
Abstract.
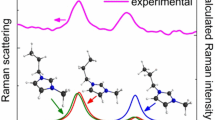
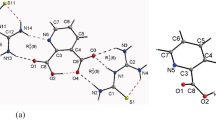
The structure and vibrational frequencies of an aromatic lithium sulfonyl imide, i.e., lithium bis(4-nitrophenylsulfonyl)imide (LiNPSI) has been studied using self-consistent ab initio Hartree–Fock and hybrid density functional methods. These calculations engender two linkage isomers, which correspond to the local minima on the potential-energy surface. In the lowest-energy isomer, the ligand binds to the metal ion through two oxygens, one from each of the different SO2 groups on the central nitrogen and forms a six-membered ring. Another LiNPSI isomer, wherein the anion coordinates through oxygen and nitrogen atoms and which is 55.9 kJmol−1 higher in energy, has also been obtained. The S–N–S bond angle in the free anion as well as in the LiNPSI complex turns out to be nearly 121°. A comparison of the vibrational spectra of the free NPSI anion and that of the LiNPSI complex reveals that the SO2 stretching vibrations at 1,239 and 1,205 cm−1 can be used to differentiate between the two linkage isomers of the complex. The stronger complexation ability of the NPSI anion, compared to that for (CF3SO2)2N− has been explained in terms of the charge density within the molecular electrostatic potential isosurface encompassing both SO2 groups of the anion.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Received: 20 February 2002 / Accepted: 25 March 2002 / Published online: 3 June 2002
Rights and permissions
About this article
Cite this article
Gejji, S., Agrawal, P. & Dhumal, N. Ab initio structure and vibrational frequencies of lithium aromatic sulfonyl imide salts. Theor Chem Acc 107, 351–356 (2002). https://doi.org/10.1007/s00214-002-0339-9
Issue Date:
DOI: https://doi.org/10.1007/s00214-002-0339-9