
Abstract.
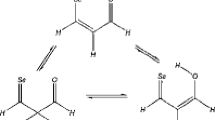
The structural and energetic changes associated with C–N bond rotation in a squaric acid derivative as well as in formamide, 3-aminoacrolein and vinylamine have been studied theoretically using ab initio molecular orbital methods. Geometry optimizations at the MP2(full)/6-31+G* level confirmed an increase in the C–N bond length and a smaller decrease in the C=O length on going from the equilibrium geometry to the twisted transition state. Other geometrical changes are also discussed. Energies calculated at the QCISD(T)/6-311+G** level, including zero-point-energy correction, show barrier heights decreasing in the order formamide, squaric acid derivative, 3-aminoacrolein and vinylamine. The origin of the barriers were examined using the atoms-in-molecules approach of Bader and the natural bond orbital population analysis. The calculations agree with Pauling's resonance model, and the main contributing factor of the barrier is assigned to the loss of conjugation on rotating the C–N bond. Finally, molecular interaction potential calculations were used to study the changes in the nucleophilicity of N and O (carbonyl) atoms upon C–N rotation, and to obtain a picture of the abilities of the molecules to act in nonbonded interactions, in particular hydrogen bonds. The molecular interaction potential results confirm the suitability of squaramide units for acting as binding units in host–guest chemistry.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Received: 13 March 2002 / Accepted: 23 June 2002 / Published online: 21 August 2002
Rights and permissions
About this article
Cite this article
Deyà, P., Frontera, A., Suñer, G. et al. Internal rotation in squaramide and related compounds. A theoretical ab initio study. Theor Chem Acc 108, 157–167 (2002). https://doi.org/10.1007/s00214-002-0373-7
Issue Date:
DOI: https://doi.org/10.1007/s00214-002-0373-7