
Abstract
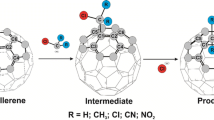
The potential energy surfaces for the addition reactions of N-heterocyclic carbene with C60 and C70 are characterized in detail, using density functional theory (M06-2X). These theoretical investigations strongly suggest that the one-point attack in formation of the single-bonded adduct is the most favorable reaction pathway at room temperature, rather than the production of the cycloadduct with a three-membered ring, from both kinetic and thermodynamic viewpoints. Also, this theoretical work indicates that the dispersion interactions cannot make the contributions to determine the regioselective formations for these NHC–fullerene Lewis acid–base adducts. These theoretical conclusions are consistent with the available experimental observations.





Similar content being viewed by others


Notes
The LANL2DZdp basis sets for C, N, and H were obtained from the Extensible Computational Chemistry Environment Basis Set Database (http://www.emsl.pnl.gov/forms/basisform.html), as and distributed by the Molecular Science Computing Facility, Environmental and Molecular Sciences Laboratory.
Vibrational frequency calculations at the RHF/LANL2DZ level were used to characterize all the stationary points as either minima (no imaginary frequencies) or transition states (one imaginary frequency). Then these stationary points were further calculated at the M06-2X/LANL2DZdp level using the opt=readfc keyword. Due to the limitation of both available CPU time and memory size, the M06-2X zero-point energy could not be applied for all of the IDipp–C60 and IDipp–C70 systems in the present work. That is, because frequencies were not calculated for all species at the M06-2X/LANL2DZdp level of theory, zero-point energy corrections were not performed. Nevertheless, the addition of these corrections would not change our conclusions.
The four types of geometrical results are found as follows: (i) the 6–6-bridged mono-adduct of C60 with a closed trans-annular bond, (ii) the 5–6-bridged molecule with an open trans-annular structure, (iii) the 6–6-bridged compound with an open trans-annular structure and (iv) the 5–6-bridged compound with a closed trans-annular bond. See reference [3].
The pyramidalization angles are measured to be 11.9°, 11.9°, 11.4°, 10.2° and 8.7° for C(a), C(b), C(c), C(d), and C(e), respectively [7].
References
Prato M, Wudl F (1995) In: Taylor R (ed) The chemistry of fullerenes, vol 4. Word Scientific, New York, pp 151–173
Osterodt J, Vögtle F (1996) J Chem Soc Chem Commun 547
Lan C-Y, Su M-D (2007) J Phys Chem A 111:6232
Yamada M, Akasaka T, Nagase S (2013) Chem Rev 113:7209
Li H, Risko C, Seo JH, Campbell C, Wu G, Brédas JL, Bazan GC (2011) J Am Chem Soc 133:12410
Iglesias-Sigüenza J, Alcarazo M (2012) Angew Chem Int Ed 51:1523
Jin P, Nagase S (2013) RSC Adv 3:10177
Zhao Y, Truhlar DG (2008) Acc Chem Res 41:157
Su M-D (2014) Inorg Chem 53:5080
Catherine EC, Timothy OF, John MB, Brian JW, Thomas MG, Lee SS (2001) J Phys Chem A 105:8111
Wiberg KB (1989) Chem Rev 89:975
Frisch MJ et al (2013) Gaussian 09, revision D.01, Gaussian, Inc., Wallingford CT
Hawkins JM, Meyer A, Lewis TA, Loren S, Hollander FJ (1991) Science 252:312
Hawkins JM, Loren S, Meyer A, Nunlist R (1991) J Am Chem Soc 113:7770
Kiley AF, Haddon RC, Meier MS, Selegue JP, Brock CP, Patrick BO, Wang G-W, Chen Y (1999) J Am Chem Soc 121:7921
Li B, Shu C, Lu X, Dunsch L, Chen Z, Dennis TJ, Shi Z, Jiang L, Wang T, Xu W, Wang C (2010) Angew Chem Int Ed 49:962
Haddon RC (1988) Acc Chem Res 21:243
Acknowledgments
The authors are grateful to the National Center for High-Performance Computing of Taiwan for generous amounts of computing time. They also thank the Ministry of Science and Technology of Taiwan for the financial support. Special thanks are also due to Reviewers 1 and 2 for very helpful suggestions and comments.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, MC., Su, MD. The addition reactions between N-heterocyclic carbenes and fullerenes (C60 and C70): a density functional study. Theor Chem Acc 134, 38 (2015). https://doi.org/10.1007/s00214-015-1629-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-015-1629-3