
Abstract
Background
Dopamine D3 receptors regulate sensorimotor gating in rats, as evidenced by changes in prepulse inhibition (PPI) of startle after acute administration of D3 agonists and antagonists. In this study, we tested the effects of the D3-preferential agonist, pramipexole, on PPI in normal men and Sprague–Dawley rats.
Materials and Methods
Acoustic startle and PPI were tested in clinically normal men, comparing the effects of placebo vs. 0.125 mg (n = 20) or placebo vs. 0.1875 mg (n = 20) pramipexole, in double blind, crossover designs. These measures were also tested in male Sprague–Dawley rats using a parallel design [vehicle vs. 0.1 mg/kg (n = 8), vehicle vs. 0.3 mg/kg (n = 8) or vehicle vs. 1.0 mg/kg pramipexole (n = 8)]. Autonomic and subjective measures of pramipexole effects and several personality instruments were also measured in humans.
Results
Pramipexole increased drowsiness and significantly increased PPI at 120-ms intervals in humans; the latter effect was not moderated by baseline PPI or personality scale scores. In rats, pramipexole causes a dose-dependent reduction in long-interval (120 ms) PPI, while low doses actually increased short-interval (10–20 ms) PPI. Effects of pramipexole on PPI in rats were independent of baseline PPI and changes in startle magnitude.
Conclusion
The preferential D3 agonist pramipexole modifies PPI in humans and rats. Unlike indirect DA agonists and mixed D2/D3 agonists, pramipexole increases long-interval PPI in humans, in a manner that is independent of baseline PPI and personality measures. These findings are consistent with preclinical evidence for differences in the D2- and D3-mediated regulation of sensorimotor gating.
Similar content being viewed by others


Introduction
Prepulse inhibition (PPI) is an operational measure of sensorimotor gating, in which a startle response to an intense stimulus is automatically suppressed by a weak lead stimulus (Graham 1975). In humans, PPI is typically assessed using electromyographic measures of the blink response, while in rodents, PPI is typically assessed by measuring whole-body startle. PPI deficits are found in several neuropsychiatric disorders (Braff et al. 1978; cf. Braff et al. 2001; cf. Swerdlow et al. 2008), and the biology of these deficits has been the focus of intense study in humans and animal models.
In rats, dopamine (DA) agonists generally reduce PPI, although these effects are highly strain-sensitive (Swerdlow et al. 2004b). For example, the mixed D1/D2 agonist apomorphine (APO) disrupts PPI in albino Sprague–Dawley rats but not in pigmented ACI rats, while in hooded Long Evans rats, APO increases PPI at short prepulse intervals and reduces it at long prepulse intervals (Swerdlow et al. 2004a, b). In humans, the effects of DA agonists on PPI differ across groups distinguished by baseline PPI levels and/or personality dimensions. For example, Bitsios et al. (2005) reported PPI-disruptive effects of the DA agonists pergolide and amantadine only among men characterized by high baseline PPI levels, Giakoumaki et al. (2007) reported a reduction in PPI after administration of the mixed D2/D3 agonist ropinirole to healthy men whose baseline PPI was >30%, and we reported similar baseline-dependent effects of amphetamine on PPI in healthy men (Swerdlow et al. 2003b) and women (Talledo et al. 2009). Furthermore, amphetamine has been reported to disrupt PPI only among normal subjects with high scores on measures of novelty- or sensation-seeking (Hutchison et al. 1999; Talledo et al. 2009). In some subgroups of normal humans (e.g., those characterized by low baseline PPI or novelty seeking), DA agonists can actually increase PPI (Talledo et al. 2009). Some evidence suggests that differences in PPI sensitivity to DA agonists in rats and humans may be mediated in part by differences in brain regional activity of catechol-O-methyl transferase (Shilling et al. 2008; Roussos et al. 2008b; Talledo et al. 2009).
The present study tested the effects of pramipexole, a non-ergot preferential D3 agonist, on PPI. While the vast majority of studies of the DAergic regulation of PPI in rats and humans focus on the role of D1 and D2 receptors, more recent evidence suggests that PPI in both species may also be regulated by D3 receptors. For example, Roussos et al. (2008a) reported that PPI differed significantly across humans characterized by Gly/Gly vs. Ser/Ser variants of the D3 receptor Ser9Gly polymorphism. Pramipexole has preferential affinity for D3 vs. D2 receptors, with an in vitro D3/D2 preference of 7.8:1 (Piercey et al. 1996, Svensson et al. 1994; cf. Kvernmo et al. 2006) relative to the high affinity state of the D2 receptor. Millan et al. (2002) determined the D3/D2 preference of pramipexole to be 90:1 relative to the short isoform of the human receptor (D2S) and 160:1 relative to the long isoform (D2L). In comparison, ropinirole is relatively less preferential for D3 receptors [e.g., in vitro D3/D2 preference = 1.3:1 (cf. Kvernmo et al. 2006), with preference of 18:1 relative to D2S and 25:1 relative to D2L (Millan et al. 2002)].
Our group reported that pramipexole disrupts PPI in laboratory rats and that this effect is relatively insensitive to selective D2 blockade but is opposed by functional D3 antagonists (Weber et al. 2008, 2009). In addition, strain differences in the PPI-disruptive effects of indirect DA agonists (amphetamine, Swerdlow et al. 2003a), mixed D1/D2 agonists (APO, Swerdlow et al. 2001), and D2-preferential agonists (quinpirole, Swerdlow et al. 2001) are not detected with pramipexole (Weber et al. 2008), suggesting that its PPI-disruptive effects are mediated differently from those of non-D3 DA agonists. In this study, we examined the effects of pramipexole on PPI in normal men, also assessing the potential moderating impact of baseline PPI and personality scale scores.
Materials and methods
Human testing
The methods used in these studies were very similar to those used in studies described in recent reports (Swerdlow et al. 2003b), were approved by the UCSD Human Subjects Institutional Review Board, and were approved and supported by the National Institute of Mental Health. Forty right-handed men (Table 1) completed testing; the study involved a phone contact and three laboratory visits. Phone screening procedures were identical to those described in previous reports from our group (Swerdlow et al. 2003b).
After passing a telephone interview, subjects came to the laboratory for a screening examination, during which the senior investigator (NRS) informed subjects of the potential risks and benefits of the study. Subjects read and signed a consent form for study participation, underwent a physical examination and electrocardiogram to rule out exclusionary medical conditions, a modified structured clinical interview for DSM disorders (non-patient edition; SCID; First et al. 1997) to rule out Axis I diagnoses, and completed a urine toxicology test with exclusion for any illicit drug. Audiometry confirmed hearing threshold <40 dB(A) at 1,000 Hz. Subjects also completed a limited test of the acoustic startle reflex to screen for a minimum eyeblink startle magnitude of 50 units (1.22 µV/unit) using 118 dB(A), 40 ms noise pulses.
Subjects completed the following questionnaires: (1) the Tridimensional Personality Questionnaire (TPQ; Cloninger 1987) to assess the relationship between novelty-seeking scores (NS) and sensitivity to the effects of pramipexole on PPI, based on reports that high NS individuals are most sensitive to the PPI-disruptive effects of indirect DA agonists (Hutchison et al. 1999; Talledo et al. 2009) and the PPI-enhancing effects of antipsychotics (Swerdlow et al. 2006); (2) the Sensation Seeking Scale (SSS; Zuckerman et al. 1972), based on reported increased sensitivity to DA agonists in individuals scoring high on this measure (Hutchison et al. 1999); and (3) the Eysenck Personality Questionnaire (EPQ) (Eysenck and Eysenck 1994). Subjects who passed screening criteria were tested 6–8 days later and retested 6–8 days after their first experimental session.
The study had a two-test within-subject design for each dose of pramipexole. In other words, one group of subjects (n = 20) was tested with placebo and 0.125 mg pramipexole, and a second group of subjects (n = 20) was tested with placebo and 0.1875 mg pramipexole. On test days, subjects arrived at 0830 hours and ate a standardized breakfast, and pramipexole (0.125 or 0.1875 mg; Boehringer Ingelheim GmbH, Ingelheim, Germany) or placebo was administered at 0930 hours. Startle testing began 60 min after pill administration. Heart rate and blood pressure were determined (sitting position and brachial cuff), and subjects completed a symptom rating scale every 30–45 min; the first one occurred before pill ingestion. Symptom-rating visual analog scales (VAS) were designed to assess general somatic and psychological symptoms and level of consciousness (modified from Norris 1971; Bond and Lader 1974; Bunney et al. 1999). Subjects made a single, vertical mark representing their current state along a 100-mm line (0 mm represents “not true” and 100 mm represents “true”). Ratings assessed several states: “happy,” “queasy,” “dizzy,” “drowsy,” and perceptual sensitivity. Details of these rating scales are found in Swerdlow et al. (2002) and included prompts such as “Normal sounds seem unusually intense or loud.”
For startle testing, subjects sat upright and were directed to look straight ahead and to stay awake. Two miniature Ag/AgCl electrodes were positioned below and to the outer canthus of each eye over orbicularis oculi; ground electrode was positioned behind the left ear (R < 10 kΩ). Electromyographic activity was band-pass-filtered (1–1,000 Hz) and 60-Hz notch-filtered, and digitized, and 250 1-ms readings were recorded starting at startle stimulus onset. Acoustic startle stimuli were delivered by Telephonics (TDH-39-P, Maico) headphones. A background 70 dB(A) white noise was continuous throughout the session. Test sessions began with a 3-min acclimation period; during this period, the number of spontaneous eyeblinks were counted by a remote observer using a RadioShack security camera system (model 49-2513) (inter-observer R = 0.97). This was followed by 42 trials with six conditions repeated in pseudorandom order: a 118-dB(A), 40-ms noise burst alone (pulse alone) and the same 118-dB(A), 40-ms noise burst preceded 10, 20, 30, 60, or 120 ms by a prepulse (5-ms burst) 16 dB over background. A variable inter-trial interval averaged 20 s (15–25 s). The test session was structured identically to that described in our previous studies of DA agonist effects on PPI in humans (Swerdlow et al. 2003b; Talledo et al. 2009). On completion of this startle test, additional autonomic and subjective rating measurements were obtained, as were additional “pilot” psychophysiological measures, including a visual latent inhibition task. Data from these subsequent tests are not included in this analysis.
The primary reasons for disqualification were that subjects had low screening startle magnitude (n = 22), withdrew from testing prior to the second test day (n = 3), or had a SCID-based diagnosis (n = 14; First et al. 1997); others included positive urine toxicology for illicit drugs (n = 2).
PPI was defined as [100-(100 × magnitude on prepulse trial/magnitude on pulse alone trial)]. Screening PPI across all 40 subjects was normally distributed [mean (SD)% across all intervals (10–120 ms) = 9.33 (20.25); median = 7.68; skewness = 0.27; kurtosis = 0.17]. Startle magnitude, latency, and PPI were analyzed with mixed-design analyses of variance (ANOVAs), with trial type and pramipexole dose (placebo vs. active) as within-subject factors, and dose groups [i.e., subjects tested with placebo vs. 0.125 mg pramipexole (“0.125 mg group”) and subjects tested with placebo vs. 0.1875 mg pramipexole (“0.1875 mg group”)] as the between-subject factor. Separate analyses were then pursued on each dose group. No consistent drug interactions were noted with eye side (left vs. right), and thus, main effects of eye side and interactions are not reported. “Baseline PPI” was defined as the mean %PPI for 60- and 120-ms prepulse intervals on the screening day, as described previously (Swerdlow et al. 2009). Personality scales previously found to be related to drug effects on PPI were treated as continuous variables: the total novelty-seeking score (NS) from the TPQ, total and disinhibition subscale scores from the SSS, and extraversion subscale scores from the EPQ. VAS ratings and autonomic measures were treated as continuous variables and were analyzed with mixed-design ANOVAs, focusing on difference scores (change from pre-pill baseline to time of PPI testing). Specific post-hoc comparisons were made with one-factor ANOVAs or the Fisher’s protected least significant difference test. Alpha was 0.05. In most cases, post-hoc comparisons were limited to tests of specific a priori hypotheses (e.g., that DA agonist effects on PPI would be dependent on baseline PPI) or planned comparisons (e.g., relationship of PPI pramipexole sensitivity to personality measures).
Rodent testing
The methods used in this study were in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications no. 80-23) and approved by the UCSD Animal Subjects Committee (protocol S01221). Adult male Sprague–Dawley rats (225–250 g; Harlan Laboratories, San Diego, CA, USA) were housed in groups of two to three animals per cage and maintained on a reversed light/dark schedule with water and food available ad libitum. Rats were handled within 2 days of arrival. Testing occurred during the dark phase. Pramipexole (Toronto Research Chemicals, North York, Ontario, Canada) was dissolved in saline vehicle and administered subcutaneously in doses of 0 or 0.1 mg/kg (n = 8), 0 or 0.3 mg/kg (n = 8), or 0 or 1.0 mg/kg (n = 8), with the same parallel within-subject dose design used in humans (doses from Weber et al. 2008). Startle chambers (San Diego Instruments, San Diego, CA, USA) were housed in a sound-attenuated room and consisted of a Plexiglas cylinder 8.2 cm in diameter resting on a 12.5 × 25.5 cm Plexiglas frame within a ventilated enclosure. Noise bursts were presented via a speaker mounted 24 cm above the cylinder. A piezoelectric accelerometer mounted below the Plexiglas frame detected and transduced motion from within the cylinder. Stimulus delivery was controlled by the SR-LAB microcomputer and interface assembly, which also digitized (0–4095), rectified, and recorded stabilimeter readings. One hundred 1-ms readings were collected beginning at stimulus onset. Startle amplitude was defined as the average of the 100 readings.
Approximately 7 days after shipment arrival, rats were exposed to a short “matching” startle session. They were placed in the startle chambers for a 5-min acclimation period with a 70 dB(A) background noise and then exposed to a total of 17 P-alone trials (40 ms–120 dB(A) noise bursts) that were interspersed with three PP12dB + P-alone trials [P-alone preceded 100 ms (onset-to-onset) by a 20-ms noise burst of 12dB above background]. Rats were assigned to dose order groups based on average %PPI from the matching session to ensure similar baseline PPI levels between groups. Four days later, rats were injected with pramipexole (vehicle or active dose) and, 15 min later, placed in the startle chambers for a 5-min acclimation period with a 70-dB(A) background noise. They were then exposed to a series of trial types within a test session identical to that used in testing humans (see above, “Human testing”). Three days later, testing was repeated, with pramipexole dose reversed. Statistical analyses of startle magnitude and PPI were structured identically to those used in humans.
Results
Human testing
Demographic variables and personality scale scores for all subjects are seen in Table 1.
Effects of pramipexole on autonomic and self-rating measures are seen in Fig. 1 and Table 2. The only robust evidence of bioactivity of these doses of pramipexole came in significant increases in self-rated “drowsiness” (Fig. 1). ANOVA of the change in ratings from pre-pill baselines for all subjects revealed a significant effect of pramipexole (F = 8.16, df 1,38, p < 0.007), no significant effect of dose group (F < 1), and no significant interaction of pramipexole × dose group (F = 1.04). Analysis limited to 0.125 mg pramipexole revealed no significant effect of pramipexole on drowsiness (F = 2.94, df 1,19, ns), while analysis limited to 0.1875 mg pramipexole revealed a significant increase in drowsiness at the time of PPI testing [mean (SEM) increase in VAS after pramipexole = 23.6 (6.02) vs. placebo = 5.05 (5.50); F = 5.27, df 1,19, p < 0.035]. In contrast, no significant effects of pramipexole were detected for either dose in measures of heart rate, blood pressure, pupil diameter, oral temperature, or spontaneous blink rate or in self-rated measures of dizziness, queasiness, happiness, or sensory sensitivity (Table 2).

Self-rated drowsiness VAS scores after placebo (open circles), 0.125 mg or 0.1875 mg pramipexole (closed circles), compared to pre-pill baselines (mean ± SEM). Time of PPI testing is indicated by arrow. Number sign Significantly greater increase in drowsiness at time of PPI testing, pramipexole vs. placebo (p < 0.007) by ANOVA; post-hoc individual comparisons detected significant effects only for the 0.1875 mg dose (p < 0.035)
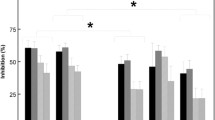
Pramipexole tended to diminish startle magnitude on pulse trials, but this effect did not reach statistical significance. Across all subjects, ANOVA of startle magnitude on all trial types revealed no significant main effect of pramipexole (F = 2.94, df 1,38, 0.10 > p > 0.05) or dose group (F < 1), and no pramipexole × dose group interaction (F = 2.12, df 1,38, ns). There was a significant effect of trial type (reflecting the startle-reducing effects of prepulses: F = 33.14, df 5,190, p < 0.0001), but no other significant two- or three-way interactions. Inspection of the data (Fig. 2) suggested no effect of 0.125 mg pramipexole on startle magnitude, and this was confirmed by ANOVA (placebo vs. 0.125 mg, F < 1; main effect of trial type, F = 15.42, df 5,95, p < 0.0001; no significant two-way interactions). However, 0.1875 mg pramipexole did appear to reduce startle magnitude, and this was also confirmed by ANOVA (placebo vs. 0.1875 mg, F = 5.40, df 1,19, p = 0.005; main effect of trial, F = 18.32, df 5,95, p < 0.0001). The interaction of dose × trial type was also significant (F = 2.43, df 5,95, p < 0.045), suggesting different effects of pramipexole on startle magnitude across the different trial types. Post hoc comparisons revealed that pramipexole did not significantly reduce startle magnitude on pulse-alone trials (F = 2.06, df 1,19, ns), but did significantly reduce startle magnitude on 120-ms prepulse trials (F = 11.97, df 1,19, p < 0.003; Fig. 2a). To examine the independence of pramipexole effects on pulse alone vs. 120-ms prepulse trials, a subgroup of 17 subjects was created by eliminating three subjects whose pulse-alone startle magnitude was most reduced by pramipexole. In this way, the remaining group (n = 17) exhibited absolutely no startle-reducing effects of pramipexole on pulse-alone trials [F<<1; mean (SEM) startle magnitude placebo vs. 0.1875 mg pramipexole = 117.20 (15.90) vs. 116.04 (17.75)] but nonetheless exhibited significant startle-reducing effects of pramipexole on 120-ms prepulse trials (F = 8.46, df 1,16, p = 0.01; Fig. 3a).

Effects of pramipexole on startle magnitude (mean ± SEM) (a) and %PPI (mean ± SEM) (b) across all trial conditions in groups treated with placebo vs. 0.125 mg pramipexole (n = 20) or placebo vs. 0.1875 mg pramipexole (n = 20). Open circle Placebo; closed circle active dose. Asterisk Startle-reducing effects of 0.1875 mg pramipexole on prepulse + pulse trials at 120-ms prepulse intervals (p < 0.003); number sign PPI-enhancing effects of pramipexole at 120-ms prepulse intervals (p < 0.05)

Effects of pramipexole on startle magnitude (mean ± SEM) (a) and %PPI (mean ± SEM) (b) across all trial conditions in a subgroup of subjects treated with placebo vs. 0.1875 mg pramipexole (n = 17) in whom pramipexole caused no change in pulse alone startle magnitude. Open circle Placebo; closed circle active dose. Asterisk Startle reducing effects of pramipexole on prepulse + pulse trials at 120 ms prepulse intervals (p = 0.01); number sign PPI-enhancing effects of pramipexole at 120-ms prepulse intervals (p = 0.005)
Consistent with the selective startle-reducing effects of pramipexole on 120-ms prepulse trials, pramipexole significantly increased PPI at 120-ms intervals (Fig. 2b). Across all subjects, ANOVA revealed no main effect of pramipexole (F < 1) or dose group (F < 1) and no pramipexole × dose group interaction (F < 1). There was a significant effect of trial type (F = 51.76, df 4,152, p < 0.0001) and a significant interaction of pramipexole × dose group × trial type (F = 2.53, df 4,152, p < 0.05). Post hoc comparisons revealed significant PPI-enhancing effects of pramipexole on 120-ms prepulse trials (main effect of pramipexole, F = 4.12, df 1,38, p < 0.05). This effect did not differ by dose group (F < 1) nor was there an interaction of pramipexole × dose group (F < 1), though the PPI-enhancing effects of 0.1875 mg appeared to be more robust than those of 0.125 mg (d = 0.48 vs. 0.20). We examined whether the PPI-enhancing effects of 0.1875 mg pramipexole were independent of the modest startle-reducing effects of this dose on pulse-alone trials, using the same subgroup of 17 subjects (described above) for whom this dose of pramipexole did not reduce pulse-alone startle magnitude. The PPI-enhancing effects of 0.1875 mg pramipexole were highly significant in this subgroup (F = 10.53, df 1,16, p = 0.005; d = 0.79; Fig. 3b). Thus, pramipexole increased PPI at 120-mg prepulse intervals, particularly at the 0.1875 mg dose, and this effect was independent of changes in pulse-alone startle magnitude.
Spearman rank correlations assessed relationships between the PPI-enhancing effects of 0.1875 mg pramipexole at 120-ms prepulse intervals [calculated as a difference score: PPI (pramipexole) minus PPI (placebo)] and scores for novelty-seeking (TPQ total NS scale), sensation-seeking (SSS total score and DIS subscale score), and extraversion (EPQ-E subscale score). No correlations approached statistical significance (−0.27 < R s < 0.05). Simple regression analyses revealed no correlation between these PPI-enhancing effects of pramipexole at 120-ms prepulse intervals and baseline startle magnitude (r = 0.02) or the impact of pramipexole on drowsiness (r = 0.03).
Pramipexole had no significant effects on startle latency, latency facilitation (reduction in latency caused by prepulses), or habituation (Table 3).
Rodent testing
The highest dose of pramipexole (1.0 mg/kg) reduced startle magnitude on pulse-alone trials (Fig. 4a). ANOVA of startle magnitude across all dose groups revealed no significant effect of group (F < 1), a significant effect of pramipexole dose (vehicle vs. active dose; F = 10.91, df 1,21, p < 0.004), and no interaction of group × dose (F = 1.45, df, 2,21, ns). Post-hoc comparison revealed significant startle-reducing effects limited to the 1.0 mg/kg group (p < 0.035).

Effects of pramipexole on startle magnitude (inset; mean ± SEM) and %PPI (mean ± SEM) across all trial conditions in three groups of rats administered placebo vs. 0.1 mg/kg pramipexole (n = 8), placebo vs. 0.3 mg/kg pramipexole (n = 8) or placebo vs. 1.0 mg/kg mg pramipexole (n = 8). Open circle Placebo; closed circle active dose. Insert, asterisk startle reducing effects of 1.0 mg/kg pramipexole (p < 0.035). Main figure, number sign PPI-enhancing effects of pramipexole at 10–20-ms prepulse intervals for 0.1 and 0.3 mg/kg doses (p = 0.02); asterisk PPI-reducing effects of 0.3 and 1.0 mg/kg pramipexole (p < −0.008–0.03)
Pramipexole had dose- and interval-dependent effects on PPI in rats (Fig. 4b). ANOVA across all groups revealed near significant effects of group (F = 3.32, df 2,21, p < 0.056) and dose (F = 3.28, df 1,21, p < 0.085), a near-significant interaction of group × dose (F = 3.24, df 2,21, p < 0.06), significant effects of prepulse interval (F = 25.70, df 4,84, p < 0.0001), and a significant interaction of dose × interval (F = 3.60, df 4,84, p < 0.01), but no significant three-way interaction. In three rats from the high-dose group, mean startle values on prepulse + pulse trials for some prepulse intervals greatly exceeded startle values on pulse alone trials, yielding large negative values for %PPI (i.e., prepulse potentiation) and thus accounting for the large error bars demonstrated in Fig. 4b; capping these values (e.g., at −100%) did not alter the statistical outcome. Inspection of the data revealed dose-dependent PPI-reducing effects of pramipexole at the 120-ms prepulse interval and PPI-increasing effects of lower doses of pramipexole at short (10–20 ms) prepulse intervals. Post-hoc comparisons for the 120-ms prepulse interval revealed significant effects of group (F = 4.15, df 2,21, p < 0.035) and dose (F = 15.04, df 1,21, p < 0.001) and a significant group × dose interaction (F = 3.45, df 2,21, p = 0.05). Pramipexole significantly reduced 120 ms PPI at the 0.3 and 1.0 mg/kg doses (p < 0.008 and 0.03, respectively) but not at the 0.1 mg/kg dose. Post-hoc comparisons for the 10–20-ms intervals revealed significant PPI-increasing effects of the 0.1 and 0.3 mg/kg doses (main effect of dose, F = 6.81, df 1,14, p = 0.02; no effect of group, F < 1; no dose × group interaction, F < 1).
While significant PPI-reducing and PPI-increasing effects of pramipexole were detected at doses that did not significantly reduce startle magnitude on pulse alone trials, we nonetheless examined whether drug effects on startle magnitude and PPI could be more completely dissociated. This was done in two ways. First, difference scores (vehicle minus active dose) were calculated for pramipexole effects on pulse-alone startle magnitude for each rat, and median splits were used to divide each dose group into rats with the least vs. most startle-reducing effects of pramipexole. Using this median split as a grouping factor, ANOVAs confirmed both the PPI-reducing effects of 0.3 and 1.0 mg/kg doses at 120-ms prepulse intervals and PPI-enhancing effects of 0.1 and 0.3 mg/kg doses at 10–20-ms intervals, with no significant interactions of dose × median splits (all F’s < 1.75, 1, 1, and 1, respectively). Next, simple regression analyses revealed no significant correlations between startle magnitude difference scores (i.e., the effect of pramipexole on startle magnitude) and PPI difference scores (i.e., the effect of pramipexole on PPI) at either the short (10–20 ms) or long (120 ms) prepulse intervals (r’s < 0.30, ns).
Lastly, we examined the potential relationship between baseline PPI (during the dose-matching session) and pramipexole effects on PPI. Baseline PPI did not correlate significantly with the effect of pramipexole on PPI at either the short (10–20 ms) or long (120 ms) prepulse intervals (r’s < 0.18, ns).
Discussion
In the present study, pramipexole, a D3-preferential agonist, increased long interval PPI in clinically normal men, and had biphasic dose effects on PPI in rats—increasing short interval PPI at low doses and decreasing long interval PPI at higher doses. Unlike the PPI-enhancing or reducing effects of the indirect DA agonist amphetamine (Hutchison et al. 1999; Talledo et al. 2009), the effects of pramipexole were not moderated by personality measures of novelty-seeking, sensation-seeking, disinhibition, or extraversion; unlike the PPI-modulatory effects of either amphetamine (Swerdlow et al. 2003b; Talledo et al. 2009), direct D2/D3 agonists (Bitsios et al. 2005), or the NMDA antagonist/DA releaser memantine (Swerdlow et al. 2009), these effects of pramipexole were independent of baseline PPI levels.
The fact that pramipexole effects on PPI differ from those of other DA agonists is consistent with preclinical evidence in rats and humans. In rats, PPI-reducing effects of pramipexole do not differ across strains that exhibit pronounced differences in sensitivity to the PPI-disruptive effects of amphetamine (Swerdlow et al. 2003b; Talledo et al. 2009) and mixed D2/D3 agonists (Swerdlow et al. 2001, 2004a, b, c; Weber et al. 2008). Furthernore, unlike amphetamine and mixed D2/D3 agonists, the PPI-disruptive effects of pramipexole in rats are relatively insensitive to selective D2 receptor blockade but are opposed by compounds with selective functional antagonism at D3 receptors (Weber et al. 2009). Evidence for differences in the PPI-modifying effects of pramipexole vs. other DA agonists in humans comes from findings that personality dimensions and/or baseline PPI moderate the PPI-disruptive effects of amphetamine (Hutchison et al. 1999; Talledo et al. 2009) and mixed D2/D3 agonists (Bitsios et al. 2005) but not those of pramipexole (present study); this may suggest that these moderating effects primarily reflect activity at D2 and not D3 DA receptors.
Pramipexole-induced PPI increases in humans were limited to the 120-ms prepulse intervals. Reflex inhibition at these longer intervals can be enhanced by attention directed at a continuous prepulse (Filion et al. 1993). While the present study utilized discrete (vs. continuous) prepulses and no attentional instructions were given, it is nonetheless possible that the mechanisms responsible for pramipexole’s enhancement of “attentionally sensitive” inhibition reflect its action on brain mechanisms that regulate attention. No significant correlations were detected between increases in self-rated drowsiness and PPI, but the present VAS measures may not be adequately sensitive to detect meaningful inter-individual differences in alertness and are not designed to assess attentional states. In a recent study with ten clinically normal adults, higher doses of pramipexole (0.25–0.50 mg) caused sedation and impaired cognitive performance in sedation-sensitive tasks (Hamidovic et al. 2008). However, because directed attention is associated with higher PPI levels in normal humans (Filion et al. 1993), it would be counter-intuitive to explain the observed PPI increases based on attention-impairing effects of pramipexole.
Confirmation that the present doses of pramipexole were bioactive came from increases in self-rated drowsiness, consistent with a number of reports with higher doses of pramipexole (Samuels et al. 2006a, b; 2007; Hamidovic et al. 2008). The lack of pramipexole effects on measures, such as “queasiness” or “happiness,” and on autonomic measures (e.g., pupil dilation), suggests that the doses in this study were physiologically low (Samuels et al. 2006a, b; 2007; Hamidovic et al. 2008) and raises the possibility that different effects on PPI might have been detected at higher doses. Such a prediction would be consistent with the present study in rodents, which demonstrated an inverted U dose function for pramipexole’s effects on PPI (i.e., it increased PPI at low doses and reduced PPI at high doses). Conceivably, low doses of pramipexole might preferentially activate presynaptic receptors or a combination of pre- and post-synaptic receptors, making it difficult to determine whether the observed increase in PPI reflected functional decreases or increases in forebrain D3 stimulation (Samuels et al. 2006b).
The nature of most studies using systemic drug administration is that the precise mechanisms of drug effects remain a matter of speculation. If we posit that the PPI-enhancing effects of pramipexole in humans reflect activation of post-synaptic forebrain D3 receptors, it would be difficult to account for PPI deficits observed in schizophrenia (Braff et al. 1978) or several other neuropsychiatric disorders (cf. Braff et al. 2001; Swerdlow et al. 2008) based solely on overactivity of D3 neurotransmission. On the other hand, if we posit that these effects of pramipexole reflect its action at presynaptic DA receptors that reduce forebrain DA “tone” (Samuels et al. 2006b), then the observed increases in PPI might be loosely consistent with PPI-enhancing effects of DA receptor antagonists in previous reports (Swerdlow et al. 2006; Vollenweider et al. 2006; Csomor et al. 2008). A strong caveat to such a connection—between PPI-enhancement by putative presynaptic effects of pramipexole and post-synaptic receptor-blocking effects of antipsychotics—is that these effects of antipsychotics are moderated by both personality dimensions (Swerdlow et al. 2006) and baseline PPI (Vollenweider et al. 2006; Csomor et al. 2008), neither of which moderate the present effects of pramipexole. Certainly, one might imagine that the PPI-enhancing effects of drugs acting pre- vs. post-synaptically could both reflect reduced DAergic “tone,” yet be moderated by different biological factors. It is also conceivable that different patterns of pramipexole effects might have emerged had the 40 subjects in this study been stratified based on different polymorphisms for the D3 receptor (Roussos et al. 2008a).
With the absence a clear understanding of whether the present drug effects reflect pre- vs. post-synaptic actions of pramipexole (or some combination thereof) and a lack of genetic information on D3 receptor polymorphisms among our subjects, we can still conclude from the present data that: (1) D3 receptors do appear to regulate PPI in clinically normal humans; (2) activation of these receptors by low doses of pramipexole enhances long-interval PPI; and (3) these effects appear to be independent of physiological variables that moderate other forms of DAergic regulation of PPI. It would be reasonable to consider whether such an effect could be used as a basis for predicting therapeutic sensitivity to D3 stimulation in clinical populations.
References
Bitsios P, Giakoumaki SG, Frangou S (2005) The effects of dopamine agonists on prepulse inhibition in healthy men depend on baseline PPI values. J Psychopharmacol 182:144–152
Bond AJ, Lader MH (1974) The use of analogue scales in rating subjective feelings. Br J Med Psychol 47:211–218
Braff DL, Geyer MA, Swerdlow NR (2001) Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology 156:234–258
Braff D, Stone C, Callaway E, Geyer M, Glock I, Bali L (1978) Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology 15:339–343
Bunney WE Jr, Hetrick WP, Bunney BG, Patterson JV, Jin Y, Potkin SG, Sandman CA (1999) Structured interview for assessing perceptual anomalies (SIAPA). Schizophr Bull 25:577–952
Cloninger CR (1987) A systematic method for clinical description and classification of personality variants—a proposal. Arch Gen Psychiatry 44:573–588
Csomor PA, Stadler RR, Feldon J, Yee BK, Geyer MA, Vollenweider FX (2008) Haloperidol differentially modulates prepulse inhibition and P50 suppression in healthy humans stratified for low and high gating levels. Neuropsychopharmacology 33:497–512
Eysenck HJ, Eysenck SBG (1994) Manual of the Eysenck personality questionnaire: comprising the EPQ-revised (EPQ-R) & EPQ-R short scale. EdITS, San Diego, CA
Filion DL, Dawson ME, Schell AM (1993) Modification of the acoustic startle-reflex eyeblink: a tool for investigating early and late attentional processes. Biol Psychol 35:185–200
First MB, Spitzer RL, Gibbon M, Williams JBW (1997) Structured clinical interview for DSM-IV—clinical version (SCID-CV) (user’s guide and interview). American Psychiatric Press, Washington, DC
Giakoumaki SG, Roussos P, Frangou S, Bitsios P (2007) Disruption of prepulse inhibition of the startle reflex by the preferential D(3) agonist ropinirole in healthy males. Psychopharmacology (Berl) 194:289–295
Graham F (1975) The more or less startling effects of weak prestimuli. Psychophysiology 12:238–248
Hamidovic A, Kang UJ, de Wit H (2008) Effects of low to moderate acute doses of pramipexole on impulsivity and cognition in healthy volunteers. J Clin Psychopharmacol 28:45–51
Hutchison KE, Wood MD, Swift R (1999) Personality factors moderate subjective and psychophysiological responses to d-amphetamine in humans. Exp Clin Psychopharmacol 7:493–501
Kvernmo T, Härtter S, Burger E (2006) A review of the receptor-binding and pharmacokinetic properties of dopamine agonists. Clin Ther 28:1065–1078
Millan MJ, Maiofiss L, Cussac D, Audinot V, Boutin JA, Newman-Tancredi A (2002) Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J Pharmacol Exp Ther 303:791–804
Norris H (1971) The action of sedation on brain-stem oculomotor systems in man. Neuropharmacology 10:181–191
Piercey MF, Hoffmann WE, Smith MW, Hyslop DK (1996) Inhibition of dopamine neuron firing by pramipexole, a dopamine D3 receptor-preferring agonist: comparison to other dopamine receptor agonists. Eur J Pharmacol 312:35–44
Roussos P, Giakoumaki SG, Bitsios P (2008a) The dopamine D(3) receptor Ser9Gly polymorphism modulates prepulse inhibition of the acoustic startle reflex. Biol Psychiatry 64:235–240
Roussos P, Giakoumaki SG, Rogdaki M, Pavlakis S, Frangou S, Bitsios P (2008b) Prepulse inhibition of the startle reflex depends on the catechol O-methyltransferase Val158Met gene polymorphism. Psychol Med 38:1651–1658
Samuels ER, Hou RH, Langley RW, Szabadi E, Bradshaw CM (2006a) Comparison of pramipexole and amisulpride on alertness, autonomic and endocrine functions in healthy volunteers. Psychopharmacology (Berl) 187:498–510
Samuels ER, Hou RH, Langley RW, Szabadi E, Bradshaw CM (2006b) Comparison of pramipexole and modafinil on arousal, autonomic, and endocrine functions in healthy volunteers. J Psychopharmacol 20:756–770
Samuels ER, Hou RH, Langley RW, Szabadi E, Bradshaw CM (2007) Comparison of pramipexole with and without domperidone co-administration on alertness, autonomic, and endocrine functions in healthy volunteers. Br J Clin Pharmacol 64:591–602
Shilling PD, Saint Marie RL, Shoemaker JM, Swerdlow NR (2008) Strain differences in the gating-disruptive effects of apomorphine: Relationship to gene expression in nucleus accumbens signaling pathways. Biol Psychiatry 63:748–758
Svensson K, Carlsson A, Huff RM, Kling-Petersen T, Waters N (1994) Behavioral and neurochemical data suggest functional differences between dopamine D2 and D3 receptors. Eur J Pharmacol 263:235–243
Swerdlow NR, Talledo J, Sutherland AN, Nagy D, Shoemaker JM (2006) Antipsychotic effects on prepulse inhibition in normal ‘low gating’ humans and rats. Neuropsychopharmacology 31:2011–2021
Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL (2008) Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology 199:331–388
Swerdlow NR, van Bergeijk DP, Bergsma F, Weber E, Talledo J (2009) The effects of memantine on prepulse inhibition. Neuropsychopharmacology 34:1854–1864
Swerdlow NR, Shoemaker JM, Platten A, Pitcher L, Goins J, Crain S (2003a) Heritable differences in the effects of amphetamine but not DOI on startle gating in albino and hooded outbred rat strains. Pharmacol Biochem Behav 75:191–197
Swerdlow NR, Stephany N, Wasserman LC, Talledo J, Shoemaker J, Auerbach PP (2003b) Amphetamine effects on prepulse inhibition across species: replication and parametric extension. Neuropsychopharmacology 28:640–650
Swerdlow NR, Shoemaker JM, Platten A, Pitcher L, Goins J, Auerbach PP (2004a) Heritable differences in the dopaminergic regulation of sensorimotor gating: I. Apomorphine effects on startle gating in albino and hooded outbred rat strains and their F1 and N2 progeny. Psychopharmacology 174:441–451
Swerdlow NR, Shoemaker JM, Auerbach PP, Pitcher L, Goins J, Platten A (2004b) Heritable differences in the dopaminergic regulation of sensorimotor gating: II. Temporal, pharmacologic and generational analyses of apomorphine effects on prepulse inhibition. Psychopharmacology 174:452–462
Swerdlow NR, Shoemaker JM, Crain S, Goins J, Onozuka K, Auerbach PP (2004c) Sensitivity to drug effects on prepulse inhibition in inbred and outbred rat strains. Pharmacol Biochem Behav 77:291–302
Swerdlow NR, Platten A, Kim YK, Gaudet I, Shoemaker J, Pitcher L, Auerbach P (2001) Sensitivity to the dopaminergic regulation of prepulse inhibition in rats: evidence for genetic, but not environmental determinants. Pharmacol Biochem Behav 70:219–226
Swerdlow NR, Stephany N, Shoemaker JM, Ross L, Wasserman LC, Talledo J, Auerbach PP (2002) Effects of amantadine and bromocriptine on startle and sensorimotor gating: parametric studies and cross-species comparisons. Psychopharmacology 164:82–92
Talledo JA, Sutherland Owens AN, Schortinghuis T, Swerdlow NR (2009) Amphetamine effects on startle gating in normal women and female rats. Psychopharmacology 204:165–175
Vollenweider FX, Barro M, Csomor PA, Feldon J (2006) Clozapine enhances prepulse inhibition in healthy humans with low but not with high prepulse inhibition levels. Biol Psychiatry 60:597–603
Weber M, Chang W, Breier M, Ko D, Swerdlow NR (2008) Heritable strain differences in sensitivity to the startle gating-disruptive effects of D2 but not D3 receptor stimulation. Behav Pharmacol 19:786–795
Weber M, Chang WL, Park PE, Durbin JP, Luedtke RL, Mach RH, Swerdlow NR (2009). Using prepulse inhibition to detect functional D3 receptor antagonism: effects of WC10 and WC44. Pharmacol Biochem Behav (in press)
Zuckerman M, Bone RN, Neary R, Mangelsdorff D, Brustman B (1972) What is the sensation seeker? personality trait and experience correlates of the sensation-seeking scales. J Consult Clin Psychol 39:308–321
Acknowledgements
This study was supported by MH58903. The authors are grateful to Maria Bongiovanni for manuscript preparation and Dr. Martin Weber for technical assistance.
Disclosures
In the past 3 years, NRS has had grant support from Pfizer Pharmaceuticals and Allergan, Inc. and was a paid consultant to Sanofi/Aventis.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Swerdlow, N.R., Lelham, S.A., Sutherland Owens, A.N. et al. Pramipexole effects on startle gating in rats and normal men. Psychopharmacology 205, 689–698 (2009). https://doi.org/10.1007/s00213-009-1577-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-009-1577-5