
Abstract.
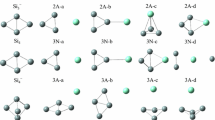
The possible geometrical structures and relative stability of (SiS2) n (n=1–6) silicon–sulfur clusters are explored by means of density functional theory quantum chemical calculations. The effects of polarization functions and electron correlation are included in these calculations. The electronic structures and vibrational spectra of the most stable geometrical structures of (SiS2) n are analyzed by the same method. As a result, the regularity of the (SiS2) n cluster growth is obtained, and the calculation may used for predicting the formation mechanism of the (SiS2) n cluster.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Received: 17 November 1999 / Accepted: 3 November 2000 / Published online: 3 May 2001
Rights and permissions
About this article
Cite this article
Wang, SF., Feng, JK., Sun, CC. et al. Theoretical study of silicon–sulfur clusters (SiS2)n (n = 1–6). Theor Chem Acc 106, 163–170 (2001). https://doi.org/10.1007/s002140100249
Issue Date:
DOI: https://doi.org/10.1007/s002140100249