
Abstract
A series of sulfanilamide Schiff base derivatives (1 to 15) have been designed as potential antitubulin agents depending on the chemical structures of combretastatine A-4 and isoquinoline sulfamate (antimitotic agents under investigation). The designed compounds were synthesized by microwave chemical synthesis, their purity was confirmed by melting point and HPLC and chemical structures were determined by FT-IR, UV, and 1H and 13C-NMR spectroscopic techniques. The synthesized compounds have been docked in the colchicine binding site of β-tubulin using molecular modeling programs and the antitumor activities were screened on human breast and lung cancer cells by cell counting assay. Some tested compounds showed potent and selective activity against breast cancer (MCF-7) with IC50 range of 90 to 166 μM. With regarding broad-spectrum activity, compounds 4, 8, and 13 have shown potent antitumor activity against human breast and human lung cells with IC50 range of 96 to 140 μM. The obtained results suggest that the sulfanilamide Schiff base derivatives might potentially constitute an interesting novel class of anticancer agents, which deserve further studies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The term “cancer” comprises a large group of diseases that can affect almost any part of the body and is characterized by rapid creation of abnormal cells that grow beyond their usual boundaries (Amvrosieva et al. 2006; Al Rashood et al. 2006; Eckhardt 2002; Medina et al. 1998). Cancer is the leading cause of death worldwide, as it accounted for 7.6 million deaths in 2008 and is projected to continue rising with an estimated 11 million deaths in 2030 (Asai et al. 2012). Many cancer patients have received chemotherapeutic agents during the course of their disease, a treatment based on cell destruction by growth interruption. Antimitotic agents constitute an important class of the current anticancer chemotherapeutic agents that act by preventing polymerization or depolymerization of microtubules (motor of mitotic stage of cell cycle) by binding to its tubulin subunits (Desai and Mitchison 1997; Gadde and Heald 2004; Prota et al. 2013). The vital role of tubulin in the cell division cycle together with the fact that aberrant cell division is the feature of cancer has made tubulin and microtubules prime targets for cancer therapy. Tubulin contains three main sites for attacks, namely vinca alkaloids binding sites, taxane binding sites, and the colchicine binding site (Chang et al. 2004; Pfisterer et al. 2010). A number of compounds react in the same manner like colchicine as combretastatine A-4, indanocine, T138067, and isoquinoline sulfamate. Some of these agents are now under study in clinical trials (Dowlati et al. 2002; Cortes and Baselga 2007; Thomas 2009; Thomas and Abbruzzese 2005).
Owing to their high affinity to the colchicine binding site, the natural product, combretastatin A-4, is regarded as one of the most promising antineoplastic and cancer vascular-targeting agents which are under investigation. Combretastatin A-4 (Fig. 1), as potent inhibitors of tubulin assembly and due to its success in phase II clinical trials (Hsieh et al. 2005; Lu et al. 2012), has made it an attractive lead molecule for the development of anticancer agents, and promote synthesis of novel derivatives (Hori et al. 2010; Mitrus et al. 2009; Cushman et al. 1991).

Chemical structures of Combretastatine A-4 and Sulfanilamide-imines derivatives
In the present study, we designed 15 compounds with sulfanilamide-imine functionalities (Fig. 1) based on structural similarity with combretastatin A-4, and we hypothesized that sulfanilamide-imines series may fit and interact at the colchicine active site and may become a promising new tubulin-binding drug candidate for treating various malignancies.
Experimental protocols
Molecular modeling tools
Two different molecular modeling tools have been used to predict the interaction of the synthesized compounds at the colchicine active site, namely Discovery Studio Visualizer and AutoDock 4.2. The protein structure (tubulin) used in the current study was downloaded from the protein data bank (PDB) (Nogales et al. 1998) as bi units co-crystallized with combretastatine A-4. The molecular modeling study was performed in several steps using the above mentioned software; these include protein preparation, ligand preparation, and definition of active site, docking, and analysis of the docking results. PDBQT file format, which is an extended PDB format, is used in this part of our work.
Preparation of the protein
The protein structure was prepared using Discovery Studio Visualizer and AutoDock 4.2, where the water molecules removed from the surface of the protein and the non-polar hydrogen atoms were removed as well. The protein in AutoDock 4.2 is treated as a rigid molecule.
Preparation of the ligand
The ligand structure is initially drawn with ACD/ChemSketch chemical structure drawing software and then transferred to Discovery Studio Visualizer to obtain standard 3D structures. The ligand chemical structure optimization was performed using the AutoDock 4.2, Gasteiger charges optimization and determination of all rotatable bonds of the ligand was performed.
Definition of active site
The protein, using AutoGrid procedure of AutoDock 4.2, is fixed in a three-dimensional grid and a probe atom is located at each grid point. The active site determined with the grid box and it must be large enough to accommodate all the possible docked compounds. AutoGrid affinity grids are calculated for each type of atom in the ligand, typically carbon, oxygen, nitrogen, and hydrogen.
Docking
This part was performed using AutoDock 4.2, where all rotatable bonds of the ligand were randomized at the beginning of the docking and treated as completely flexible during the docking runs. Bond lengths, angles, and torsions associated with non-rotatable bonds in the ligand were held. For each independent Lamarckian genetic algorithm run, a maximum number of 100,000 operations were performed. The docking energy (Table 1) for the possible interaction between the targeted microtubules with the compounds under investigation was calculated and presented by AutoDock 4.2. Low value of the docking energy indicated the possible high drug/target interaction, while the high docking energy reflects the possible weak activity against the targeted microtubules. The Discovery Studio Visualizer is used for viewing and analyzing the possible type on interaction as it can easily detect the hydrogen bonds, lipophilic, and dipolar interactions.
Chemical synthesis of the Schiff’s bases
Materials and solvents
All chemical materials and solvents used in the chemical synthesis of Schiff bases were of highest purity and used without further purification, purchased from Sigma/Aldrich chemical company (UK) or Fluka analytical company, UK.
Instruments
Synthesis of compounds was performed in a microwave-closed system (milestone start E 2,450 MHz, Italy). Thin layer chromatographic (TLC) analysis was performed on pre-coated aluminum plates (silica gel 60778, Fluka analytical, UK). TLC spots were visualized with both short (254 nm) and long (366 nm) UV light. Melting points were measured in open capillary tubes on an Electrothermal SMP30 melting point apparatus (Stuart, UK). The IR spectra of samples were recorded in region 4,000–400 cm−1 by Varian FT-IR spectrophotometer 660 (Varian, Australia), UV-visible spectra were recorded by Cary 5000 UV\VIS\NIR spectrophotometer (Varian, Australia), using 1-cm quartz cuvettes. 1H-NMR spectra (400 MHz) recorded in dimethyl sulfoxide (DMSO) by employing tetramethylsilane as an internal standard on Bruker Avance 400 MHz NMR Spectrometer (Bruker, France).
General procedure for synthesis of Schiff bases (1a-15a) by microwave synthesis
Equimolar amounts (0.01 M) of sulfanilamide and substituted aldehydes were triturated to form uniform mixture using clean and dry Teflon vessels (Kundu et al. 2009; Venugopala and Jayashree 2008). In case of solid aldehydes, minimum amounts of ethanol (10–15 ml) were used to dissolve the reaction mixture. This mixture was subjected to microwave irradiation at 350–400 W power for about 0.5–1 min with maximum heating of 60 °C. The optimum reaction time was determined depending on the reaction competition using TLC and the appropriate solvent system. The reaction mixture was allowed to cool and the crude solid product was collected through vacuum filtration and washed several times with ethanol, and finally recrystallized from absolute ethanol. The achieved crystals were dried and their melting points were determined. The chemical purity where investigated by high performance liquid chromatography (HPLC) (Waters, UK) using chloroform/methanol (80:20) or acetonitrile/methanol (90:10) as mobile phases and Luna C18 (2) as a column (length 25 cm, inner diameter 4.6 mm, particle size 5 μm), while the chemical structure for all the synthesized compounds was elucidated using several spectroscopic techniques including IR, 1H NMR, 13C NMR, and UV spectra the following abbreviations for the NMR are used: singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m).
4-(Benzylidene-amino)-benzenesulfonamide (1)
FT-IR (cm−1): 3,292 (N–H str), 1,619 (HC=N), 3,002 (C–H aromatic), 1,574 (C=C aromatic), 1,150 (S=O asym), 1,331 (S=O sym). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 8.65 (s, 1H, HC=N), 8–7.40 (m, 9H, Ar–H). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 162.77 (1C, *C=N), 154.35 (1C, C=*C–N), 153.56 (1C, C=*C–SO2NH2), 141.14 (1C, Ar), 132.65 (1C, Ar), 128.96 (2C, Ar), 128.90 (2C, Ar), 126.92 (2C, Ar) 121.24 (2C, Ar). λ max (nm): 298. MP = 191–192 °C (literature = 190 °C) (Baluja et al. 2006), % yield = 83 %.
4-(3-Phenyl-propylideneamino)-benzene sulfonamide (2)
FT-IR (cm−1): 3,176(C–H aromatic), 1,621 (HC=N), 1,575 (C=C aromatic), 3,306 (N–H str), 1,150 (S=O asym), 1,330 (S=O sym). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 9.70 (d, 1H, HC=N), 8.45–7.51 (m, 9H, Ar–H), 7.10 (d, 1H, Ar–HC=C), 6.60 (d, 1H, Ar–C=CH). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 164.02 (1C, *C=N), 154.30 (1C, C=*C–N), 145.62 (1C, C=*C–SO2NH2), 140.92 (1C, Ar), 135.12 (1C, Ar–*C=C), 129.83 (2C, Ar), 128.91 (2C, Ar), 127.95 (2C, Ar), 127.69 (1C, Ar), 126.84 (2C, Ar), 121.02 (1C, Ar–C=*C). λ max (nm) = 231, 295. MP = 203 °C, % yield = 90 %.
4-[(2-Hydroxy-benzylidene)-amino]-benzenesulfonamide (3)
FT-IR (cm−1): 1,614 (HC=N), 3,239 (N–H str), 1,151 (S=O asym), 1,307 (S=O sym), 3,335 (O–H str), 1,570 (phenolic OH). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 12.7 (s, 2H, NH2), 9.67 (s, 1H, HC=N), 7.90–7.40 (m, 8H, Ar–H). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 164.97 (1C, *C=N), 160.21(1C, *C–OH), 151.19 (1C, C=*C–N), 141.93 (1C, C=*C–SO2NH2), 133.89 (1C, Ar), 132.51 (1C, Ar), 127.04 (2C, Ar), 121.79 (2C, Ar), 119.33 (1C, Ar), 119.27 (1C, Ar), 116.68 (1C, Ar). λ max (nm): 350. MP = 212 °C (literature = 212 °C) (Bharat et al. 2007), % yield = 88 %.
4-[(2,3-Dihydroxy-benzylidene)-amino]-benzenesulfonamide (4)
FT-IR (cm−1): 3,248 (N–H str), 1,617 (HC=N), 1,572 (C=C aromatic), 1,148 (S=O asym), 1,328 (S=O sym), 1,209 (C–O phenolic), 1,273 (OH). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 8.55 (s, 1H, HC=N), 7.95–7.1 (m, 7H, Ar–H). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 165.4 (1C, *C=N), 150.54 (1C, C=*C–N), 149.1 (1C, *C–OH), 145.5 (1C, *C–OH), 141.53 (1C, C=*C–SO2NH2), 127 (2C, Ar), 121.52 (2C, Ar), 119.30 (2C, Ar), 118.90 (2C, Ar). MP = 243 °C, % yield = 78 %.
4-{[(2-Hydroxyphenyl)methylidene]amino}benzenesulfonamide (5)
FT-IR (cm−1): 3,246 (N–H str), 1,618 (HC=N), 1,156 (S=O asym), 1,275 (S=O sym). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 12 (s, 1H, OH), 8.95 (s, 1H, HC=N), 7.92–6.92 (m, 7H, Ar–H), 3.42 (s, 3H, OCH3). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 164.26 (1C, *C=N), 154.29 (1C, C=*C–N), 151.92 (1C, *C–OCH3), 151.56 (1C, *C–OH), 141.82 (1C, C=*C–SO2NH2), 127.06 (2C, Ar), 121.71 (2C, Ar), 121.25 (1C, Ar), 119.21 (1C, Ar), 117.64 (1C, Ar), 114.54 (1C, Ar), 55.49 (1C, OCH3). λ max (nm): 286, 373. MP = 180 °C, % yield = 78 %.
4-[(2-Hydroxy-naphthalen-1-ylmethylene)-amino]-benzenesulfonamide (6)
FT-IR (cm−1): 3,023 (C–H aromatic), 3,165 (N–H str), 1,621 (HC=N), 1,587 (C=C aromatic), 1,310 (S=O sym), 1,292 (OH). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 15.5 (s, 2H, NH2), 10.82 (s, 1H, HC=N), 9.70–7.00 (m, 10H, Ar–H). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 156.38 (1C, *C=N), 146.62 (1C, C=*C–N), 141.34 (1C, C=*C–SO2NH2), 137.70 (1C, *C–OH), 133.07 (1C, Ar), 129.07 (1C, Ar), 128.28 (2C, Ar), 127.39 (1C, Ar), 127.16 (1C, Ar), 126.74 (1C, Ar), 123.79 (1C, Ar), 122.17 (1C, Ar), 120.71 (1C, Ar), 120.50 (1C, Ar), 112.37 (1C, Ar), 108.74 (1C, Ar). λ max (nm): 302, 383, 441, 464. MP = 274 °C, % yield = 85 %.
4-[(4-Chloro-benzylidene)-amino]-benzene sulfonamide (7)
FT-IR (cm−1): 3,289 (N–H str), 3,001 (C–H aromatic), 1,620 (HC=N), 1,581 (C=C aromatic), 1,150 (S=O asym), 1,331 (S=O sym). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 8.67 (s, 1H, HC=N), 8.02–7.40 (m, 8H, Ar–H). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 161.62 (1C, *C=N), 154 (1C, C=*C–N), 141.33 (1C, C=*C–SO2NH2), 136.57 (1C, *C–Cl), 134.42 (1C, Ar), 130.57 (2C, Ar), 129.06 (2C, Ar), 126.92 (2C, Ar), 121.30 (2C, Ar). MP = 181–182 °C (literature = 184 °C) (Santosh et al. 2010), % yield = 80 %.
4-[(3-Ethoxy-4-hydroxy-benzylidene)-amino]-benzenesulfonamide (8)
FT-IR (cm−1): 3,271 (N–H str), 1,598 (HC=N), 1,518 (C=C aromatic), 1,145 (S=O asym), 1,330 (S=O sym), 1,286 (C–O phenolic), 1,192 (OH). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 9.76 (s, 2H, SO2NH2), 8.46 (s, 1H, HC=N), 7.85–6.6 (m, 7H, Ar–H), 5.83 (q, 2H, OCH2*CH3), 1.96 (t, 3H, OCH2CH3*). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 162.13 (1C, *C=N), 154.83 (1C, C=*C–N), 151.88 (1C, *C–OH), 150.91 (1C, C*–OCH2CH3), 145.15 (1C, C=*C–SO2NH2), 140.05 (1C, Ar), 129.93 (2C, Ar), 127.39 (1C, Ar), 124.55 (2C, Ar), 121.14 (1C, Ar), 115.42 (1C, Ar), 63.70 (1C, O*CH2CH3), 16.46 (1C, OCH2*CH3). λ max (in nanometer): 205, 371. MP = 185 °C, % yield = 90 %.
4-[(4-Dimethylamino-benzylidene)-amino]-benzenesulfonamide (9)
FT-IR (cm−1): 3,278 (N–H str), 1,569(HC=N), 1,518(C=C aromatic), 1,143(S=O asym), 1,329 (S=O sym). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 9.67(s, 2H, NH2), 8.43 (s, 1H, N=CH), 8.83–6.60 (m, 8H, Ar–H), 6.10 (s, 6H, N(CH3)2). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 162.70 (1C, *C=N), 158.20 (1C, C=*C–N), 146.80 (1C, *C–N(CH3)2), 137.00 (1C, C=*C–SO2NH2), 129.80 (2C, Ar), 127.80 (2C, Ar), 123.00 (1C, Ar), 120.70 (2C, Ar), 113.20 (2C, Ar), 43.60 (2C, N(CH3)2). MP = 204–205 °C (literature = 202 °C) (Santosh et al. 2010), % yield = 75 %.
4-[(4-Methoxy-benzylidene)-amino]-benzenesulfonamide (10)
FT-IR (cm−1): 3,272 (N–H str), 2,913 (C–H aromatic), 1,606 (HC=N), 1,569 (C=C aromatic), 1,147 (S=O asym), 1,315 (S=O sym), 1,265 (C–O phenolic). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 8.60 (s, 1H, HC=N), 7.90–7.00 (m, 8H, Ar–H), 3.80 (s, 3H, OCH3). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 162.28 (1C, *C=N), 161.85 (1C, *C–OCH3), 154.67 (1C, C=*C–N), 140.72 (1C, C=*C–SO2NH2), 130.85 (1C, Ar), 128.49 (2C, Ar), 126.89 (2C, Ar), 121.18 (2C, Ar), 114.33 (2C, Ar), 55.44 (OCH3). λ max (nm): 275. MP = 205 °C (literature = 206 °C) (Baluja et al. 2006), % yield = 83 %.
4-[(3-Methoxy-benzylidene)-amino]-benzene sulfonamide (11)
FT-IR (cm−1): 3,329 (N–H str), 3,175 (C–H aromatic), 1,618 (HC=N), 1,579 (C=C aromatic), 1,296 (C–O phenolic), 1,150 (S=O asym), 1,329 (S=O sym). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 8.62 (s, 1H, HC=N), 7.85–7.15 (m, 8H, Ar–H), 3.84 (s, 3H, OCH3). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 162.67 (1C, *C=N), 159.53 (1C, *C–OCH3), 154.25 (1C, C=*C–N), 141.18 (1C, C=*C–SO2NH2), 137 (1C, Ar), 130.02 (2C, Ar), 126.91 (1C, Ar), 121.96 (2C, Ar), 121.24 (1C, Ar), 118.30 (1C, Ar), 112.67 (1C, Ar), 55.23 (1C, OCH3). λ max (nm): 278, 320. MP = 161 °C, % yield = 77 %.
4-[(4-Nitro-benzylidene)-amino]-benzenesulfonamide (12)
FT-IR (cm−1): 3,265 (N–H str), 3,105 (C–H aromatic), 1,591 (C=C aromatic), 1,628 (HC=N), 1,598 (NO2), 1,145 (S=O asym), 1,341 (S=O sym), 1,277 (OH). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 12.70 (s, 2H, NH2), 8.39 (s, 1H, HC=N), 7.50–8.22 (m, 8H, Ar–H). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 162.7 (1C, *C=N), 156.4 (1C, C=*C–N), 150.7 (1C, *C–NO2), 137.8 (1C, C=*C–SO2NH2), 137.3 (1C, Ar), 129.9 (2C, Ar), 126.8 (2C, Ar), 123.7 (2C, Ar), 122.3 (2C, Ar). λ max (nm): 271. MP = 193 °C, % yield = 92 %.
4-{[(2-methoxy-4-nitrophenyl)methylidene]amino}benzenesulfonamide(13)
FT-IR (cm−1): 3,289 (N–H str), 3,029 (C–H aromatic), 1,620 (HC=N), 1,579 (C=C aromatic), 1,258 (C–O phenolic), 1,334 (S=O sym), 1,153 (S=O asym), 1,378 (NO2). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 8.90 (s, 1H, HC=N), 8.25–7.25 (m, 7H, Ar–H), 4.05 (s, 3H, OCH3). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 159.41 (1C, *C=N), 156.02 (1C, *C–OCH3), 153.86 (1C, C=*C–N), 150.33 (1C, *C–NO2), 141.73 (1C, C=*C–SO2NH2), 141.03 (1C, Ar), 128.98 (1C, Ar), 128.16 (2C, Ar), 127.30 (1C, Ar), 126.93 (1C, Ar), 121.32 (2C, Ar), 115.59 (1C, Ar), 107.15 (1C, Ar), 56.61 (1C, OCH3). MP = 224 °C, % yield = 92 %.
4-[(Furan-2-ylmethylene)-amino]-benzene sulfonamide (14)
FT-IR (cm−1): 2,978 (C–H aromatic), 3,284 (N–H str), 1,623 (HC=N), 1,578 (C=C aromatic), 1,147 (S=O asym), 1,325 (S=O sym). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 8.46 (s, 1H, HC=N), 8.10–6.75 (m, 7H, Ar–H). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 154.00 (1C, *C=N), 151.55 (1C, C=*C–N), 150.15 (1C, O–*C–C=N), 147.08 (1C, O–C*=C), 141.10 (1C, C=*C–SO2NH2), 126.93 (2C, Ar), 121.24 (2C, Ar), 118.42 (1C, Ar), 112.74 (1C, Ar). λ max (nm): 302. MP = 191 °C, % yield = 70 %.
4-[(Thiophen-2-ylmethylene)-amino]-benzenesulfonamide (15)
FT-IR (cm−1): 1,605 (HC=N), 3,285 (N–H str), 1,149 (S=O asym), 1,321 (S=O sym). 1H-NMR (400 MHz, DMSO-d6): δ [ppm] 8.82 (d, 1H, HC=N), 7.90–6.60 (m, 7H, Ar–H). 13C-NMR (400 MHz, DMSO-d6): δ [ppm] 155.79 (1C, *C=N), 153.54 (1C, C=*C–N), 141.85 (1C, S–*C–C=N), 141.03 (1C, C=*C–SO2NH2), 134.62 (1C, S–C*=C), 132.04 (1C, Ar), 128.35 (2C, Ar), 127.30 (1C, Ar), 126.85 (1C, Ar), 121.27 (2C, Ar). MP = 208 °C, % yield = 89 %.
Biological activities testing
Antitumor activity
Cell lines and culturing
Human breast (MCF7, Cell Line human breast adenocarcinoma) and lung carcinoma (A549, Cell Line human lung carcinoma) were purchased from the European Collection of Cell Culture, UK. The cells were stored in a liquid nitrogen storage facility at the National Medical Research Centre (Zawia/Libya). The cells were grown in Dulbecco’s Minimum essential medium supplemented with 10 % fetal bovine serum containing antibiotics (streptomycin, penicillin–G, kanamycin, amphotericin B). All cell culture work was carried out in a sterile tissue culture laboratory under a laminar flow hood. The frozen vials of the cells were held at room temperature for 1 min before being placed in 37 °C incubator for a further minute until thawed. The outside of the ampoule was sprayed with 70 % ethanol before the cells were poured into a flask containing trypsin-EDTA.
Cell culturing
The cells were passaged every 4 to 7 days using trypsin-EDTA and fed three times a week. Cells were split once confluence was reached and 1 × 105 cells were placed into 25-ml tissue culture flasks. To split the cells, the medium was removed, the cells washed with Hank’s Buffered Salt Solution and gently harvested with 1-ml detachment trypsin neutralizing solution. Then cells were poured into a sterile falcon tube and centrifuged at 1,300 rpm for 5 min. The supernatant was removed and the pellet gently resuspended in an appropriate volume of medium. This mixture was then placed into fresh culture flasks at 1:2 split ratios. Cells (5 × 106 cells/ml) were plated in 1-ml cell suspension concentration in RPMI1640 supplement. The cells were allowed to attach to the bottom for 24 h and finally harvested. After 48 h, the cells were treated with aseptic samples which were sterilized by passing through 0.22-μm Millipore filter and cisplatin was used as positive control. Untreated cells were used as negative controls. Each plate was incubated for another 72 h at 37 °C in a humidified incubator with 5 % CO2. The duplicate plates were then used for screening the chemical synthesized antitumor agents.
Evaluation of cytotoxicity
All synthesized compounds were tested for their cytotoxicity on lung and breast carcinoma in our cell culture lab at the National Medical Research Center, Zawia, Libya. The compounds were dissolved in DMSO (1 ml) and tested on cells at a concentration of 200 μg/ml for 72 h. DMSO was used as negative control, after this period the cell culture was examined under phase contrast inverted microscope.
Statistical analysis
The experimental data were expressed as mean ± SEM. The significance of differences among the various treated groups and control group were analyzed by means of one–way ANOVA. The level of significance was set at p <0.05. IC50 (inhibitory concentration which caused 50 % inhibition) were estimated using linear regression method of the percent of cell viability against the concentration of the tested compounds using Microsoft Excel software program.
Results and discussion
Design of sulfanilamide-imine scaffold
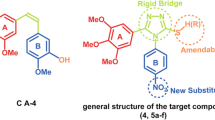
A great variety of compounds have been reported in the literature (Desai and Mitchison, 1997; Gadde and Heald 2004) as inhibitors for polymerization or depolymerization of microtubules by binding to the colchicine binding site of tubulin β-subunit. Combretastatine A-4 (Fig. 2a) and isoquinoline sulfamate (Fig. 2b) are examples of the most interesting compounds under investigation as anti-microtubules which are currently in clinical trials. They were used to design structurally related sulfanilamide-imine derivatives (Fig. 2c) as possible antitubulin agents. The main structural features used in the current study (red in Fig. 2) are two phenyl rings separated by azomethene group rather than the alkene moiety in combretastatine A-4. In addition, sulfonamide functionality of isoquinoline sulfamate is used instead the methoxy group of combretastatine A-4. Fifteen sulfanilamide-imine derivatives have been designed and then synthesized by varying the R group(s) on the second benzene ring.

Chemical structures of Combretastatine A-4, Isoquinoline sulfamate and Sulfanilamide-imines derivatives
Molecular modeling study
All the targeted compounds were docked to the single β-tubulin colchicine active site (Fig. 3a, e.g., compound 6 and colchicine) and the docking energy was calculated. At the same time, the possible interaction of the designed compounds with the colchicine active site of tubulin unit was studied. The docking results showed that most of the synthesized sulfanilamide-imines theoretically fit and interact with the colchicine active site in the same manner as combretastatine A-4 with extra binding to the active site via the sulfonamide moiety. Interestingly, the docking results showed that most of the high score compounds are planar and occupy the same part of the active site (Fig. 3b). These flexible molecules are of lower docking energy, which is in total agreement with the biological results achieved for the same compounds. The sulfonamide group in the tested compounds occupies the same part used by the same group of isoquinoline sulfamate. They further showed greater reactivity at the active site and a high ability to form four different hydrogen bonds with amino acids of the colchicine active site, namely histidine 94, histidine 119, threonine 199 and threonine 200 (Fig. 3a). The hydrophobic pocket of the active site, which accommodates the lipophilic moieties of the tested compounds, has large size. This can be concluded based on the comparison of naphthyl side chain of compound 6 and colchicine phenyl group, as they represented in different positions at the hydrophobic pocket. Additionally, increasing the lipophilicity of the substituents at this position leads to decreasing of docking energy and possibly increased the interaction with the targeted microtubules.

a Interaction of compound 6 (red) and colchicine (green) at the colchicine active site. b Planarity of compound 6 (red) and colchicine (green) at the colchicine active site
Chemical synthesis
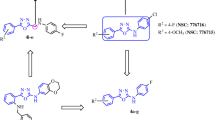
The sulfanilamide-imines (1–15) were synthesized by condensation of sulfanilamide with the starting aldehydes in equimolar amounts using microwave method for chemical synthesis (Scheme 1). The optimum microwave power (in Watt) and the corresponding reaction time were determined depending on the completion of the reaction which is monitored by TLC using the appropriate solvent system. Highly purified crystals of all compounds were obtained via successive recrystallization using absolute ethanol and their purity was determined by HPLC using either chloroform/methanol (80:20) or acetonitrile/methanol (90:10) as a mobile phase. The percentage of the achieved dried yield of the synthetic compounds was high (70–93 %) for most of the synthesized compounds.

Chemical synthesis of sulfanilamide-imines (1–15)
Chemical structures of the synthesized compounds were determined on the basis of their FT-IR, UV, 1H NMR, and 13C NMR spectroscopic analysis. The synthesized compounds were in agreement with those found in the literature (Zevzikoviene et al. 2012; Kaya et al. 2012; Yang et al. 2010; Scozzafava et al. 2000; Popescu et al. 1999). The IR spectra of the synthesized compounds confirmed the presence of C=N stretching bands at 1,500–1,690 cm−1 and the absence of C=O at 1,700 cm−1 for the starting aldehyde. The bands due to symmetric stretching SO2 groups that are shifted to lower frequencies, while NH disappeared or was hidden under the broad band at 3,450–3,300 cm−1 of the Schiff’s base. 1H NMR spectra of the synthesized compounds were recorded in DMSO-d6, and the chemical shifts (δ), expressed in parts per million (ppm) downfield from tetramethylsilane. The signals for the methene protons of the azomethine group, −N=C (H) were observed between 9.6 and 10.82 ppm.
In vitro antitumor evaluation
The antitumor activity of all compounds against breast and lung cell lines was determined using cell counting assay and cisplatin as a reference drug control. Each cell line was incubated with four concentrations of each compound. The response parameter (IC50) was calculated for each cell line (Table 1). The predicted binding free energy as shown in Table 1 is proportional to the in vitro cytotoxicity results. The antitumor drug discovery screen has been designed to distinguish between broad-spectrum antitumor compounds and tumor selective agents. The active analogs showed a characteristic potential model of selectivity as well as broad-spectrum antitumor activity. With regard to selectivity against individual cell lines, compounds 2, 3, 4, 5, 6, 7, 8, 9, 12, and 13 had a positive impact against cell line human breast cancer MCF-7 with IC50 values range of 90–166 μM compared to Combretastatine A4 (IC50 2.0 μM) and colchicine (IC50 3.2 μM)(De Martino et al. 2004) (Table 1). Compounds with binding free energy bigger than −5.23 were ineffective against human breast cancer cells. The lung human cell line proved to be sensitive toward compounds 4, 8, and 13 with a IC50 concentration range of 120–140 μM compared to Combretastatine A4 (IC50 2.0 μM) and colchicine (IC50 3.2 μM)(De Martino et al. 2004) with binding free energy ranging from −5.87 to −6.23. Whereas, compounds 1, 10, 11, 14, and 15 proved to be ineffective against breast cancer cells while compounds 1, 2, 3, 5, 6, 7, 9, 10, 11, 12, 14, and 15 also proved to be ineffective against lung cancer cells. These results indicate that cytotoxicity is generally correlated with binding affinity for the colchicine site. However, some results were not fully explained in terms of binding affinity, suggesting that others factors, such as specific effects of the compound on the tubulin molecule, may play a role in the cytotoxic activities of these derivatives.
The activity of the tested compounds could be correlated to structure variations and modifications. By studying the variation in selectivity of the tested compounds over the two cell lines, 11 compounds showed significant inhibition of the breast cancer cells (MCF-7) and only 3 compounds affected lung cells. The agreement between the inhibition of breast and lung cancer cells could be correlated to a similar inhibitory mechanism related to the common structural features, while the variation in selectivity for breast compared to lung cells is probably caused by the differences in the substitution pattern in sulfanilamide Schiff base derivatives. These variations could be further correlated to the lipophilic substitution pattern on sulfanilamide Schiff base. The high activity of compounds 4, 8, and 13 in breast cancer over lung cancer cells may be attributed to the occurrence of OH, OCH3, and NO2 moieties which may be important for hydrogen bonding at the receptor site as described in the docking section.
When the chemical structure of the synthesized compounds was correlated with their anticancer activity, a number of structure activity relationships could be derived. Table 1 shows that no anticancer activity is achieved either against lung or breast carcinoma when the R substituent is unsubstituted phenyl group (compound 1). However, the introduction of an extra double bond between the R substituent and the azomethine moiety leads to strong anti-breast cancer activity of compound 2. Introduction of a hydrogen bond donor group, such as the hydroxyl group (−OH) in the ortho position of the phenyl group of the inactive compound 1, leads to strong activity against breast cancer cells as found for compound 3 (Table 1). Introduction of a methoxy group (−OCH3) at the same phenyl group leads to a small decrease of activity as in compound 5, while an additional hydroxyl group promotes anticancer activity against both breast and lung cancer cells, as found for compound 4. Increasing the lipophilicity of compound 3 by substitution of the phenyl with a naphthyl group leads to a small decrease in the activity as in compound 6. The para position of phenyl group is important for activity as the introduction of chloro-, hydroxyl-, dimethylamino- or nitro- substituents improves activity as in compounds 7, 8, 9, and 12, respectively. The methoxy group at the meta or para position showed no activity as in compounds 10 and 11. The presence of methoxy- or ethoxy- groups, however, appears to be important for the activity on both types of cancer cells as in compounds 8 and 13. This may be attributed to the effect of both groups on the pharmacokinetics compared to mono-substituted compounds. Finally, the isosteric substitution of the phenyl group of compound 1 with thiophenyl or furfuryl rings showed no activity as in compounds 14 and 15.
Conclusion
All the biological results complied with the molecular modeling studies to a considerable extent. Sulfanilamide-imine derivatives 4, 8, and 13 proved to be the most active compounds in this study on both breast carcinoma and lung carcinoma cells, whereas sulfanilamide-imine derivatives 2, 3, 5, 6, 7, 9, and 12 were active only against breast carcinoma cell as compared to the standard drug cisplatin. The active compound analogs could be considered as useful templates for further development to obtain more potent antitumor agent(s).
Future work
The exact mechanism of colchicines on microtubule dynamics is unclear; however, it is known to first bind to a specific site on the β-tubulin subunit of the tubulin heterodimer. The tubulin-colchicine complexes then become incorporated into the microtubule ends, leading to changes in the rate and extent of growing and shortening of microtubules. Tubulin-binding assays will be used as a screening tool to identify our synthesized compounds is able to interact with tubulin at the colchicine binding domain.
References
Al Rashood ST, Aboldahab IA, Nagi MN, Abouzeid LA, Abdel-Aziz AA, Abdel-Hamide SG, Youssef KM, Al Obaid AM, El Subbagh HI (2006) Synthesis, dihydrofolate reductase inhibition, antitumor testing, and molecular modeling study of some new 4(3H)-quinazolinone analogs. Bioorg Med Chem 14:8608–8621
Amvrosieva TV, Paklonskaya NV, Biazruchka AA, Kazinetz ON, Bohush ZF, Fisenko EG (2006) Enteroviral infection outbreak in the Republic of Belarus: principal characteristics and phylogenetic analysis of etiological agents. Cent Eur J Public Health 14:67–73
Asai N, Aoshima M, Ohkuni Y, Otsuka Y, Kaneko N (2012) Should blood cultures be performed in terminally ill cancer patients? Indian J Palliat Care 18:40–44
Baluja S, Solanki A, Kachhadia N (2006) Evaluation of biological activities of some Schiff bases and metal complexes. J Iranian Chem Soc 3:312–317
Bharat BS, Prasanna KP, Sabuj S (2007) Synthesis and evaluation of antimicrobial activity of metal complexes of 4-(2′-hydroxyphenylimino)phenysulphonamide. Iranian J Pharm Sci 3:245–250
Chang JY, Chang CY, Kuo CC, Chen LT, Wein YS, Kuo YH (2004) Salvinal, a novel microtubule inhibitor isolated from Salvia miltiorrhizae Bunge (Danshen), with antimitotic activity in multidrug-sensitive and -resistant human tumor cells. Mol Pharmacol 65:77–84
Cortes J, Baselga J (2007) Targeting the microtubules in breast cancer beyond taxanes: the epothilones. Oncologist 12:271–280
Cushman M, Nagarathnam D, Gopal D, Chakraborti AK, Lin CM, Hamel E (1991) Synthesis and evaluation of stilbene and dihydrostilbene derivatives as potential anticancer agents that inhibit tubulin polymerization. J Med Chem 34:2579–2588
De Martino G, La Regina G, Coluccia A, Edler MC, Barbera MC, Brancale A, Wilcox E, Hamel E, Artico M, Silvestri R (2004) Arylthioindoles, potent inhibitors of tubulin polymerization. J Med Chem 47(25):6120–6123
Desai A, Mitchison TJ (1997) Microtubule polymerization dynamics. Annu Rev Cell Dev Biol 13:83–117
Dowlati A, Robertson K, Cooney M, Petros WP, Stratford M, Jesberger J, Rafie N, Overmoyer B, Makkar V, Stambler B, Taylor A, Waas J, Lewin JS, McCrae KR, Remick SC (2002) A phase I pharmacokinetic and translational study of the novel vascular targeting agent combretastatin a-4 phosphate on a single-dose intravenous schedule in patients with advanced cancer. Cancer Res 62:3408–3416
Eckhardt S (2002) Recent progress in the development of anticancer agents. Curr Med Chem Anticancer Agents 2:419–439
Gadde S, Heald R (2004) Mechanisms and molecules of the mitotic spindle. Curr Biol 14:R797–R805
Hori K, Nishihara M, Shiraishi K, Yokoyama M (2010) The combretastatin derivative (Cderiv), a vascular disrupting agent, enables polymeric nanomicelles to accumulate in microtumors. J Pharm Sci 99:2914–2925
Hsieh HP, Liou JP, Mahindroo N (2005) Pharmaceutical design of antimitotic agents based on combretastatins. Curr Pharm Des 11:1655–1677
Kaya M, Basar E, Cakir E, Tunca E, Bulbul M (2012) Synthesis and characterization of novel dioxoacridine sulfonamide derivatives as new carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 27:509–514
Kundu A, Shakil NA, Saxena DB, Kumar J, Walia S (2009) Microwave assisted solvent-free synthesis and biological activities of novel imines (Schiff bases). J Environ Sci Health B 44:428–434
Lu Y, Chen J, Xiao M, Li W, Miller DD (2012) An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res 29:2943–2971
Medina JC, Shan B, Beckmann H, Farrell RP, Clark DL, Learned RM, Roche D, Li A, Baichwal V, Case C, Baeuerle PA, Rosen T, Jaen JC (1998) Novel antineoplastic agents with efficacy against multidrug resistant tumor cells. Bioorg Med Chem Lett 8:2653–2656
Mitrus I, Sochanik A, Cichon T, Szala S (2009) Combination of combretastatin A4 phosphate and doxorubicin-containing liposomes affects growth of B16-F10 tumors. Acta Biochim Pol 56:161–165
Nogales E, Wolf SG, Downing KH (1998) Structure of the alpha beta tubulin dimer by electron crystallography. Nature 391:199–203
Pfisterer PH, Wolber G, Efferth T, Rollinger JM, Stuppner H (2010) Natural products in structure-assisted design of molecular cancer therapeutics. Curr Pharm Des 16:1718–1741
Popescu A, Simion A, Scozzafava A, Briganti F, Supuran CT (1999) Carbonic anhydrase inhibitors. Schiff bases of some aromatic sulfonamides and their metal complexes: towards more selective inhibitors of carbonic anhydrase isozyme IV. J Enzyme Inhib 14:407–423
Prota AE, Bargsten K, Zurwerra D, Field JJ, Diaz JF, Altmann KH, Steinmetz MO (2013) Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 339:587–590
Santosh K, Niranjan MS, Chaluvaraju KC, Jamakhandi CM, Dayanand K (2010) Synthesis and antimicrobial study of some Schiff bases of sulfonamides. J Curr Pharm Res 39–42
Scozzafava A, Banciu MD, Popescu A, Supuran CT (2000) Carbonic anhydrase inhibitors: synthesis of Schiff bases of hydroxybenzaldehydes with aromatic sulfonamides and their reactions with arylsulfonyl isocyanates. J Enzyme Inhib 15:533–546
Thomas M (2009) Molecular targeted therapy for hepatocellular carcinoma. J Gastroenterol 44(Suppl 19):136–141
Thomas MB, Abbruzzese JL (2005) Opportunities for targeted therapies in hepatocellular carcinoma. J Clin Oncol 23:8093–8108
Venugopala KN, Jayashree BS (2008) Microwave-induced synthesis of Schiff bases of aminothiazolyl bromocoumarins as antibacterials. Indian J Pharm Sci 70:88–91
Yang DC, Yan JF, Xu J, Ye F, Zhou ZW, Zhang WY, Fan L, Chen X (2010) Synthesis and investigation on antidiabetic activity of 4-(1-aryl-3-oxo-5-phenylpentylamino) benzenesulfonamide. Yao Xue Xue Bao 45:66–71
Zevzikoviene A, Zevzikovas A, Tarasevicius E, Pavlonis A, Dirse V (2012) Synthesis and in vitro antimicrobial study of 4-thiazolidinone containing sulfanilamide. Acta Pol Pharm 69:911–915
Acknowledgments
The authors would like to express their gratitude and thanks to Dr Saad A. Zaied for providing the cell lines. We are also thankful to Sir Ashor Al-Fazany for his valuable technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Mohamed, S.S., Tamer, A.R., Bensaber, S.M. et al. Design, synthesis, molecular modeling, and biological evaluation of sulfanilamide-imines derivatives as potential anticancer agents. Naunyn-Schmiedeberg's Arch Pharmacol 386, 813–822 (2013). https://doi.org/10.1007/s00210-013-0883-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-013-0883-y