuroradiologist to identify some and exclude other conditions, leading the clinician to a more appropriate genetic testing to ultimately achieve a diagnosis.
References
Manto M, Gandini J, Feil K, Strupp M (2020) Cerebellar ataxias: an update. Curr Opin Neurol 33(1):150–160. https://doi.org/10.1097/WCO.0000000000000774
Beaudin M, Matilla-Duenas A, Soong BW, Pedroso JL, Barsottini OG, Mitoma H, Tsuji S, Schmahmann JD, Manto M, Rouleau GA, Klein C, Dupre N (2019) The classification of autosomal recessive cerebellar ataxias: a consensus statement from the Society for Research on the Cerebellum and Ataxias Task Force. Cerebellum 18(6):1098–1125. https://doi.org/10.1007/s12311-019-01052-2
de Silva RN, Vallortigara J, Greenfield J, Hunt B, Giunti P, Hadjivassiliou M (2019) Diagnosis and management of progressive ataxia in adults. Pract Neurol 19(3):196–207. https://doi.org/10.1136/practneurol-2018-002096
Renaud M, Tranchant C, JVT M, Mochel F, Synofzik M, van de Warrenburg B, Pandolfo M, Koenig M, Kolb SA, Anheim M, Group RW (2017) A recessive ataxia diagnosis algorithm for the next generation sequencing era. Ann Neurol 82(6):892–899. https://doi.org/10.1002/ana.25084
Heidelberg D, Ronsin S, Bonneville F, Hannoun S, Tilikete C, Cotton F (2018) Main inherited neurodegenerative cerebellar ataxias, how to recognize them using magnetic resonance imaging? J Neuroradiol 45(5):265–275. https://doi.org/10.1016/j.neurad.2018.05.005
Bird TD (1993) Hereditary ataxia overview. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews((R)). Seattle (WA),
Schols L, Bauer P, Schmidt T, Schulte T, Riess O (2004) Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 3(5):291–304. https://doi.org/10.1016/S1474-4422(04)00737-9
Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, Zoghbi HY (1993) Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 4(3):221–226. https://doi.org/10.1038/ng0793-221
Schols L, Amoiridis G, Buttner T, Przuntek H, Epplen JT, Riess O (1997) Autosomal dominant cerebellar ataxia: phenotypic differences in genetically defined subtypes? Ann Neurol 42(6):924–932. https://doi.org/10.1002/ana.410420615
Zoghbi HY, Pollack MS, Lyons LA, Ferrell RE, Daiger SP, Beaudet AL (1988) Spinocerebellar ataxia: variable age of onset and linkage to human leukocyte antigen in a large kindred. Ann Neurol 23(6):580–584. https://doi.org/10.1002/ana.410230609
Guerrini L, Lolli F, Ginestroni A, Belli G, Della Nave R, Tessa C, Foresti S, Cosottini M, Piacentini S, Salvi F, Plasmati R, De Grandis D, Siciliano G, Filla A, Mascalchi M (2004) Brainstem neurodegeneration correlates with clinical dysfunction in SCA1 but not in SCA2. A quantitative volumetric, diffusion and proton spectroscopy MR study. Brain 127(Pt 8):1785–1795. https://doi.org/10.1093/brain/awh201
Pedroso JL, Barsottini OG (2013) Spinal cord atrophy in spinocerebellar ataxia type 1. Arq Neuropsiquiatr 71(12):977. https://doi.org/10.1590/0004-282X20130187
Mandelli ML, De Simone T, Minati L, Bruzzone MG, Mariotti C, Fancellu R, Savoiardo M, Grisoli M (2007) Diffusion tensor imaging of spinocerebellar ataxias types 1 and 2. AJNR Am J Neuroradiol 28(10):1996–2000. https://doi.org/10.3174/ajnr.A0716
Namekawa M, Honda J, Shimazaki H (2015) "Hot cross bun" sign associated with SCA1. Intern Med 54(7):859–860. https://doi.org/10.2169/internalmedicine.54.3460
Nakayama T, Nakayama K, Takahashi Y, Ohkubo K, Tobe H, Soma M, Ozawa Y, Kanmatsuse K, Nakamura M, Hironaga T, Makizumi Y, Nagura H (2001) Case of spinocerebellar ataxia type 1 showing high intensity lesions in the frontal white matter on T2-weighted magnetic resonance images. Med Sci Monit 7(2):299–303
Martins CR Jr, Martinez ARM, de Rezende TJR, Branco LMT, Pedroso JL, Barsottini OGP, Lopes-Cendes I, Franca MC Jr (2017) Spinal cord damage in spinocerebellar ataxia type 1. Cerebellum 16(4):792–796. https://doi.org/10.1007/s12311-017-0854-9
Pulst SM (1993) Spinocerebellar ataxia type 2. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews((R)). Seattle (WA),
Velazquez-Perez LC, Rodriguez-Labrada R, Fernandez-Ruiz J (2017) Spinocerebellar ataxia type 2: clinicogenetic aspects, mechanistic insights, and management approaches. Front Neurol 8:472. https://doi.org/10.3389/fneur.2017.00472
Orozco Diaz G, Nodarse Fleites A, Cordoves Sagaz R, Auburger G (1990) Autosomal dominant cerebellar ataxia: clinical analysis of 263 patients from a homogeneous population in Holguin, Cuba. Neurology 40(9):1369–1375. https://doi.org/10.1212/wnl.40.9.1369
Geschwind DH, Perlman S, Figueroa CP, Treiman LJ, Pulst SM (1997) The prevalence and wide clinical spectrum of the spinocerebellar ataxia type 2 trinucleotide repeat in patients with autosomal dominant cerebellar ataxia. Am J Hum Genet 60(4):842–850
Charles P, Camuzat A, Benammar N, Sellal F, Destee A, Bonnet AM, Lesage S, Le Ber I, Stevanin G, Durr A, Brice A, French Parkinson’s Disease Genetic Study G (2007) Are interrupted SCA2 CAG repeat expansions responsible for parkinsonism? Neurology 69 (21):1970-1975. https://doi.org/10.1212/01.wnl.0000269323.21969.db
Furtado S, Farrer M, Tsuboi Y, Klimek ML, de la Fuente-Fernandez R, Hussey J, Lockhart P, Calne DB, Suchowersky O, Stoessl AJ, Wszolek ZK (2002) SCA-2 presenting as parkinsonism in an Alberta family: clinical, genetic, and PET findings. Neurology 59(10):1625–1627. https://doi.org/10.1212/01.wnl.0000035625.19871.dc
Payami H, Nutt J, Gancher S, Bird T, McNeal MG, Seltzer WK, Hussey J, Lockhart P, Gwinn-Hardy K, Singleton AA, Singleton AB, Hardy J, Farrer M (2003) SCA2 may present as levodopa-responsive parkinsonism. Mov Disord 18(4):425–429. https://doi.org/10.1002/mds.10375
Engel KC, Anderson JH, Gomez CM, Soechting JF (2004) Deficits in ocular and manual tracking due to episodic ataxia type 2. Mov Disord 19(7):778–787. https://doi.org/10.1002/mds.20121
Dohlinger S, Hauser TK, Borkert J, Luft AR, Schulz JB (2008) Magnetic resonance imaging in spinocerebellar ataxias. Cerebellum 7(2):204–214. https://doi.org/10.1007/s12311-008-0025-0
Lastres-Becker I, Rub U, Auburger G (2008) Spinocerebellar ataxia 2 (SCA2). Cerebellum 7(2):115–124. https://doi.org/10.1007/s12311-008-0019-y
Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I, Kimura J, Narumiya S, Kakizuka A (1994) CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 8(3):221–228. https://doi.org/10.1038/ng1194-221
Paulson H, Shakkottai V (1993) Spinocerebellar ataxia type 3. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews((R)). Seattle (WA),
Gan SR, Shi SS, Wu JJ, Wang N, Zhao GX, Weng ST, Murong SX, Lu CZ, Wu ZY (2010) High frequency of Machado-Joseph disease identified in southeastern Chinese kindreds with spinocerebellar ataxia. BMC Med Genet 11:47. https://doi.org/10.1186/1471-2350-11-47
Wang YG, Du J, Wang JL, Chen J, Chen C, Luo YY, Xiao ZQ, Jiang H, Yan XX, Xia K, Pan Q, Tang BS, Shen L (2009) Six cases of SCA3/MJD patients that mimic hereditary spastic paraplegia in clinic. J Neurol Sci 285(1-2):121–124. https://doi.org/10.1016/j.jns.2009.06.027
Schols L, Haan J, Riess O, Amoiridis G, Przuntek H (1998) Sleep disturbance in spinocerebellar ataxias: is the SCA3 mutation a cause of restless legs syndrome? Neurology 51(6):1603–1607. https://doi.org/10.1212/wnl.51.6.1603
Zawacki TM, Grace J, Friedman JH, Sudarsky L (2002) Executive and emotional dysfunction in Machado-Joseph disease. Mov Disord 17(5):1004–1010. https://doi.org/10.1002/mds.10033
Sudarsky L, Corwin L, Dawson DM (1992) Machado-Joseph disease in New England: clinical description and distinction from the olivopontocerebellar atrophies. Mov Disord 7(3):204–208. https://doi.org/10.1002/mds.870070303
Eichler L, Bellenberg B, Hahn HK, Koster O, Schols L, Lukas C (2011) Quantitative assessment of brain stem and cerebellar atrophy in spinocerebellar ataxia types 3 and 6: impact on clinical status. AJNR Am J Neuroradiol 32(5):890–897. https://doi.org/10.3174/ajnr.A2387
Meira AT, Arruda WO, Ono SE, Neto AC, Raskin S, Camargo CHF, Teive HAG (2019) Neuroradiological findings in the spinocerebellar ataxias. Tremor Other Hyperkinet Mov (N Y) 9. https://doi.org/10.7916/tohm.v0.682
Yamada S, Nishimiya J, Nakajima T, Taketazu F (2005) Linear high intensity area along the medial margin of the internal segment of the globus pallidus in Machado-Joseph disease patients. J Neurol Neurosurg Psychiatry 76(4):573–575. https://doi.org/10.1136/jnnp.2004.040279
Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC (1997) Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat Genet 15(1):62–69. https://doi.org/10.1038/ng0197-62
Geschwind DH, Perlman S, Figueroa KP, Karrim J, Baloh RW, Pulst SM (1997) Spinocerebellar ataxia type 6. Frequency of the mutation and genotype-phenotype correlations. Neurology 49(5):1247–1251. https://doi.org/10.1212/wnl.49.5.1247
Gomez CM, Thompson RM, Gammack JT, Perlman SL, Dobyns WB, Truwit CL, Zee DS, Clark HB, Anderson JH (1997) Spinocerebellar ataxia type 6: gaze-evoked and vertical nystagmus, Purkinje cell degeneration, and variable age of onset. Ann Neurol 42(6):933–950. https://doi.org/10.1002/ana.410420616
Jodice C, Mantuano E, Veneziano L, Trettel F, Sabbadini G, Calandriello L, Francia A, Spadaro M, Pierelli F, Salvi F, Ophoff RA, Frants RR, Frontali M (1997) Episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) due to CAG repeat expansion in the CACNA1A gene on chromosome 19p. Hum Mol Genet 6(11):1973–1978. https://doi.org/10.1093/hmg/6.11.1973
Casey HL, Gomez CM (1993) Spinocerebellar ataxia type 6. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews((R)). Seattle (WA),
Hashimoto T, Sasaki O, Yoshida K, Takei Y, Ikeda S (2003) Periodic alternating nystagmus and rebound nystagmus in spinocerebellar ataxia type 6. Mov Disord 18(10):1201–1204. https://doi.org/10.1002/mds.10511
Moscovich M, Okun MS, Favilla C, Figueroa KP, Pulst SM, Perlman S, Wilmot G, Gomez C, Schmahmann J, Paulson H, Shakkottai V, Ying S, Zesiewicz T, Kuo SH, Mazzoni P, Bushara K, Xia G, Ashizawa T, Subramony SH (2015) Clinical evaluation of eye movements in spinocerebellar ataxias: a prospective multicenter study. J Neuroophthalmol 35(1):16–21. https://doi.org/10.1097/WNO.0000000000000167
Yabe I, Sasaki H, Takeichi N, Takei A, Hamada T, Fukushima K, Tashiro K (2003) Positional vertigo and macroscopic downbeat positioning nystagmus in spinocerebellar ataxia type 6 (SCA6). J Neurol 250(4):440–443. https://doi.org/10.1007/s00415-003-1020-5
Filla A, Mariotti C, Caruso G, Coppola G, Cocozza S, Castaldo I, Calabrese O, Salvatore E, De Michele G, Riggio MC, Pareyson D, Gellera C, Di Donato S (2000) Relative frequencies of CAG expansions in spinocerebellar ataxia and dentatorubropallidoluysian atrophy in 116 Italian families. Eur Neurol 44(1):31–36. https://doi.org/10.1159/000008189
Storey E, du Sart D, Shaw JH, Lorentzos P, Kelly L, McKinley Gardner RJ, Forrest SM, Biros I, Nicholson GA (2000) Frequency of spinocerebellar ataxia types 1, 2, 3, 6, and 7 in Australian patients with spinocerebellar ataxia. Am J Med Genet 95(4):351–7. https://doi.org/10.1002/1096-8628(20001211)95:4<351::aid-ajmg10>3.0.co;2-r
La Spada AR (1993) Spinocerebellar ataxia type 7. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews((R)). Seattle (WA),
Benton CS, de Silva R, Rutledge SL, Bohlega S, Ashizawa T, Zoghbi HY (1998) Molecular and clinical studies in SCA-7 define a broad clinical spectrum and the infantile phenotype. Neurology 51(4):1081–1086. https://doi.org/10.1212/wnl.51.4.1081
Giunti P, Stevanin G, Worth PF, David G, Brice A, Wood NW (1999) Molecular and clinical study of 18 families with ADCA type II: evidence for genetic heterogeneity and de novo mutation. Am J Hum Genet 64(6):1594–1603. https://doi.org/10.1086/302406
Hugosson T, Granse L, Ponjavic V, Andreasson S (2009) Macular dysfunction and morphology in spinocerebellar ataxia type 7 (SCA 7). Ophthalmic Genet 30(1):1–6. https://doi.org/10.1080/13816810802454081
To KW, Adamian M, Jakobiec FA, Berson EL (1993) Olivopontocerebellar atrophy with retinal degeneration. An electroretinographic and histopathologic investigation. Ophthalmology 100(1):15–23. https://doi.org/10.1016/s0161-6420(93)31702-1
Lebre AS, Brice A (2003) Spinocerebellar ataxia 7 (SCA7). Cytogenet Genome Res 100(1-4):154–163. https://doi.org/10.1159/000072850
Bang OY, Lee PH, Kim SY, Kim HJ, Huh K (2004) Pontine atrophy precedes cerebellar degeneration in spinocerebellar ataxia 7: MRI-based volumetric analysis. J Neurol Neurosurg Psychiatry 75(10):1452–1456. https://doi.org/10.1136/jnnp.2003.029819
Lee YC, Liu CS, Wu HM, Wang PS, Chang MH, Soong BW (2009) The ‘hot cross bun’ sign in the patients with spinocerebellar ataxia. Eur J Neurol 16(4):513–516. https://doi.org/10.1111/j.1468-1331.2008.02524.x
Ikeda Y, Dalton JC, Moseley ML, Gardner KL, Bird TD, Ashizawa T, Seltzer WK, Pandolfo M, Milunsky A, Potter NT, Shoji M, Vincent JB, Day JW, Ranum LP (2004) Spinocerebellar ataxia type 8: molecular genetic comparisons and haplotype analysis of 37 families with ataxia. Am J Hum Genet 75(1):3–16. https://doi.org/10.1086/422014
Juvonen V, Hietala M, Kairisto V, Savontaus ML (2005) The occurrence of dominant spinocerebellar ataxias among 251 Finnish ataxia patients and the role of predisposing large normal alleles in a genetically isolated population. Acta Neurol Scand 111(3):154–162. https://doi.org/10.1111/j.1600-0404.2005.00349.x
Ayhan F, Ikeda Y, Dalton JC, Day JW, Ranum LPW (1993) Spinocerebellar ataxia type 8. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews((R)). Seattle (WA),
Day JW, Schut LJ, Moseley ML, Durand AC, Ranum LP (2000) Spinocerebellar ataxia type 8: clinical features in a large family. Neurology 55(5):649–657. https://doi.org/10.1212/wnl.55.5.649
Maschke M, Oehlert G, Xie TD, Perlman S, Subramony SH, Kumar N, Ptacek LJ, Gomez CM (2005) Clinical feature profile of spinocerebellar ataxia type 1-8 predicts genetically defined subtypes. Mov Disord 20(11):1405–1412. https://doi.org/10.1002/mds.20533
Silveira I, Alonso I, Guimaraes L, Mendonca P, Santos C, Maciel P, Fidalgo De Matos JM, Costa M, Barbot C, Tuna A, Barros J, Jardim L, Coutinho P, Sequeiros J (2000) High germinal instability of the (CTG)n at the SCA8 locus of both expanded and normal alleles. Am J Hum Genet 66(3):830–840. https://doi.org/10.1086/302827
Lilja A, Hamalainen P, Kaitaranta E, Rinne R (2005) Cognitive impairment in spinocerebellar ataxia type 8. J Neurol Sci 237(1-2):31–38. https://doi.org/10.1016/j.jns.2005.05.008
Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10(14):1441–1448. https://doi.org/10.1093/hmg/10.14.1441
Shah AG, Friedman MJ, Huang S, Roberts M, Li XJ, Li S (2009) Transcriptional dysregulation of TrkA associates with neurodegeneration in spinocerebellar ataxia type 17. Hum Mol Genet 18(21):4141–4152. https://doi.org/10.1093/hmg/ddp363
Stevanin G, Fujigasaki H, Lebre AS, Camuzat A, Jeannequin C, Dode C, Takahashi J, San C, Bellance R, Brice A, Durr A (2003) Huntington’s disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain 126(Pt 7):1599–1603. https://doi.org/10.1093/brain/awg155
Schneider SA, Walker RH, Bhatia KP (2007) The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test. Nat Clin Pract Neurol 3(9):517–525. https://doi.org/10.1038/ncpneuro0606
Filla A, De Michele G, Cocozza S, Patrignani A, Volpe G, Castaldo I, Ruggiero G, Bonavita V, Masters C, Casari G, Bruni A (2002) Early onset autosomal dominant dementia with ataxia, extrapyramidal features, and epilepsy. Neurology 58(6):922–928. https://doi.org/10.1212/wnl.58.6.922
Grundmann K, Laubis-Herrmann U, Dressler D, Vollmer-Haase J, Bauer P, Stuhrmann M, Schulte T, Schols L, Topka H, Riess O (2004) Mutation at the SCA17 locus is not a common cause of primary dystonia. J Neurol 251(10):1232–1234. https://doi.org/10.1007/s00415-004-0520-2
Rolfs A, Koeppen AH, Bauer I, Bauer P, Buhlmann S, Topka H, Schols L, Riess O (2003) Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17). Ann Neurol 54(3):367–375. https://doi.org/10.1002/ana.10676
Loy CT, Sweeney MG, Davis MB, Wills AJ, Sawle GV, Lees AJ, Tabrizi SJ (2005) Spinocerebellar ataxia type 17: extension of phenotype with putaminal rim hyperintensity on magnetic resonance imaging. Mov Disord 20(11):1521–1523. https://doi.org/10.1002/mds.20529
Delatycki MB, Corben LA (2012) Clinical features of Friedreich ataxia. J Child Neurol 27(9):1133–1137. https://doi.org/10.1177/0883073812448230
Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, Cavalcanti F, Monros E, Rodius F, Duclos F, Monticelli A, Zara F, Canizares J, Koutnikova H, Bidichandani SI, Gellera C, Brice A, Trouillas P, De Michele G, Filla A, De Frutos R, Palau F, Patel PI, Di Donato S, Mandel JL, Cocozza S, Koenig M, Pandolfo M (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271(5254):1423–1427. https://doi.org/10.1126/science.271.5254.1423
Durr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M (1996) Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med 335(16):1169–1175. https://doi.org/10.1056/NEJM199610173351601
Tsou AY, Paulsen EK, Lagedrost SJ, Perlman SL, Mathews KD, Wilmot GR, Ravina B, Koeppen AH, Lynch DR (2011) Mortality in Friedreich ataxia. J Neurol Sci 307(1-2):46–49. https://doi.org/10.1016/j.jns.2011.05.023
Cook A, Giunti P (2017) Friedreich’s ataxia: clinical features, pathogenesis and management. Br Med Bull 124(1):19–30. https://doi.org/10.1093/bmb/ldx034
Parkinson MH, Boesch S, Nachbauer W, Mariotti C, Giunti P (2013) Clinical features of Friedreich’s ataxia: classical and atypical phenotypes. J Neurochem 126(Suppl 1):103–117. https://doi.org/10.1111/jnc.12317
Reetz K, Dogan I, Hohenfeld C, Didszun C, Giunti P, Mariotti C, Durr A, Boesch S, Klopstock T, Rodriguez de Rivera Garrido FJ, Schols L, Giordano I, Burk K, Pandolfo M, Schulz JB, Group ES (2018) Nonataxia symptoms in Friedreich ataxia: report from the Registry of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS). Neurology 91(10):e917–e930. https://doi.org/10.1212/WNL.0000000000006121
Mascalchi M (2013) The cerebellum looks normal in Friedreich ataxia. AJNR Am J Neuroradiol 34(2):E22. https://doi.org/10.3174/ajnr.A3480
Stefanescu MR, Dohnalek M, Maderwald S, Thurling M, Minnerop M, Beck A, Schlamann M, Diedrichsen J, Ladd ME, Timmann D (2015) Structural and functional MRI abnormalities of cerebellar cortex and nuclei in SCA3, SCA6 and Friedreich’s ataxia. Brain 138(Pt 5):1182–1197. https://doi.org/10.1093/brain/awv064
Pagani E, Ginestroni A, Della Nave R, Agosta F, Salvi F, De Michele G, Piacentini S, Filippi M, Mascalchi M (2010) Assessment of brain white matter fiber bundle atrophy in patients with Friedreich ataxia. Radiology 255(3):882–889. https://doi.org/10.1148/radiol.10091742
Koeppen AH, Kuntzsch EC, Bjork ST, Ramirez RL, Mazurkiewicz JE, Feustel PJ (2013) Friedreich ataxia: metal dysmetabolism in dorsal root ganglia. Acta Neuropathol Commun 1:26. https://doi.org/10.1186/2051-5960-1-26
Deistung A, Stefanescu MR, Ernst TM, Schlamann M, Ladd ME, Reichenbach JR, Timmann D (2016) Structural and functional magnetic resonance imaging of the cerebellum: considerations for assessing cerebellar ataxias. Cerebellum 15(1):21–25. https://doi.org/10.1007/s12311-015-0738-9
Lindig T, Bender B, Kumar VJ, Hauser TK, Grodd W, Brendel B, Just J, Synofzik M, Klose U, Scheffler K, Ernemann U, Schols L (2019) Pattern of cerebellar atrophy in Friedreich’s ataxia-using the SUIT template. Cerebellum 18(3):435–447. https://doi.org/10.1007/s12311-019-1008-z
Solbach K, Kraff O, Minnerop M, Beck A, Schols L, Gizewski ER, Ladd ME, Timmann D (2014) Cerebellar pathology in Friedreich’s ataxia: atrophied dentate nuclei with normal iron content. Neuroimage Clin 6:93–99. https://doi.org/10.1016/j.nicl.2014.08.018
Bouchard JP, Barbeau A, Bouchard R, Bouchard RW (1978) Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci 5(1):61–69
Dougherty SC, Harper A, Al Saif H, Vorona G, Haines SR (2018) A Chromosomal deletion and new frameshift mutation cause ARSACS in an African-American. Front Neurol 9:956. https://doi.org/10.3389/fneur.2018.00956
Okamoto K, Tokiguchi S, Furusawa T, Ishikawa K, Quardery AF, Shinbo S, Sasai K (2003) MR features of diseases involving bilateral middle cerebellar peduncles. AJNR Am J Neuroradiol 24(10):1946–1954
Richter AM, Ozgul RK, Poisson VC, Topaloglu H (2004) Private SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) families from Turkey. Neurogenetics 5(3):165–170. https://doi.org/10.1007/s10048-004-0179-y
Girard M, Lariviere R, Parfitt DA, Deane EC, Gaudet R, Nossova N, Blondeau F, Prenosil G, Vermeulen EG, Duchen MR, Richter A, Shoubridge EA, Gehring K, McKinney RA, Brais B, Chapple JP, McPherson PS (2012) Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc Natl Acad Sci U S A 109(5):1661–1666. https://doi.org/10.1073/pnas.1113166109
Parfitt DA, Michael GJ, Vermeulen EG, Prodromou NV, Webb TR, Gallo JM, Cheetham ME, Nicoll WS, Blatch GL, Chapple JP (2009) The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum Mol Genet 18(9):1556–1565. https://doi.org/10.1093/hmg/ddp067
Vermeer S, van de Warrenburg BP, Kamsteeg EJ, Brais B, Synofzik M (1993) Arsacs. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews((R)). Seattle (WA),
Vermeer S, Meijer RP, Pijl BJ, Timmermans J, Cruysberg JR, Bos MM, Schelhaas HJ, van de Warrenburg BP, Knoers NV, Scheffer H, Kremer B (2008) ARSACS in the Dutch population: a frequent cause of early-onset cerebellar ataxia. Neurogenetics 9(3):207–214. https://doi.org/10.1007/s10048-008-0131-7
Prodi E, Grisoli M, Panzeri M, Minati L, Fattori F, Erbetta A, Uziel G, D’Arrigo S, Tessa A, Ciano C, Santorelli FM, Savoiardo M, Mariotti C (2013) Supratentorial and pontine MRI abnormalities characterize recessive spastic ataxia of Charlevoix-Saguenay. A comprehensive study of an Italian series. Eur J Neurol 20(1):138–146. https://doi.org/10.1111/j.1468-1331.2012.03815.x
Synofzik M, Soehn AS, Gburek-Augustat J, Schicks J, Karle KN, Schule R, Haack TB, Schoning M, Biskup S, Rudnik-Schoneborn S, Senderek J, Hoffmann KT, MacLeod P, Schwarz J, Bender B, Kruger S, Kreuz F, Bauer P, Schols L (2013) Autosomal recessive spastic ataxia of Charlevoix Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis 8:41. https://doi.org/10.1186/1750-1172-8-41
Biswas A, Varman M, Yoganathan S, Subhash PK, Mani S (2018) Teaching NeuroImages: autosomal recessive spastic ataxia of Charlevoix-Saguenay: Typical MRI findings. Neurology 90(14):e1271–e1272. https://doi.org/10.1212/WNL.0000000000005252
Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonca P, Costa M, Barros J, Yanagisawa T, Watanabe M, Ikeda Y, Aoki M, Nagata T, Coutinho P, Sequeiros J, Koenig M (2001) The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet 29(2):189–193. https://doi.org/10.1038/ng1001-189
Le Ber I, Moreira MC, Rivaud-Pechoux S, Chamayou C, Ochsner F, Kuntzer T, Tardieu M, Said G, Habert MO, Demarquay G, Tannier C, Beis JM, Brice A, Koenig M, Durr A (2003) Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain 126(Pt 12):2761–2772. https://doi.org/10.1093/brain/awg283
Coutinho P, Barbot C (1993) Ataxia with oculomotor apraxia type 1. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews((R)). Seattle (WA),
Mariani LL, Rivaud-Pechoux S, Charles P, Ewenczyk C, Meneret A, Monga BB, Fleury MC, Hainque E, Maisonobe T, Degos B, Echaniz-Laguna A, Renaud M, Wirth T, Grabli D, Brice A, Vidailhet M, Stoppa-Lyonnet D, Dubois-d’Enghien C, Le Ber I, Koenig M, Roze E, Tranchant C, Durr A, Gaymard B, Anheim M (2017) Comparing ataxias with oculomotor apraxia: a multimodal study of AOA1, AOA2 and AT focusing on video-oculography and alpha-fetoprotein. Sci Rep 7(1):15284. https://doi.org/10.1038/s41598-017-15127-9
Ronsin S, Hannoun S, Thobois S, Petiot P, Vighetto A, Cotton F, Tilikete C (2019) A new MRI marker of ataxia with oculomotor apraxia. Eur J Radiol 110:187–192. https://doi.org/10.1016/j.ejrad.2018.11.035
Duquette A, Roddier K, McNabb-Baltar J, Gosselin I, St-Denis A, Dicaire MJ, Loisel L, Labuda D, Marchand L, Mathieu J, Bouchard JP, Brais B (2005) Mutations in senataxin responsible for Quebec cluster of ataxia with neuropathy. Ann Neurol 57(3):408–414. https://doi.org/10.1002/ana.20408
Nicolaou P, Georghiou A, Votsi C, Middleton LT, Zamba-Papanicolaou E, Christodoulou K (2008) A novel c.5308_5311delGAGA mutation in Senataxin in a Cypriot family with an autosomal recessive cerebellar ataxia. BMC Med Genet 9:28. https://doi.org/10.1186/1471-2350-9-28
Anheim M, Monga B, Fleury M, Charles P, Barbot C, Salih M, Delaunoy JP, Fritsch M, Arning L, Synofzik M, Schols L, Sequeiros J, Goizet C, Marelli C, Le Ber I, Koht J, Gazulla J, De Bleecker J, Mukhtar M, Drouot N, Ali-Pacha L, Benhassine T, Chbicheb M, M’Zahem A, Hamri A, Chabrol B, Pouget J, Murphy R, Watanabe M, Coutinho P, Tazir M, Durr A, Brice A, Tranchant C, Koenig M (2009) Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain 132(Pt 10):2688–2698. https://doi.org/10.1093/brain/awp211
Frismand S, Salem H, Panouilleres M, Pelisson D, Jacobs S, Vighetto A, Cotton F, Tilikete C (2013) MRI findings in AOA2: cerebellar atrophy and abnormal iron detection in dentate nucleus. Neuroimage Clin 2:542–548. https://doi.org/10.1016/j.nicl.2013.03.018
Pfeffer G, Pyle A, Griffin H, Miller J, Wilson V, Turnbull L, Fawcett K, Sims D, Eglon G, Hadjivassiliou M, Horvath R, Nemeth A, Chinnery PF (2015) SPG7 mutations are a common cause of undiagnosed ataxia. Neurology 84(11):1174–1176. https://doi.org/10.1212/WNL.0000000000001369
Hewamadduma CA, Hoggard N, O’Malley R, Robinson MK, Beauchamp NJ, Segamogaite R, Martindale J, Rodgers T, Rao G, Sarrigiannis P, Shanmugarajah P, Zis P, Sharrack B, McDermott CJ, Shaw PJ, Hadjivassiliou M (2018) Novel genotype-phenotype and MRI correlations in a large cohort of patients with SPG7 mutations. Neurol Genet 4(6):e279. https://doi.org/10.1212/NXG.0000000000000279
Casari G, Marconi R (1993) Spastic paraplegia 7. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews((R)). Seattle (WA),
Gatti RA, Berkel I, Boder E, Braedt G, Charmley P, Concannon P, Ersoy F, Foroud T, Jaspers NG, Lange K et al (1988) Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature 336(6199):577–580. https://doi.org/10.1038/336577a0
Modell V, Orange JS, Quinn J, Modell F (2018) Global report on primary immunodeficiencies: 2018 update from the Jeffrey Modell Centers Network on disease classification, regional trends, treatment modalities, and physician reported outcomes. Immunol Res 66(3):367–380. https://doi.org/10.1007/s12026-018-8996-5
Amirifar P, Ranjouri MR, Yazdani R, Abolhassani H, Aghamohammadi A (2019) Ataxia-telangiectasia: a review of clinical features and molecular pathology. Pediatr Allergy Immunol 30(3):277–288. https://doi.org/10.1111/pai.13020
Crawford TO, Skolasky RL, Fernandez R, Rosquist KJ, Lederman HM (2006) Survival probability in ataxia telangiectasia. Arch Dis Child 91(7):610–611. https://doi.org/10.1136/adc.2006.094268
Perucca G, Leboucq N, Roubertie A, Rivier F, Menjot N, Valentini C, Bonafe A (2016) Role of neuroimaging in the diagnosis of hereditary cerebellar ataxias in childhood. J Neuroradiol 43(3):176–185. https://doi.org/10.1016/j.neurad.2016.03.006
Lin DD, Barker PB, Lederman HM, Crawford TO (2014) Cerebral abnormalities in adults with ataxia-telangiectasia. AJNR Am J Neuroradiol 35(1):119–123. https://doi.org/10.3174/ajnr.A3646
Burck U, Goebel HH, Kuhlendahl HD, Meier C, Goebel KM (1981) Neuromyopathy and vitamin E deficiency in man. Neuropediatrics 12(3):267–278. https://doi.org/10.1055/s-2008-1059657
Meier R, Tomizaki T, Schulze-Briese C, Baumann U, Stocker A (2003) The molecular basis of vitamin E retention: structure of human alpha-tocopherol transfer protein. J Mol Biol 331(3):725–734. https://doi.org/10.1016/s0022-2836(03)00724-1
Hentati F, El-euch G, Bouhlal Y, Amouri R (2012) Ataxia with vitamin E deficiency and abetalipoproteinemia. Handbook of Clinical Neurology, vol 103. Elsevier. https://doi.org/10.1016/B978-0-444-51892-7.00018-8
Gabsi S, Gouider-Khouja N, Belal S, Fki M, Kefi M, Turki I, Ben Hamida M, Kayden H, Mebazaa R, Hentati F (2001) Effect of vitamin E supplementation in patients with ataxia with vitamin E deficiency. Eur J Neurol 8(5):477–481. https://doi.org/10.1046/j.1468-1331.2001.00273.x
Jayadev S, Bird TD (2013) Hereditary ataxias: overview. Genet Med 15(9):673–683. https://doi.org/10.1038/gim.2013.28
Schuelke M (1993) Ataxia with vitamin E deficiency. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews((R)). Seattle (WA),
Benomar A, Yahyaoui M, Meggouh F, Bouhouche A, Boutchich M, Bouslam N, Zaim A, Schmitt M, Belaidi H, Ouazzani R, Chkili T, Koenig M (2002) Clinical comparison between AVED patients with 744 del A mutation and Friedreich ataxia with GAA expansion in 15 Moroccan families. J Neurol Sci 198(1-2):25–29. https://doi.org/10.1016/s0022-510x(02)00057-6
Mariotti C, Gellera C, Rimoldi M, Mineri R, Uziel G, Zorzi G, Pareyson D, Piccolo G, Gambi D, Piacentini S, Squitieri F, Capra R, Castellotti B, Di Donato S (2004) Ataxia with isolated vitamin E deficiency: neurological phenotype, clinical follow-up and novel mutations in TTPA gene in Italian families. Neurol Sci 25(3):130–137. https://doi.org/10.1007/s10072-004-0246-z
Clayton P (2016) Disorders of bile acid synthesis. Inborn metabolic diseases - diagnosis and treatment, 6th edn. Springer Berlin Heidelberg. https://doi.org/10.1007/978-3-662-49771-5_33
Mignarri A, Magni A, Del Puppo M, Gallus GN, Bjorkhem I, Federico A, Dotti MT (2016) Evaluation of cholesterol metabolism in cerebrotendinous xanthomatosis. J Inherit Metab Dis 39(1):75–83. https://doi.org/10.1007/s10545-015-9873-1
Federico A, Dotti MT, Gallus GN (1993) Cerebrotendinous xanthomatosis. In: Adam MP, Ardinger HH, Pagon RA et al. (eds) GeneReviews((R)). Seattle (WA),
Salen G, Steiner RD (2017) Epidemiology, diagnosis, and treatment of cerebrotendinous xanthomatosis (CTX). J Inherit Metab Dis 40(6):771–781. https://doi.org/10.1007/s10545-017-0093-8
Mignarri A, Dotti MT, Federico A, De Stefano N, Battaglini M, Grazzini I, Galluzzi P, Monti L (2017) The spectrum of magnetic resonance findings in cerebrotendinous xanthomatosis: redefinition and evidence of new markers of disease progression. J Neurol 264(5):862–874. https://doi.org/10.1007/s00415-017-8440-0
Beaudin M, Klein CJ, Rouleau GA, Dupre N (2017) Systematic review of autosomal recessive ataxias and proposal for a classification. Cerebellum Ataxias 4:3. https://doi.org/10.1186/s40673-017-0061-y
Vermeer S, Hoischen A, Meijer RP, Gilissen C, Neveling K, Wieskamp N, de Brouwer A, Koenig M, Anheim M, Assoum M, Drouot N, Todorovic S, Milic-Rasic V, Lochmuller H, Stevanin G, Goizet C, David A, Durr A, Brice A, Kremer B, van de Warrenburg BP, Schijvenaars MM, Heister A, Kwint M, Arts P, van der Wijst J, Veltman J, Kamsteeg EJ, Scheffer H, Knoers N (2010) Targeted next-generation sequencing of a 12.5 Mb homozygous region reveals ANO10 mutations in patients with autosomal-recessive cerebellar ataxia. Am J Hum Genet 87(6):813–819. https://doi.org/10.1016/j.ajhg.2010.10.015
Miskovic ND, Domingo A, Dobricic V, Max C, Braenne I, Petrovic I, Grutz K, Pawlack H, Tournev I, Kalaydjieva L, Svetel M, Lohmann K, Kostic VS, Westenberger A (2016) Seemingly dominant inheritance of a recessive ANO10 mutation in romani families with cerebellar ataxia. Mov Disord 31(12):1929–1931. https://doi.org/10.1002/mds.26816
Balreira A, Boczonadi V, Barca E, Pyle A, Bansagi B, Appleton M, Graham C, Hargreaves IP, Rasic VM, Lochmuller H, Griffin H, Taylor RW, Naini A, Chinnery PF, Hirano M, Quinzii CM, Horvath R (2014) ANO10 mutations cause ataxia and coenzyme Q(1)(0) deficiency. J Neurol 261(11):2192–2198. https://doi.org/10.1007/s00415-014-7476-7
Chamova T, Florez L, Guergueltcheva V, Raycheva M, Kaneva R, Lochmuller H, Kalaydjieva L, Tournev I (2012) ANO10 c.1150_1151del is a founder mutation causing autosomal recessive cerebellar ataxia in Roma/Gypsies. J Neurol 259(5):906–911. https://doi.org/10.1007/s00415-011-6276-6
Nanetti L, Sarto E, Castaldo A, Magri S, Mongelli A, Rossi Sebastiano D, Canafoglia L, Grisoli M, Malaguti C, Rivieri F, D’Amico MC, Di Bella D, Franceschetti S, Mariotti C, Taroni F (2019) ANO10 mutational screening in recessive ataxia: genetic findings and refinement of the clinical phenotype. J Neurol 266(2):378–385. https://doi.org/10.1007/s00415-018-9141-z
Hall DA, O’Keefe JA (2012) Fragile x-associated tremor ataxia syndrome: the expanding clinical picture, pathophysiology, epidemiology, and update on treatment. Tremor Other Hyperkinet Mov (N Y) 2. https://doi.org/10.7916/D8HD7TDS
Kalus S, King J, Lui E, Gaillard F (2016) Fragile X-associated tremor/ataxia syndrome: an under-recognised cause of tremor and ataxia. J Clin Neurosci 23:162–164. https://doi.org/10.1016/j.jocn.2015.08.010
Muzar Z, Lozano R (2014) Current research, diagnosis, and treatment of fragile X-associated tremor/ataxia syndrome. Intractable Rare Dis Res 3(4):101–109. https://doi.org/10.5582/irdr.2014.01029
Bacalman S, Farzin F, Bourgeois JA, Cogswell J, Goodlin-Jones BL, Gane LW, Grigsby J, Leehey MA, Tassone F, Hagerman RJ (2006) Psychiatric phenotype of the fragile X-associated tremor/ataxia syndrome (FXTAS) in males: newly described fronto-subcortical dementia. J Clin Psychiatry 67(1):87–94. https://doi.org/10.4088/jcp.v67n0112
Hagerman PJ, Hagerman RJ (2004) The fragile-X premutation: a maturing perspective. Am J Hum Genet 74(5):805–816. https://doi.org/10.1086/386296
Hall DA, Birch RC, Anheim M, Jonch AE, Pintado E, O’Keefe J, Trollor JN, Stebbins GT, Hagerman RJ, Fahn S, Berry-Kravis E, Leehey MA (2014) Emerging topics in FXTAS. J Neurodev Disord 6(1):31. https://doi.org/10.1186/1866-1955-6-31
Hall DA, Robertson E, Shelton AL, Losh MC, Mila M, Moreno EG, Gomez-Anson B, Martinez-Cerdeno V, Grigsby J, Lozano R, Hagerman R, Maria LS, Berry-Kravis E, O’Keefe JA (2016) Update on the clinical, radiographic, and neurobehavioral manifestations in FXTAS and FMR1 premutation carriers. Cerebellum 15(5):578–586. https://doi.org/10.1007/s12311-016-0799-4
Acknowledgements
Open access funding provided by Università degli Studi di Napoli Federico II within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
S.C. received fees for speaking from Genzyme and Shire and fess for adv.board from Amicus and Takeda. All other authors declare that they have no conflict of interest.
Ethical approval
This report is a literature review and does not require approval by an ethical committee.
Informed consent
All patients gave written informed consent to use their brain MRI scans for scientific purposes.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key points
- Brain MRI exam plays a central role in the diagnostic assessment of patients with cerebellar ataxia, being usually characterized by the presence of cerebellar atrophy. Nonetheless, a proper assessment of imaging and clinical data is crucial to identify some conditions and exclude others, addressing the neurologist to a more appropriate genetic testing
- We propose a review of the main clinical and conventional imaging findings of the most common hereditary degenerative ataxias, to highlight the main features of these conditions
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cocozza, S., Pontillo, G., De Michele, G. et al. Conventional MRI findings in hereditary degenerative ataxias: a pictorial review. Neuroradiology 63, 983–999 (2021). https://doi.org/10.1007/s00234-021-02682-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00234-021-02682-2