
Abstract.
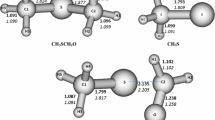
Selective bond dissociation energies for CH3SH and CH3CH2SH radical cations were evaluated with G1, G2, G2MP2, B3LYP, BLYP, and SVWN computational methods. It was determined that both G2 and CBSQ evaluate very accurate bond dissociation energies for thiol radical cations, while gradient-corrected BLYP computes the best energies of three employed DFT methods. For the CH3CH2SH radical cation, new, higher than previously estimated selective bond dissociation energies were suggested.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Received: 10 September 1997 / Accepted: 9 September 1998 / Published online: 11 November 1998
Rights and permissions
About this article
Cite this article
Jursic, B. High level ab initio and density functional theory study of bond selective dissociation of CH3SH and CH3CH2SH radical cations. Theor Chem Acc 100, 329–332 (1998). https://doi.org/10.1007/s002140050394
Issue Date:
DOI: https://doi.org/10.1007/s002140050394