
Abstract
Exposure to a variety of toxins and/or infectious agents leads to disease, degeneration and death, often characterised by circumstances in which cells or tissues do not merely die and cease to function but may be more or less entirely obliterated. It is then legitimate to ask the question as to whether, despite the many kinds of agent involved, there may be at least some unifying mechanisms of such cell death and destruction. I summarise the evidence that in a great many cases, one underlying mechanism, providing major stresses of this type, entails continuing and autocatalytic production (based on positive feedback mechanisms) of hydroxyl radicals via Fenton chemistry involving poorly liganded iron, leading to cell death via apoptosis (probably including via pathways induced by changes in the NF-κB system). While every pathway is in some sense connected to every other one, I highlight the literature evidence suggesting that the degenerative effects of many diseases and toxicological insults converge on iron dysregulation. This highlights specifically the role of iron metabolism, and the detailed speciation of iron, in chemical and other toxicology, and has significant implications for the use of iron chelating substances (probably in partnership with appropriate anti-oxidants) as nutritional or therapeutic agents in inhibiting both the progression of these mainly degenerative diseases and the sequelae of both chronic and acute toxin exposure. The complexity of biochemical networks, especially those involving autocatalytic behaviour and positive feedbacks, means that multiple interventions (e.g. of iron chelators plus antioxidants) are likely to prove most effective. A variety of systems biology approaches, that I summarise, can predict both the mechanisms involved in these cell death pathways and the optimal sites of action for nutritional or pharmacological interventions.
Similar content being viewed by others

Introduction
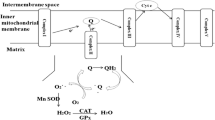
As a transition metal that can exist in several valencies, and that can bind up to six ligands, iron is an important component of industrial catalysts in the chemical industry (Hagen 2006), especially for redox reactions. Its catalysis of specific reactions requires rather exact architectures at the catalytic centre, and indeed much of the art and science of catalyst production involves determining and synthesising them. Nearly half of all enzymes are metalloproteins (Waldron et al. 2009), and iron is also of considerable importance in biology as a component of all kinds of metalloproteins (Andreini et al. 2008, 2009) from haemoglobin to cytochromes, as well as in the directed evolution of novel enzyme activities (Pordea and Ward 2008; Que and Tolman 2008; Turner 2009). When serving in enzymes, the iron is normally safely liganded, and any reactions catalysed are usually fairly specific. However, as is widely recognised, iron can also have a dark side (Kell 2009a), in that when it is not properly liganded (Graf et al. 1984), and in the ferrous form, it can react with hydrogen peroxide (produced by mitochondria (e.g. Brennan and Kantorow 2009; Fato et al. 2008; Orrenius et al. 2007) or (per)oxidases (Bedard and Krause 2007; Cave et al. 2006) via the Fenton reaction (Goldstein et al. 1993; Kruszewski 2003; Toyokuni 2002; Wardman and Candeias 1996; Winterbourn 1995), leading to the very reactive and damaging hydroxyl radical (OH•)

The Haber-Weiss and Fenton reactions combine using poorly liganded iron in a catalytic cycle to produce the very damaging hydroxyl radical. Poorly liganded iron can also be liberated via the destruction of haem and other iron-containing substances. Peroxynitrite anion (ONOO−) is produced by the reaction of superoxide and nitric oxide (NO•) which when protonated (pH ca 6.5–6.8) decomposes to OH• and NO2

Some small molecules that are derived from the oxidative attack of hydroxyl and other radicals on cellular macromolecules and that can act as biomarkers of oxidative stress, including that mediated by iron

Very strong relationship between serum ferritin concentrations and urinary concentrations of the DNA damage/oxidative stress marker 8-hydroxy-2′-deoxyguanosine. Data are replotted from Fig. 1 of Hori et al. (2010)

A mind map (Buzan 2002) setting out the structure of this review. To read this start at “1 o’clock” and move outwards and clockwise

Some of the interactions between the prion protein in its two main conformations, reactive oxygen species and iron dysregulation. This diagram is based on Fig. 12 of (Singh et al. 2010b), and illustrates in particular the autocatalytic nature of the ROS- and iron-dependent conversion of PrPC to PrPSc and the neurotoxicity of the latter

Iron catalyses the formation of the hydroxyl radical (and thence other kinds of ROS) that can react with proteins and lipids to denature them, leading to insoluble plaques and other fibrotic structures that can themselves bind/entrap the iron that caused their formation. This bound iron can cause further hydroxyl radical formation such that the process is autocatalytic. Eventually this overwhelms cellular defences, leading to cell death (with further release of iron)

Large (17-fold) accumulation of EPR-detectable iron in atherosclerotic plaques relative to healthy non-atherosclerotic (‘intima’) controls. The difference is highly significant (P = 0.0001). Data are replotted from those in Fig. 1b of Stadler et al. (2004)

The major events accompanying sepsis and the Systemic Inflammatory Response Syndrome. Note in particular the positive feedback by which the release of pro-inflammatory cytokines and ROSs leads to the release of poorly liganded iron (e.g. from ferritin and haem) that causes the release of further inflammatory cytokines and ROSs. In principle, iron chelators could interfere with this vicious cycle

A diagram, based very loosely on the narrative in (Kohanski et al. 2007), illustrating how the autocatalytic activity of the reactions of superoxide, peroxide and the hydroxyl radical involving poorly liganded iron can exert a positive feedback leading to the death of cells

‘Bow-tie’ models of cellular networks. There are many examples of cellular networks in which a large variety of possible initial events leads to complex sequelae, but these are mediated via a comparatively small number of ‘intermediate’ reactions. While in the present case it is suggested that these in part involve complex positive feedbacks, the general ‘bow-tie’ idea does allow one to recognise that despite the many different possible inputs, a broadly unitary kind of mechanism of action—here involving iron dysregulation—can reasonably be invoked to explain the multiple causes that can lead to cell death and destruction

The three main iron chelators approved for clinical use

The extent of cell damage caused by the unliganded iron-catalysed production of ROSs and RNSs is determined by many factors, some of which promote and some of which act against it. While the existence of hormesis (see text) means that the see-saw illustrated here is an imperfect metaphor, the diagram serves to illustrate the complexity of the problem and the need for a systems biology approach to its solution

The steady-state concentrations of a molecule depend on the rates of production and removal of the molecule in question. Both lowering creation and increasing removal represents a particularly effective strategy relative to doing just one of these alone. Thus if C is the hydroxyl radical we can lower its concentration by decreasing A and/or B and by increasing the rates of the reaction to D and E. The latter (decrease of concentration of C) may sometimes better be effected by increasing the activities of enzymes yet further downstream

The relationship between ‘forward’ and ‘inverse’ methods of systems biology. The comparison of two ‘inverse’ systems (one a control and one treated with a toxin) in terms of the estimation of which parameters have changed most allows one to infer the sites or modes of action of that toxin [In a related manner we also discriminate forward and reverse genetics, including chemical genetics (Kell 2006a, b)].
Superoxide (also produced by mitochondria) can react with ferric iron in the Haber-Weiss reaction (Kehrer 2000) to produce Fe(II) again, thereby effecting redox cycling of the iron (Fig. 1):
Ascorbate (vitamin C) can also replace \( \hbox{O}_2^{\bullet -} \) for reducing the Fe(III) to Fe(II) (Hershko and Weatherall 1988), as can other reducing agents, and indeed too low a redox poise leads to DNA damage (e.g. Li and Marbán 2010; Seifried et al. 2007).
The hydroxyl radical is exceptionally reactive and damaging to cellular components, and, for instance, can liberate further Fe(II) from iron-sulphur centres and other iron-containing compounds such as ferritin (Arosio et al. 2009), thereby driving reaction (1) in an autocatalytic, runaway kind of reaction. This kind of phenomenon has the potential to overwhelm any kinds of attempts at repair, and inflammation and oxidative stress are the hallmarks of each of the conditions I summarise. Related reactions include peroxynitrite production (from the reaction of NO and superoxide) (Babior 2000; Beckman et al. 1990; Beckman and Koppenol 1996; Goldstein and Merényi 2008; Koppenol et al. 1992; Murphy et al. 1998; Pacher et al. 2007; Pavlovic and Santaniello 2007; Pryor and Squadrito 1995; Radi et al. 2001, 2002; Rubbo and O’Donnell 2005; Rubbo et al. 2009; Smith et al. 1997b; Squadrito and Pryor 1998; Szabo 1996; Szabó et al. 2007; Torreilles et al. 1999; White et al. 1994; Zimmet and Hare 2006). These can lead to nitrotyrosine (Beckman 1996; Goldstein and Merényi 2008; Herce-Pagliai et al. 1998) (a reaction catalysed by poorly liganded iron, Beckman et al. 1992), or nitro-fatty acid (Aslan et al. 2001; O’Donnell and Freeman 2001) production or protein cysteine nitrosylation (Lancaster 2008; Landino 2008; Vaz and Augusto 2008) that can provide a means of their detection downstream. Some of these are shown in Fig. 2. A key point here is that despite the widespread and uncritical use of the term ROS to describe any ‘Reactive Oxygen Species’, most such as superoxide and peroxide are not terribly reactive, in contrast to the hydroxyl radical (and peroxynitrite) which is, and unliganded iron is required for hydroxyl radical production in the Fenton reaction. Hence the focus on unliganded iron rather than the more nebulous ROSs, albeit that (su)peroxide is necessarily involved.
Clearly the occurrence of these activities leads to the (more or less irreversible) formation of a variety of substances that can act as biomarkers of these activities (Kell 2009a), and such oxidised compounds are represented by both small and macro-molecules. For instance, 8-hydroxy-2′-deoxyguanosine (8-OHdG) (ChEBI 40304) also referred to as its tautomer 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-oxodG) (HMDB03333) is produced when the hydroxyl radical reacts with DNA and damages it (e.g. Bal and Kasprzak 2002; Burrows and Muller 1998; Cooke et al. 2003, 2008; Lloyd et al. 1998; Loft and Poulsen 1996; Orhan et al. 2004; Shi et al. 2003; Toyokuni and Sagripanti 1996; Valavanidis et al. 2009; Valko et al. 2005), and body-iron status correlates rather strongly with its production or urinary excretion (Agarwal et al. 2004; Broedbaek et al. 2009; Fujita et al. 2007a, 2009; Gackowski et al. 2002; Hori et al. 2010; Kang et al. 1998; Kuo et al. 2008; Maruyama et al. 2007; Nakano et al. 2003; Toyokuni and Sagripanti 1996; Tuomainen et al. 2007; Valavanidis et al. 2005). The data in (Hori et al. 2010) relating urinary 8-OHdG levels to serum ferritin concentrations are particularly striking, and I have digitised them (cf. Pettifer et al. 2009 for pointers to automated ways of doing this in the future) and replotted some of them in Fig. 3.
Ferritin levels (a common measure of body iron stores) are also widely associated with disease development/ severity/ poor outcomes (e.g. Alissa et al. 2007; Armand et al. 2007; Bartzokis et al. 2007; Braun et al. 2004; Bugianesi et al. 2004; Chen et al. 2006; Choi et al. 2005; Double et al. 2000; Fargion et al. 2001; Fernández-Real et al. 2002; Ford and Cogswell 1999; Forouhi et al. 2007; He et al. 2007; Hubel et al. 2004; Ishizaka et al. 2005; Jehn et al. 2004, 2007; Kaur et al. 2007; Kiechl et al. 1997; Lee et al. 2006f; Lim et al. 2001; Rayman et al. 2002; Rouault 2006; Salonen et al. 1992; Sheth and Brittenham 2000; Tuomainen et al. 1998; Valenti et al. 2007; Wilson et al. 2003; You et al. 2003; You and Wang 2005) (Adamkiewicz et al. 2009; Baune et al. 2010; Busca et al. 2010; DePalma et al. 2010; Ferrara et al. 2009; Ferrucci et al. 2010; Gamberini et al. 2008; Goodall et al. 2008; Inati et al. 2010; Kaysen 2009; Knovich et al. 2009; Kolberg et al. 2009; Lecube et al. 2008; Lim et al. 2010; Mateo-Gallego et al. 2010; McNeill et al. 2008; Menke et al. 2009; Qureshi et al. 2008; Rajpathak et al. 2009; Sharifi et al. 2008; Skinner et al. 2010; Song et al. 2009; Storey et al. 2009; Sun et al. 2008; Tsimikas et al. 2009; Valenti et al. 2010; Walker et al. 2010; Wang et al. 2010b; Yoneda et al. 2010; Zandman-Goddard and Shoenfeld 2008), though note that most assays are for the protein itself, and not for the full molecule including any iron that it may sequester effectively (Balla et al. 1992; Hintze and Theil 2006; Theil 2007) or otherwise.
Although iron is most commonly bivalent, Fe(II), or trivalent, Fe(III), this simple statement does not remotely cover the relevant chemistry and speciation that are necessary to recognise what forms of ‘iron’ may be safe and which are likely to catalyse damaging reactions. The first issue is that at neutral pH Fe(III) is more or less insoluble, and in aerobic environments it is necessary to chelate the otherwise ‘free’ Fe(III) with appropriate chelators or siderophores (especially for microbes, e.g. Andrews et al. 2003; Barry and Challis 2009; Cornelis and Andrews 2010; de Carvalho and Fernandes 2010; Haas et al. 2008; Johnson 2008; Miethke and Marahiel 2007; Raymond et al. 2003; Sandy and Butler 2009; Wandersman and Delepelaire 2004; Winkelmann 2002). These siderophores—secreted as are other bacterial pheromones (Kell et al. 1995)—typically have extremely tight binding constants (K f > 1030, e.g. Clifton et al. 2009; Loomis and Raymond 1991) and can solubilise and sequester iron such that it can be internalised via suitable transporter molecules within the plasma membrane (Stintzi et al. 2000).
Iron contains up to six individual chelation sites, arranged octahedrally, and ligands can typically satisfy them partially (i.e. some ligands are bidentate or tridentate and need three or two molecules for full liganding/activity) or fully i.e. are hexadentate. Since iron cannot be transmuted into any other substance, the only way to stop the damaging activity of free or partially liganded ‘iron’ is to ensure that all of its six possible liganding sites are satisfied , whether by endogenous chelators or those added from the diet or as pharmaceuticals. Put another way, it is not simply enough to know that ‘iron’ is present at an adequate level but that it is available in a suitably liganded form. Anaemia can be caused by poor liganding as well as by an actual shortage of ‘iron’ itself. Note too that partial chelation in the presence of an antioxidant agent such as ascorbate (vitamin C) can in fact make ascorbate (or other reducing agent) act as a pro-oxidant and thus actually promote the production of OH• radicals in the presence of inappropriately or inadequately liganded Fe(II) (Allen and Cornforth 2009; Fábián and Csordás 2003; Halliwell 2009; Hininger et al. 2005; Lachili et al. 2001; Long et al. 2000; Miller et al. 1990; Reif 1992; Sugihara et al. 1999). This very likely explains the often and indeed surprisingly disappointing clinical results obtained when using antioxidants alone (Bjelakovic et al. 2008; Giustarini et al. 2009; Kell 2009a; Miller et al. 2005).
The above facts are well known (e.g. Halliwell and Gutteridge 2006; Weinberg 2004), and I discussed them at length in a recent and wide-ranging review (Kell 2009a). My purpose here is to look more closely at the evidence that they are part of the sequelae of a number of predispositions to cellular and organismal death (whether by necrosis or apoptosis) that follow from a large variety of initial ‘insults’ or distal causes, whether of genetic or environmental origin (or both). While, as a systems property, many other cellular processes contribute measurably to any specific activity, I suggest, in all the disparate cases I review, that it is the binding of poorly liganded iron (mainly bivalent) to inappropriate cellular structures that serves to generate this catalytic activity, albeit that the overall manifestations may differ at a physiological level, and I here review what evidence is available. This basic suggestion can be tested explicitly more or less easily. I suggest further that it is this ongoing, autocatalytic activity based on positive feedback that is responsible for the really large-scale damage, leading to organismal death, that can occur in affected cells, tissues, organs and organisms. This analysis also suggests an important role for iron chelation as part of combination approaches in the acute and chronic therapy of these conditions. An overview of this article is given as a Mind Map (Buzan 2002) in Fig. 4.
Note that I do recognise that other transition and polyvalent metal ions (Aln+, Crn+, Cun+, Mnn+, Sen+, Znn+ etc.) may also contribute to the kinds of process I describe. However, for reasons of simplicity, focus, and because of the natural abundance of this metal in biological systems, it is ‘iron’ on which I shall concentrate. As previously (Kell 2009a), I use the term ‘iron’ to include iron of any valencies or speciation, unless specified otherwise.
Since the role of iron is obvious in cases of primary diseases of iron overload, such as hereditary haemochromatosis (e.g. Camaschella and Merlini 2005; Ellervik et al. 2007; Gan et al. 2010; Limdi and Crampton 2004; Mair and Weiss 2009; Marx 2002; McLaren and Gordeuk 2009; Pantopoulos 2008; Papanikolaou and Pantopoulos 2005; Pietrangelo 2006; Weiss 2010), thalassaemias (e.g. Borgna-Pignatti et al. 2005; Camaschella and Merlini 2005; Cao and Galanello 2010; Cohen et al. 2004; Gattermann 2009; Lam et al. 2008; Li et al. 2010; Mohkam et al. 2008; Peng et al. 2008; Taher et al. 2010; Vichinsky et al. 2005) and myelodysplastic syndrome (e.g. Cazzola et al. 2008; Cuijpers et al. 2010; Dreyfus 2008; Gattermann 2008; Greenberg 2006; Greenberg et al. 2008; Jabbour et al. 2009; Jädersten and Hellström-Lindberg 2010; Leitch 2007; Mahesh et al. 2008; Malcovati 2009; Porter et al. 2008; Wimazal et al. 2009), I largely ignore this literature. However, I recognise that the sequelae of iron overload, especially various kinds of organ failure, share many similarities to those I describe below, consistent with the role of iron in these other cases where its involvement has been less widely recognised. Clearly the commonality of any specific effect with those of known iron overload (Dever et al. 2010) might give strong hints for the involvement of iron in specific toxicological processes.
The literature survey at the time of initial submission for review extended to 14 June, 2010.
To start our survey, we look at the consequences of an acute cerebral infarction or stroke.
Stroke
Stroke is a term used to describe the destruction of brain cells, typically following a temporary blockage in blood flow (ischaemia-reperfusion injury) or an intra-cranial haemorrhage. The extent of damage varies considerably, and can be exacerbated because affected cells can release inflammatory cytokines that activate other cells in a similar way, providing a positive autocatalytic effect and leading to spreading of the damage. It is this secondary spreading that is especially damaging, but because it is slower (4–7days), it also affords the opportunity for therapeutic intervention (Qureshi et al. 2009). It is now clear that iron is intimately involved (Armengou and Davalos 2002; Bailey et al. 2006; Bishop and Robinson 2001; Chang et al. 2005; Dávalos, et al. 1994; Demougeot et al. 2004; Ferro and Dávalos 2006; Garoufi et al. 2006; Gillum et al. 1996; Lee et al. 2006e; Marniemi et al. 2005; Mascitelli and Pezzetta 2006; Mehta et al. 2004; Mu et al. 2005; Nakamura et al. 2006; Saxena et al. 2005; Selim and Ratan 2004; Switzer et al. 2006; Wagner et al. 2003; Wu et al. 2003; Zuliani et al. 2006; Adams 2007; Altamura et al. 2009; Assenza et al. 2009; Basak et al. 2008; Bosomtwi et al. 2008; Carbonell and Rama 2007; Cho et al. 2007; Ekblom et al. 2007; Hanson et al. 2009; Heckl 2007; Helal 2008; Jolkkonen et al. 2007; Justicia et al. 2008; Kaushal and Schlichter 2008; Kim et al. 2008a; Kobayashi et al. 2008; Lou et al. 2009; Maguire et al. 2007; Mazumdar et al. 2007; Mehdiratta et al. 2008; Millan et al. 2007, 2008; Millerot-Serrurot et al. 2008; Nighoghossian et al. 2008; O’Rourke et al. 2008; Ratan et al. 2008; Ross and Meschia 2009; Saleh et al. 2007; Santhosh et al. 2009; Verduzco and Nathan 2009; Walters and Rye 2009; Weng et al. 2008), whether by release from (ferritin in) cells or from the haem of haemoglobin. Iron chelators have thus shown promise in decreasing the sequelae of an initial stroke-inducing event (Demougeot et al. 2004; Ferro and Dávalos 2006; Hurn et al. 1995; Kompala et al. 1986; Mu et al. 2005; Patt et al. 1990; Prass et al. 2002; Selim and Ratan 2004; Soloniuk et al. 1992; White et al. 1988; Chen-Roetling et al. 2009; Gu et al. 2009; Hanson et al. 2009; Hua et al. 2008; Mazumdar et al. 2007; Méthy et al. 2008; Millerot-Serrurot et al. 2008; Mirre et al. 2010; Okauchi et al. 2009, 2010; Ratan et al. 2008; Robinson et al. 2009; Selim 2009; Verduzco and Nathan 2009), providing further evidence for the primary importance of iron in causing injury.
While stroke is a sudden occurrence, albeit with secondary consequences, a number of neurodegenerative diseases are rather more long term in their development, and we now look at several, each of which is seen to involve iron intimately.
Huntington’s disease
The pathology of HD reveals striking neurodegeneration in the corpus striatum and shrinkage of the brain, leading to its most prominent manifestation, viz. movement disorders or chorea (Bhidayasiri and Truong 2004). Huntington’s disease occurs via the addition of trinucleotide CAG repeats within exon 1 of the relevant gene, encoding (poly)glutamine (polyQ) repeats in the huntingtin protein (htt) and leading to a gain of (toxic) function (Bauer and Nukina 2009; Gusella and MacDonald 2000; Imarisio et al. 2008; Quintanilla and Johnson 2009). The number of these repeats determines both the time of onset of observable disease (more repeats meaning earlier onset) (Perutz and Windle 2001; Ross 1995; Walters and Murphy 2009), accounting for 70% of the variance (Imarisio et al. 2008), and the disease severity, implying that it is indeed the polyglutamines themselves that are the chief culprits. Some of the polyQ-containing huntingtin proteins can also aggregate to form inclusion bodies, and aggregation and neurodegeneration can in part be related (Chopra et al. 2007; Cowan et al. 2003; Michalik and Van Broeckhoven 2003; Nagai et al. 2003; Wang et al. 2005, 2009; Zhang et al. 2005a). Now the evidence for the involvement of poorly liganded iron in a wide variety of neurodegenerative diseases is overwhelming (e.g. Benarroch 2009; Bishop et al. 2010a; Brown 2009a; Friedman et al. 2007; Jellinger 1999; Ke and Qian 2003; Kell 2009a; Lee et al. 2006a; Perez and Franz 2010; Thompson et al. 2001; Youdim et al. 2004b; Zecca et al. 2004), and the question arises as to whether huntingtin (or, more likely, its degradation fragments), containing these polyQ tracts, can catalyse oxidative stress and/or hydroxyl formation directly. In an important paper, Firdaus et al. (Firdaus et al. 2006) show that that huntingtin inclusion bodies act as centres of oxidative stress and that partially purified inclusion bodies contain large amounts of oxidised proteins. Iron metabolism is deranged in gene knockdown models of Huntington’s (Henshall et al. 2009; Lumsden et al. 2007), and experiments with the iron chelator deferroxamine revealed that the oxidation, localisation and structural organisation of the inclusion bodies formed by mutant htt were indeed irondependent. They did not demonstrate the ability of these inclusion bodies to catalyse hydroxyl formation, but this could easily be shown using specific assays, such as the hydroxylation of suitable aromatics (e.g. Grootveld and Halliwell 1986; Halliwell and Gutteridge 2006; Thomas et al. 2009a). The iron chelator clioquinol is also protective (Nguyen et al. 2005).
Parkinson’s disease
Parkinson’s disease (PD) results primarily from the death of dopaminergic neurons in the substantia nigra part of the brain and is characterised in particular by the presence of intracytoplasmic inclusions from protein aggregates called Lewy bodies (LB) and at the physiological level by a variety of movement disorders (e.g. Singh et al. 2007). It is strongly linked with oxidative stress (e.g. Alam et al. 1997; Büeler 2009; Jenner 2003; Jenner and Olanow 1996; Kidd 2000; Mandel et al. 2003; Olivares et al. 2009; Seet et al. 2010; Van Laar and Berman 2009; Zhang et al. 1999). Again (e.g. Berg et al. 2001; Bharath et al. 2002; Buchanan et al. 2002; Bush 2000; Castellani et al. 2002; Double et al. 2000; Good et al. 1998; Gotz et al. 2004; Hirsch and Faucheux 1998; Jellinger 1999; Johnson 2000; Olanow and Arendash 1994; Ostrerova-Golts et al. 2000; Sofic et al. 1991; Wolozin and Golts 2002; Altamura and Muckenthaler 2009; Andersen 2004; Barapatre et al. 2010; Baudrexel et al. 2010; Becker 2010; Becker et al. 2010a, b; Berg 2006, 2007; Berg and Hochstrasser 2006; Berg et al. 2006, 2008; Brar et al. 2009; Brown 2009b; Crichton and Ward 2006; Fasano et al. 2006; Friedman et al. 2007, 2009; Ghosh et al. 2010; Hirsch 2009; Jellinger 2009; Jimenez-Del-Rio et al. 2010; Kaur and Andersen 2004; Lee and Andersen 2010; Levenson 2003; Mandemakers et al. 2007; Matusch et al. 2010; Oakley et al. 2007; Salvador 2010; Shi et al. 2010; Wayne Martin 2009; Wypijewska et al. 2010; Yeager and Coleman 2010; Zecca et al. 2004; Zhang et al. 2005, 2010b)—there is overwhelming evidence for the involvement of iron in this neurodegeneration. Why these dopaminergic neurons are especially susceptible is not entirely clear, although dopamine is capable of reacting with iron directly to form a toxic complex (Arreguin et al. 2009; Paris et al. 2005) that probably itself catalyses hydroxyl formation. Iron chelators inhibit these inimical processes (Gal et al. 2006; Perez et al. 2008; Xu et al. 2008; Youdim et al. 2004a, b; Zheng et al. 2005), including (Kaur et al. 2003; Reznichenko et al. 2010; Youdim 2003) in the case of a Parkinson-like disease induced (Blum et al. 2001; Choi et al. 2009; Gal et al. 2009; Yokoyama et al. 2008) by 1-methyl-4-phenyl-1,2,3,6-tetrahydro-pyridine (MPTP) (ChEBI 17963). The Lewy bodies contain lipid and a variety of proteins, including ubiquitin, neurofilament, various proteasomal elements and α-synuclein, which may be oxidatively modified (Double et al. 2008). Ferric iron may itself catalyse the formation of α-synuclein oligomers (Brown 2009a; Hillmer et al. 2009; Peng et al. 2010), and copper may also be involved (Wang et al. 2010c).
Melanins are polymers of polyphenols, especially of L-dopamine, although neuromelanin, which also contains oxidatively polymerised dopamine or noradrenaline with the possible involvement of cysteinylderivatives (Fedorow et al. 2005), is of special interest here. The substantia nigra also contains a substantial amount of the dark, insoluble polymeric pigment neuromelanin, and this has been shown directly to bind (and release) iron in high amounts (Double et al. 2003a, b; Fasano et al. 2006; Faucheux et al. 2003; Gerlach et al. 2003, 2008; Shamoto-Nagai et al. 2006), and thereby to produce an iron-loaded form that seems to serve as a reservoir catalysing (hydroxyl) radical formation and whose amounts correlate with PD. In this case, the in vitro experiments have been done, and incubation of human neuromelanin with iron in vitro stimulates oxidative tissue damage (Ben-Shachar et al. 1991; Double et al. 2003a; Gerlach et al. 2008).
Another dark pigment commonly observed in PD patients (and other neurodegenerative diseases) is lipofuscin (Double et al. 2008; Jung et al. 2007; Terman and Brunk 2004). Lipofuscin too consists of lipids and oxidised proteins, often crosslinked by oxidised lipid derivatives such as (E)-4-hydroxy-non-2-enal (ChEBI 58968) and can adsorb high contents of metals (including iron) of up to 2% by weight (Jung et al. 2007). It is typically formed in lysosomes from degrading mitochondria (Terman et al. 2006a), themselves rich in metalloproteins, and the inability of cells to degrade it means that (purportedly) non-dividing cells (such as brain tissue) simply accumulate it as cells age (Terman and Brunk 2004, 2006). This arguably explains why non- or rarely-dividing tissues such as brain tissues are particularly susceptible, though note the important and increasing evidence (e.g. Abrous et al. 2005; Christie and Cameron 2006; Demir et al. 2009; Fuchs and Gould 2000; Götz and Huttner 2005; Gould 2007; Imayoshi et al. 2009; Leuner et al. 2007; Lledo et al. 2006; Ohnuma and Harris 2003; Taupin 2007; Zhao et al. 2008) for considerable turnover—neurogenesis—in at least some regions of the brain. The autocatalytic nature of the process—lipofuscin loaded with iron catalyses more lipofuscin production—is especially dangerous and can (as one would predict) be inhibited using appropriate iron chelators (Persson et al. 2003). Iron chelation also assists neurogenesis (Nowicki et al. 2009).
Thus, as with Huntington’s, Parkinson’s disease is clearly characterised by all the hallmarks of hyperactive hydroxyl radical generation, leading to cell death and destruction, in this case mainly, it would seem (Chen et al. 2007; Chiueh et al. 2000; Ekshyyan and Aw 2004; Jellinger 2002; Jenner 2003; Jenner and Olanow 1996; Kermer et al. 2004; Levenson 2005; Loh et al. 2006; Mandel et al. 2005; Mattson 2006; Okouchi et al. 2007; Xu et al. 2008a; Yasuda and Mochizuki 2010), by apoptosis.
Gaucher’s disease
Gaucher’s disease is an autosomally recessive inborn error of metabolism due to deficiency of a lysosomal enzyme, glucocerebrosidase (GBA), resulting in the accumulation of glucocerebroside in large macrophages throughout the reticuloendothelial system, leading to various neuronopathies. Given that the lysosome is the site of most labile iron in the cell (Fakih et al. 2008; Gorria et al. 2008; Kurz et al. 2004, 2008a, b; Persson 2005; Tenopoulou et al. 2007; Terman et al. 2006b; Yu et al. 2003), it is also of interest that there is an increased frequency of mutations in the gene encoding GBA among patients with Parkinson’s disease (e.g. Aharon-Peretz et al. 2004; Clark et al. 2007; Gan-Or et al. 2008; Lwin et al. 2004), and vice versa (Sidransky et al. 2009). Iron dysregulation is also well established in Gaucher’s disease (Finch et al. 1986; Lee et al. 1967, 1977; Lorber 1960, 1970; Morgan et al. 1983; Schiano et al. 1993; Weisberger et al. 2004). This recognition of the role of iron dysregulation in Gaucher’s disease may offer novel therapeutic approaches.
Alzheimer’s disease
Alzheimer’s disease (AD), the commonest of the neurodegenerative disease of ageing, shares many similarities with Huntington’s and Parkinson’s diseases, not least the extensive evidence for the role of oxidative stress (e.g. Butterfield et al. 2007; Christen 2000; DiMauro and Schon 2008; Good et al. 1996; Milton 2004; Miranda et al. 2000; Moreira et al. 2009; Nunomura et al. 2001, 2006; Reddy and Beal 2008; Reddy et al. 2009; Rottkamp et al. 2000; Smith et al. 1996, 2000; Zhu et al. 2007) and of iron (e.g. Adlard and Bush 2006; Avramovich-Tirosh et al. 2007a; Becker et al. 2010b; Bishop et al. 2002; Blázquez et al. 2007; Bolognin et al. 2009b; Brar et al. 2009; Bush 2000, 2003, 2008; Casadesus et al. 2004; Castellani et al. 2007; Collingwood and Dobson 2006; Collingwood et al. 2008; Connor and Lee 2006; Ding et al. 2009; Doraiswamy and Finefrock 2004; Gerlach et al. 1994; Good et al. 1996; Hegde et al. 2009; Honda et al. 2004; Jellinger et al. 1990; Jellinger 2009; Kala et al. 1996; Lehmann et al. 2006; LeVine 1997; Lovell et al. 1998; Malecki and Connor 2002; Mandel et al. 2007; Markesbery 1997; Markesbery and Lovell 1998; Mascitelli et al. 2009; Olanow and Arendash 1994; Ong and Farooqui 2005; Quintana et al. 2006; Rival et al. 2009; Robson et al. 2004; Silvestri and Camaschella 2008; Smith et al. 1997a, 2010; Tabner et al. 2005; Thomas and Jankovic 2004; Thompson et al. 2001; Valko et al. 2005; Zatta et al. 2009; Zecca et al. 2004; Zheng et al. 2005). It is especially noteworthy that iron correlates with disease severity as measured by cognitive ability (Ding et al. 2009; Gómez Ravetti et al. 2010; Grossi et al. 2009; Lavados et al. 2008; Perez et al. 2010; Smith et al. 2010). AD is characterized by the loss of neurons in the cognitive centres of the brain and by the presence of two separate pathological lesions, extracellular β-amyloid (Aβ) plaques (e.g. Dong et al. 2003; Scott and Orvig 2009) and neurofibrillary tangles within neurons (Shcherbatykh and Carpenter 2007).
Again, there is evidence that insoluble polymers (fibrillary tangles) of proteins such as the β-amyloid can bind iron (Castellani et al. 2007; Dickens and Franz 2010; Exley 2006; Good et al. 1992; Jiang et al. 2009; Mancino et al. 2009; Rival et al. 2009; Sayre et al. 2000b; Smith et al. 2007) and thereby act in an autocatalytic manner to promote further radical production and oxidative stress. There is a also relationship between the ability to bind haem and neurotoxicity (Atamna 2009; Atamna et al. 2009), and (HFE) mutations that cause haemochromatosis increase the susceptibility to AD (Bartzokis et al. 2010; Candore et al. 2003; Combarros et al. 2003; Connor and Lee 2006; Haacke et al. 2005; Kauwe et al. 2010; Lleó et al. 2002; Moalem et al. 2000; Pulliam et al. 2003; Robson et al. 2004; Sampietro et al. 2001), especially when in the presence of the APOE4 allele (Pulliam et al. 2003). (HFE mutations also increase susceptibility to PD (Biasiotto et al. 2008; Dekker et al. 2003)—but cf. (Aamodt et al. 2007)—and to ALS (Ellervik et al. 2007)).
Importantly, iron chelators have been shown to ameliorate the development of fibril formation/neurodegeneration/dementia/AD (Amit et al. 2008; Avramovich-Tirosh et al. 2007a; Bandyopadhyay et al. 2006; Barnham et al. 2004; Biran et al. 2009; Bolognin et al. 2009a; Crapper McLachlan et al. 1991; Cuajungco et al. 2000; Faux et al. 2010; Finefrock et al. 2003; Liu et al. 2009b, c, 2010; Mancino et al. 2009; Mandel 2007, 2008; Reznichenko et al. 2006; Scott and Orvig 2009; Weinreb et al. 2009a; Zheng et al. 2005). Although this fact rarely appears in papers setting out therapeutic options for preventing (or at least ameliorating the progress of) AD, this already considerable literature suggests that trials using modern iron chelators would be of worth.
Amyotrophic lateral sclerosis (ALS, Lou Gehrig’s disease)
ALS is another neurodegenerative disease in which iron has been strongly implicated (and thus the same kinds of mechanism as described previously) (Bush 2000; Carri et al. 2003; Cozzolino et al. 2008; Goodall et al. 2008; Jeong et al. 2009; Kasarskis et al. 1995; Mattson 2004; Migliore et al. 2005; Molfino et al. 2009; Qureshi et al. 2008; Reynolds et al. 2007; Sadrzadeh and Saffari 2004; Sayre et al. 2000a; Scott and Orvig 2009; Spasojević et al. 2010; Sutedja et al. 2007; Wang et al. 2004). Again, the clear benefits of iron chelators in mouse models (Jeong et al. 2009; Petri et al. 2007) and elsewhere (Avramovich-Tirosh et al. 2007b; Bolognin et al. 2009a) would seem to merit trials in humans known to be at risk. Long-standing associations of some forms of ALS with mutations in genes coding for superoxide dismutase 1 (e.g. Dalle-Donne 2007; Pasinelli and Brown 2006; Rosen et al. 1993; Vucic and Kiernan 2009; Wijesekera and Leigh 2009) are consistent with this, and recent genetic associations (e.g. Kwiatkowski Jr. et al. 2009; Valdmanis et al. 2009; Vance et al. 2009) include one with optineurin (Maruyama et al. 2010) that together with other evidence implies an interaction with NF-κB (see later) leading to the downstream effects of apoptotic neuronal cell death.
Friedreich’s ataxia
Friedreich’s ataxia is another neurodegenerative disease caused by the insertion of a trinucleotide repeat (or occasionally a missense mutation) in the gene encoding a protein called frataxin (Adibhatla and Hatcher 2010; Babady et al. 2007; Becker and Richardson 2001; Bencze et al. 2006; Chakravarty 2003; González-Cabo et al. 2009; Palau 2001; Pandolfo 2009; Patel and Isaya 2001; Puccio 2009; Rötig et al. 2002; Santos et al. 2010; Sheftel et al. 2010). Driven not least by studies in bacteria and lower eukaryotes (Ayala-Castro et al. 2008; Becker and Richardson 2001; Bulteau et al. 2007; Cavadini et al. 2000; Cook et al. 2006; De Freitas et al. 2003; Foury 1997; Foury and Cazzalini 1997; Foury and Talibi 2001; González-Cabo et al. 2005; Irazusta et al. 2006, 2008; Knight et al. 1999; Pastore et al. 2007; Vázquez-Manrique et al. 2006; Wang and Craig 2008; cf. Seguin, et al. 2010), it is now understood that frataxin is a mitochondrial iron chaperone protein (Atkinson and Winge 2009; Ba et al. 2009; Babady et al. 2007; Bencze et al. 2007; Cavadini et al. 2002; Correia et al. 2008; Gakh et al. 2006; Haugen et al. 2010; Huang et al. 2008a; Huang et al. 2009; Karlberg et al. 2006; Kondapalli et al. 2008; Lill 2009; Lill and Muhlenhoff 2008; Lu and Cortopassi 2007; MacKenzie et al. 2008; Mandemakers et al. 2007; Marmolino and Acquaviva 2009; Napier et al. 2005; Napoli et al. 2006; O’Neill et al. 2005; Pandolfo 2009; Pandolfo and Pastore 2009; Pastore et al. 2007; Reddy 2008; Schagerlöf et al. 2008; Sparaco et al. 2009; Wilson 2006; Zanella et al. 2008) involved in the safe insertion of Fe(II) during the production of Fe–S centres in the mitochondrial respiratory chain (Anderson et al. 2005). The defect thus leads to the liberation of mitochondrial iron (Huang et al. 2009; Popescu et al. 2007; Richardson et al. 2009a). Fenton chemistry is strongly implicated (Park et al. 2002; Sturm et al. 2005), as the attenuation of H2O2 production (but not of superoxide) (Anderson et al. 2005) ameliorates the disease (Anderson et al. 2008). As would be expected, appropriate iron chelation may have therapeutic benefits (Boddaert et al. 2007; Campanella et al. 2009; Goncalves et al. 2008; Kakhlon et al. 2010; Lim et al. 2008; Marmolino and Acquaviva 2009; Santos et al. 2010; Schulz et al. 2009; Sohn et al. 2008). This very clear molecular picture underlines strongly the problems of iron release leading to cell death and destruction.
Multiple sclerosis
MS is an inflammatory demyelinating autoimmune disease affecting the CNS. While its underlying causes remain uncertain, and do not seem to have a significant genetic component (Baranzini et al. 2010; Oksenberg et al. 2008; Ramagopalan et al. 2008), ROS are certainly implicated (Adibhatla and Hatcher 2010), and antioxidant therapies may be useful (Mirshafiey and Mohsenzadegan 2009). It is at least plausible that part of the demyelination is caused by ROS and in particular hydroxyl radicals, and it is thus of much interest that iron dysregulation and/or deposition have indeed been implicated directly (Adams 1988; Bakshi et al. 2000, 2001, 2002, 2008; Bermel et al. 2005; Brass et al. 2006a, b; Ceccarelli et al. 2009, 2010; Chard and Miller 2009; Craelius et al. 1982; Drayer et al. 1987; Eissa et al. 2009; Exley et al. 2006; Filippi and Agosta 2009; Ge et al. 2007; Grimaud et al. 1995; Haacke et al. 2005, 2009, 2010; Hammond et al. 2008; He and Yablonskiy 2009; Khalil et al. 2009; LeVine and Chakrabarty 2004; Neema et al. 2007a, b, 2009a, b; Pirko et al. 2009; Sayre et al. 2005; Schenck and Zimmerman 2004; Simka and Rybak 2008; Singh and Zamboni 2009; Stankiewicz et al. 2007; Tjoa et al. 2005; Walter et al. 2009; Zamboni 2006; Zivadinov and Bakshi 2004; Zivadinov and Minagar 2009). While the extent of causality of the iron dysregulation in MS is not yet established, it is highly pertinent that iron chelators have been shown to suppress disease progression in experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (Bowern et al. 1984; Mitchell et al. 2007; Pedchenko and LeVine 1998). To say the least, the question of the involvement of iron in the human disease would seem to need pursuing with some urgency.
Age-related macular degeneration
While this review is intended to be much more circumscribed than the previous one (Kell 2009a), there are some pertinent diseases in which my understanding has improved significantly, and age-related macular degeneration is one such. Age-related macular degeneration (AMD) (Rattner and Nathans 2006) is recognised as the leading cause of blindness and visual disability in the elderly in developed countries (Fine 2005; Harvey 2003; Lotery and Trump 2007; Tielsch et al. 1995). Interestingly, despite the relatively modest impact of genome-wide association studies to date (Maher 2008; Moore et al. 2010), and notwithstanding their promise as their statistical power improves, 80% of the variation in susceptibility to AMD is said to be explainable (Collins 2010) (and see e.g. Chu et al. 2008) though cf. e.g. (Chen et al. 2010; Hadley et al. 2010; Katta et al. 2009; Neale et al. 2010; Patel et al. 2008; Sobrin et al. 2010) by just two genes concerned with inflammation combined with two environmental factors (smoking and obesity). The environmental factors can certainly be related to iron dysregulation. Indeed, as previously discussed, iron is significantly implicated in AMD (and other related diseases such as glaucoma) (Blasiak et al. 2009; Cai et al. 2000; Charkoudian et al. 2008; Chen et al. 2009; Chowers et al. 2006; Dentchev et al. 2005; Dunaief 2006; García-Castiñeiras 2010; Goralska et al. 2009a, b, c; Hahn et al. 2003, 2004; He et al. 2007; Loh et al. 2009; Richer et al. 2002; Wong et al. 2007), iron chelation may help to reverse the process (Voloboueva et al. 2007), and dietary and other antioxidants are also protective (see Tan et al. 2008; Wang et al. 2009).
Prion diseases: including transmissible spongiform encephalopathies and Creutzfeldt–Jakob disease
A particularly striking set of diseases, where the downstream events manifest as extreme cellular destruction leading to large areas of (brain) tissue disappearing completely, are represented by the prion diseases, in particular the TSEs including bovine spongiform encephalopathy (BSE) (e.g. Aguzzi et al. 2008a; Aguzzi and Heikenwälder 2006; Caughey and Baron 2006; Caughey and Lansbury 2003; Linden et al. 2008) and scrapie (e.g. Bulgin and Melson 2007; Hur et al. 2002; Roostaee et al. 2010; Wisniewski and Sigurdsson 2007), kuru (Gajdusek 2008; McLean 2008) and CJD (e.g. McLean 2008; Peden and Ironside 2004) (note too the co-location of amyloid plaques and spongiform degeneration in CJD, Ghoshal et al. 2009).
Now it is well known that prion diseases are caused in part (or at least accompanied by) the conversion of the prion protein PrP from its normal form PrPC to a conformationally distinct version PrPSc and that the latter can autocatalyse this conversion (Aguzzi and Heikenwälder 2006; Aguzzi et al. 2008b; Prusiner 1998, 2001; Tamgüney et al. 2008; Watts and Westaway 2007). However, PrPSc levels are in many cases poorly correlated with prion disease progression, suggesting that it is necessary but not sufficient; ‘something else’ is required (Caughey and Baron 2006). Equally, it is far from clear how a simple conformational change of a protein leads to the vacuolation or spongiosis (e.g. Aguzzi 2006; Aguzzi et al. 2007; Armstrong et al. 2001; Chung et al. 1999; Crozet et al. 2008; Diedrich et al. 1991; Foster et al. 2001; Julius et al. 2008; Kourie 2002; Mallucci et al. 2003; Mallucci et al. 2007; Miele et al. 2001; Sakudo and Ikuta 2009a, b; Westaway et al. 1994; Williams et al. 1997) (formation of holes) in brain tissue characteristic of late-stage prion disease (and indeed of Alzheimer’s (Erkinjuntti et al. 1996; Sakudo and Ikuta 2009a). The question then arises as to whether it is possible that PrPSc modifies iron metabolism in an unfavourable way (and/or vice versa), not least since prion proteins bear phylogenetic relationships to the ZIP family of metal ion transporters (Schmitt-Ulms et al. 2009). Building on earlier work relating prion biology with (especially) iron-induced oxidative stress (e.g. Bareggi et al. 2009; Barnham et al. 2006; Basu et al. 2007; Choi et al. 2006; Fernaeus et al. 2005a, b; Fernaeus and Land 2005; Iwamaru et al. 2008; Kim et al. 2000, 2001, 2007; Lee et al. 2007; Lehmann 2002; Milhavet and Lehmann 2002; Pamplona et al. 2008; Park et al. 2008; Petersen et al. 2005; Pollera et al. 2005; Rana et al. 2009; Stegmann and Grohmann 2003; Westergard et al. 2007; Wong et al. 2001), this is exactly what Singh and colleagues (Singh et al. 2009a, b, c, 2010a, b) have now demonstrated, which is that the ‘something else’ (Kell 2009a, b) is (as so often) an imbalance in iron metabolism. They start by showing (Singh et al. 2009c) that even normal PrPC mediates cellular iron uptake and transport and that mutant PrP forms alter cellular iron levels differentially.
PrPC binds iron tightly, and this helps effect its conversion to PrPSc (Singh et al. 2010a, b). Singh et al. (2009a) showed that PrPSc can sequester cellular iron in insoluble PrPSc–ferritin complexes, making it bio-unavailable, and that this leads to an upregulation of iron uptake via transferrin and its receptor, leading to an overall iron excess in brain tissue. This was true for both scrapie and sporadic Creutzfeldt–Jakob disease, and stresses that it is not only the total amount of Fe(II) and Fe(III) that matter but their speciation. Mice knocked out for PrP also show derangement in iron metabolism (Singh et al. 2009b), and given what we know already (Kell 2009a) this leads to many obvious predictions of other diseases (Altamura and Muckenthaler 2009; Granic et al. 2009; Reddy et al. 2009; Serra et al. 2009) to which they might be susceptible. It has not yet been shown that PrPSc–ferritin complexes catalyse OH• production directly, but if this is increased in diseased brains this continuous, autocatalytic spewing out of toxic OH• radicals would clearly be able to account for the massive damage observed. A systems biology approach by Hwang et al. (Hwang et al. 2009) also found many genes of iron metabolism modified in prion disease. There is little doubt that all these new findings about the role of iron (and other metals, e.g. Brown et al. 1997) open up many new inroads into understanding the important role of iron in prion diseases, and thereby novel therapeutic options involving both anti-oxidants and cell-permeable iron chelators. Some of these new ideas and findings are illustrated in Fig. 5.
Finally, here, it is worth pointing up the evidence for the role of iron and its metabolic dysregulation in the oxidative stress-mediated aetiology of scrapie (Choi et al. 2006; Fernaeus et al. 2005a; Fernaeus and Land 2005; Gudmundsdóttir et al. 2006; Kim et al. 2000, 2007; Pamplona et al. 2008; Singh et al. 2009a, 2010a, b; Wong et al. 2001a, b; Yun et al. 2006), a prion disease of sheep and goats (Bulgin and Melson 2007; Hur et al. 2002) and of CJD (Choi et al. 2006; Freixes et al. 2006; Petersen et al. 2005; Singh et al. 2009a, 2010a, b). As such, the use of geostatistics relating the variation of metals present in soils to prion disease prevalence is also a powerful tool here (e.g. Gudmundsdóttir et al. 2006; Imrie et al. 2009; Purdey 2000; Stevens et al. 2009).
Iron deposition and disease: cause or effect? The case of atherosclerosis
I have implied that the role of unliganded iron is causative of a variety of sequelae, but sometimes this is hard to infer as the networks in which iron is involved are multiple and complex (hence the need for a systems approach—see later), and often simple one-time snapshots of covariates do not permit the inference of causality. However, one effect of the reaction of hydroxyl radicals (their formation catalysed by unliganded iron) with proteins (or lipids) is to tend to make those proteins (or lipids) insoluble or fibrotic (Davies 2005; Fig. 6), and if one believes that this is causative, the presence of insoluble plaques should correlate with the presence of the iron that is seen to have caused their formation and thereby became entrapped (and may yet be further reactive). Because of its biomedical importance, considerable work has been performed on atherosclerotic lesions, which provide a very clear example.
Atherosclerosis is a progressive (and inflammatory) disease (Altman 2003; Binder et al. 2002; Blake and Ridker 2001; Duewell et al. 2010; Dwyer et al. 2004; Forrester 2002, 2004; Gieseg et al. 2009; Grainger 2007; Hansson 2001, 2005; Himmelfarb et al. 2002; Kibel et al. 2008; Kunsch and Medford 1999; Libby 2002; Libby et al. 2002; Madamanchi et al. 2005a, b; Mullenix et al. 2005; Nigro et al. 2006; Packard and Libby 2008; Paoletti et al. 2004; Popa et al. 2007; Rader and Daugherty 2008; Ridker et al.et al. 2004; Ross 1999; Schleicher and Friess 2007; Subramanian and Ferrante 2009; Sullivan 2009; Tan and Lip 2008; Tang et al. 2009; Taqueti et al. 2006; Tedgui and Mallat 2006; van Leuven et al. 2008; van Oostrom et al. 2004; Willerson and Ridker 2004; Young et al. 2002) characterized by the accumulation of both oxidised lipids and various fibrous elements in arteries, often as plaques (Lusis 2000; Stocker and Keaney 2004). Several lines of evidence point to the involvement of iron in these processes:
-
Both iron and oxidised lipids (de Valk and Marx 1999; Smith et al. 1992) are found in atherosclerotic lesions (Altamura and Muckenthaler 2009; Brewer 2007; Chau 2000; Fernandes de Godoy et al. 2007; Gajda et al. 2008; Halliwell 2009; Horwitz et al. 1998; Kazi et al. 2008; Lee et al. 1998; McRae et al. 2009; Rajendran et al. 2007; Ramakrishna et al. 2003; Roijers et al. 2005; Smith et al. 1992; Stadler et al. 2004; Stanley et al. 2006; Stocker and Keaney 2005; Sullivan 2009; Watt et al. 2006; Wolff et al. 2004; Yuan and Li 2008)
-
Iron depletion by dietary or other means delays the formation of such lesions (Ferrara and Taylor 2005; Halliwell 2009; Lee et al. 1999; Matthews et al. 1997; Ponraj et al. 1999; Ren et al. 2005).
-
There is a correlation between iron status and atherosclerosis (Ahluwalia et al. 2010; Chau 2000; Day et al. 2003; de Valk and Marx 1999; DePalma et al. 2010; Gozzelino et al. 2010; Howes et al. 2000; Kallianpur 2005; Kartikasari et al. 2009; Kiechl et al. 1997; Lapenna et al. 2007; Marx et al. 2008; McRae et al. 2009; Minqin et al. 2003; Ramakrishna et al. 2003; Ren et al. 2003; Salonen et al. 1992; Shah and Alam 2003; Sullivan 2007, 2009; You et al. 2003; You and Wang 2005; Zacharski et al. 2000; Zacharski and Gerhard 2003) (and zinc is protective only against subsequent calcification, Stadler et al. 2008)
-
Exogenous ferric iron is deleterious to endothelial function (Rooyakkers et al. 2002)
-
Iron chelation improves endothelial function (Altamura and Muckenthaler 2009; Duffy et al. 2001; Halliwell 2009; Ishizaka et al. 2005; Saito et al. 2005; Thomas et al. 2006).
-
Iron levels in plaques correlate with the amount of oxidised proteins therein (Stanley et al. 2006); indeed in one study (Stadler et al. 2004), the EPR-detectable iron (essentially Fe(III)) in atherosclerotic tissue was seventeen times greater than that in the equivalent healthy tissue. I redraw these data in Fig. 7.
It seems clear that the application of similar studies to other diseases in which plaque formation occurs will be of benefit. Indeed, the location of iron in plaques has also been observed, for instance, for Alzheimer’s (e.g. Collingwood et al. 2008; Connor et al. 1992; El Tannir El Tayara et al. 2006; Ghribi et al. 2006; Haacke et al. 2005; Lovell et al. 1998; Morgan et al. 2004; Quintana 2007; Quintana et al. 2006; Sayre et al. 2000b; Schrag et al. 2009), ALS (Leveugle et al. 1997), MS (Haacke et al. 2009) and Parkinson’s (see above).
Sepsis, septic shock and the systemic inflammatory response syndrome
Although prions lack nucleic acids, they are seen as infectious agents. Bacteria are more conventional infective agents, and bacterial sepsis provides a very interesting example in which iron release is the key to cell death and the multiple organ failure that normally precedes organismal death. This said, the processes leading to septic shock and ultimately death (Fig. 8) are independent of the presence of viable/culturable bacteria (as defined in Kaprelyants et al. 1993; Kell et al. 1998), i.e. in the apparent absence of infection (Goris 1990; Suntharalingam et al. 2006). This is because it is the products of bacteria, especially lipopolysaccharide (LPS), that cause the cells themselves to initiate the progressive processes of self-destruction. The sequelae are typically referred to as a systemic inflammatory response syndrome (SIRS) (e.g. Baue et al. 1998; Beal and Cerra 1994; Bolton 1996; Bone 1996; Davies and Hagen 1997; Gutteridge and Mitchell 1999; Hotchkiss and Karl 2003; Johnson and Mayers 2001; Keel and Trentz 2005; Leithead et al. 2009; Lolis and Bucala 2003; Martin et al. 2003; Nguyen et al. 2006; Protti and Singer 2006; Rittirsch et al. 2008; Ulloa et al. 2009).
It is by now well established that—while a ‘cytokine storm’ (Huang et al. 2005; Sriskandan and Altmann 2008; Stacey et al. 2009; Suntharalingam et al. 2006; Wang and Ma 2008; Woo et al. 2010) is an important part of the aetiology of sepsis/septic shock—these phenomena are associated with the hyperproduction of ROSs (Abdelrahman et al. 2005; Andrades et al. 2005; Bulger and Maier 2001; Cadenas and Cadenas 2002; Closa and Folch-Puy 2004; Crimi et al. 2006a, b; Goode and Webster 1993; Gutteridge and Mitchell 1999; Horn 1998; Javadi et al. 2005; Lagan et al. 2008; Lemineur et al. 2006; Mishra 2007; Protti and Singer 2006; Quinlan et al. 2001; Redl et al. 1993; Victor et al. 2004, 2005; Vlessis et al. 1995), and (local and circulating) free iron is raised in sepsis and related conditions (Duvigneau et al. 2008; Galley et al. 1996, 1997; Galley and Webster 1996; Ghio et al. 2003; Lagan et al. 2008), as is the iron siderophore-binding protein lipocalin-2 (Hattori et al. 2009).
The natural iron-chelating antioxidant melatonin has been found to be particularly effective in preventing septic shock (Carrillo-Vico et al. 2005; Escames et al. 2006; Reynolds et al. 2003), and a variety of suitable antioxidants have shown promise (Bekyarova et al. 2009; Cuzzocrea et al. 2001, 2004; Di Paola et al. 2006; Galley et al. 1997; Mishra 2007; Nathens et al. 2002; Sebai et al. 2009; Streck et al. 2008; Zapelini et al. 2008). Most importantly, antioxidants are especially effective when given in combination with iron chelators (Barichello et al. 2007; Ritter et al. 2004, 2006) (see also Bulucu et al. 2009; Cassol et al. 2009; Petronilho et al. 2009; Pinho et al. 2005; Teixeira et al. 2008; Valvassori et al. 2008). Erythropoietin is also protective (Abdelrahman et al. 2004; Cuzzocrea et al. 2006), as is the iron-binder lactoferrin (Tian et al. 2010) and as are anti-inflammatory statins (Durant et al. 2004; Gao et al. 2008; Kopterides and Falagas 2009). All of these observations make it very clear indeed that iron is intimately involved in the death following SIRS and sepsis, and—while not even mentioned in the guidelines (Dellinger et al. 2008)—that its effective chelation should be considered for research leading towards its inclusion as an important part of any therapy.
Bactericidal antibiotics
The classical test for if a bacterium is (or was) ‘alive’ is to analyze its ability to multiply, i.e. viability = culturability (Kell et al. 1998; Postgate 1967, 1976). Accordingly, bacterial antibiotics have long been classified into those that are bacteriostatic, i.e. stop individual cells growing but allow culturability to recover if the antibiotic is removed, and those that are bactericidal, i.e. that actually kill cells and thereby decrease the numbers of those that are culturable (capable of multiplication) even if the antibiotic is removed. Since this clearly implies that causing growth to cease is not of itself bactericidal, and anyway different antibiotics have a variety of different modes of action, it has always been a slight mystery as to what it is that makes a particular antibiotic bactericidal rather than merely bacteriostatic.
A very important paper (Kohanski et al. 2007) provides much of the answer. As one may suppose from the subject matter of this review, iron is again involved, specifically via the Fenton reaction in which poorly liganded ferrous ions reduce the comparatively harmless hydrogen peroxide to the deadly hydroxyl radical.
Based on a previous paper from the same group (Dwyer et al. 2007), Kohanski et al. (2007) (see also Davies et al. 2009; Dwyer et al. 2007, 2009; Imlay 2008; Kohanski et al. 2008; Liu et al.2009a; Wang and Zhao, 2009) reported that the three major classes of bactericidal antibiotics (exemplified by norfloxacin, ampicillin and kanamycin), regardless of drug–target interaction, utilize internal iron liberated from iron–sulphur clusters to promote Fenton-mediated hydroxyl radical formation in bacteria, that such hydroxyl radical generation contributes to the killing efficiency of these lethal drugs (probably via their known ability to damage DNA), and that in contrast antibiotics that are merely bacteriostatic do not cause the production of such hydroxyl radicals. The availability of multiple genetically marked strains allowed several predictions (such as the resistance to be expected in mutants that could not make iron-sulphur centres so effectively) to be tested successfully. Nitric oxide is correspondingly protective (Gusarov et al. 2009). This suggests, much as above, that there is a common mechanism of iron/hydroxyl-mediated cellular death underlying the action of all classes of bactericidal antibiotics and opens up many new possibilities for combination therapies based on promoting this kind of activity selectively (see Fig. 9). This said, how synergistic a combination is does depend on the exact speciation of the liganded iron; surprisingly, the iron chelator deferasirox promoted the antibacterial activity of ciprofloxacin against V. vulnificus (Neupane and Kim 2010), as did deferiprone with various azoles against Aspergillus fumigatus, while deferoxamine was antagonistic to azoles (Zarember et al. 2009). As with any complex system, the interactions can be highly dosedependent (Hegreness et al. 2008; Kohanski et al. 2010; Yeh et al. 2009).
Viruses
A typical viral infection involves the biding of virus to the cell surface followed by internalisation and other effects. Some viruses are especially virulent, and as well as inflammation cause the death of cells that they have invaded. It is obviously of interest to establish whether iron is involved in this kind of cell death (and if so whether iron chelation might contribute to a suitable therapy during the acute phase of an infection).
The above suggestions are certainly true of infections caused by influenza A (Visseren et al. 2002) (see also Akaike et al. 1996; Beck et al. 2004), herpes simplex virus (Gennero et al. 2010; Lamey and Biagioni 1995; Romeo et al. 2001), hepatitis C (Alavian and Tabatabaei 2009; Alla and Bonkovsky 2005; Ameli et al. 2008; Bassett 2007; Batts 2007; Bonkovsky 2002; Bonkovsky et al. 2006; Desai et al. 2008; Di Marco et al. 2008; Drakesmith and Prentice 2008; Fargion et al. 2002; Ferrara et al. 2009; Franchini et al. 2008; Fujita et al. 2007a, b; Fujita and Takei 2007; Girelli et al. 2009; Gürkan et al. 2005; Guyader et al. 2007; Hayashi and Yano 2002; Heathcote 2004; Kaito 2007; Kaito et al. 2006; Kato et al. 2001, 2007; Ko et al. 2007; Mahmoud et al. 2008; Mueller et al. 2006; Nahon et al. 2008; Nishina et al. 2008; Otogawa et al. 2008; Price and Kowdley 2009; Rigamonti et al. 2005; Sebastiani and Walker 2007; Sikorska et al. 2010; Sugimoto et al. 2009; Tanaka et al. 2007; Tanaka and Kiyosawa 2004; Trinder et al. 2008; Tung et al. 2003; Valenti et al. 2008; Venturini et al. 2010; Won et al. 2009) and HIV (Boelaert et al. 1996; Butensky James et al. 2009; de Monyé et al. 1999; Debebe et al. 2007; Drakesmith and Prentice 2008; Georgiou et al. 2000; Gordeuk et al. 2001, 2006; Kagu et al. 2007; Ketonen et al. 1996; McDermid et al. 2007; McDermid and Prentice 2006; McDermid et al. 2009; Meyer 2006; Northrop-Clewes 2008; Rawat et al. 2008; Savarino et al. 1999; Schreck et al. 1992; Szajerka and Jablecki 2007; Traoré and Meyer 2004, 2007; van Asbeck et al. 2001; Weinberg 2006; Ziegler et al. 2001).
Microbial, plant and animal toxins
Before considering chemical toxicants, it is worth noting that the natural world contains a very great many molecules that are toxic to cells and hence organisms. Although the mechanisms of cell death are rather rarely pursued, there is evidence for the involvement of iron in the sequelae of exposure to toxic doses of gentamicin (Petronilho et al. 2009), ochratoxin (Hasinoff et al. 1990; Omar et al. 1990; Størmer and Hoiby 1996) and ricin (Hassoun and Wang 1999; Kumar et al. 2003; Muldoon et al. 1996), as well as to certain snake venoms (Gutierrez et al. 2010).
Chemical toxicants: aniline, asbestos, dioxins, hydrazines, methyl mercury and paraquat
The most dangerous kinds of man-made toxic chemical agents, those that kill cells or organisms, exhibit a rich diversity of chemical structures and can bind to multiple cellular targets, to the extent that a unitary mode of action is not considered likely. (This said, the same was thought to be true for the mode of action of general anaesthetics or narcotics, and they turn out to have a comparatively small number of rather specific protein targets (Dobson et al. 2009; Dobson and Kell 2008; Franks 2006, 2008; Grasshoff et al. 2006)). However, this does not mean that multiple initial events do not at some point converge to a comparatively small number of crucial events of cell death (whether through necrosis or apoptosis, Vanden Berghe et al. 2009), equivalent to the ‘bow-tie’ model of cellular networks (see e.g. Csete and Doyle 2004; Ma’ayan 2009; Ma et al. 2004, 2007; Ma and Zeng 2003; Oda and Kitano 2006; Rodríguez-Caso et al. 2009; Wang and Chen 2010; Zhao et al. 2006, 2010) and (Fig. 10), and my purpose here is to highlight the evidence that iron-based mechanisms, largely based on Fenton chemistry, are involved in a number of these cases. A recent special issue on toxicants and neurodegeneration is also of interest here (Gupta and Milatovic 2009).
Acetaminophen
Acetaminophen (paracetamol in the United Kingdom) (ChEBI 46195) is a well-known analgesic that is nevertheless highly toxic at concentrations in excess of the therapeutic dose, as it is converted to the electrophilic N-acetyl-p-benzoquinone imine (NAPQI). If glutathione is in insufficient supply, the NAPQI cannot be conjugated harmlessly by glutathione and so it reacts covalently with proteins, especially mitochondrial proteins (Hinson et al. 2004; Jaeschke and Bajt 2006; Kon et al. 2004; Masubuchi et al. 2005; Reid et al. 2005), resulting in apoptotic cell death and organismal death via liver damage (e.g. Barker et al. 1977; Beckett et al. 1989; James et al. 2003; Martin-Murphy et al. 2010; Olaleye and Rocha 2008). One of the effects of mitochondrial damage is iron release, and it has long been known that iron chelators help prevent this toxicity (Sakaida et al. 1995; Schnellmann et al. 1999). Appropriate hydroxyl radical scavengers are also partly protective (Chandrasekaran et al. 2009), while lactoferrin is also protective, although not apparently only via its iron-binding capacity (Yin et al. 2010).
Aniline
As is well known, the adulteration with aniline of cooking oil in Spain showed that aniline so administered can be exceedingly toxic to humans (the ‘toxic oil syndrome’, Hill et al. 1987; Vázquez Roncero et al. 1983), especially in the lungs and spleen. It is now clear (Bomhard and Herbold 2005; Khan et al. 1995, 1999; Ma et al. 2008; Mellert et al. 2004; Wang et al. 2005, 2008, 2010) that iron is heavily involved in this via Fenton chemistry.
Asbestos
Many mutagens are carcinogens, typically by intercalating into DNA, so it was not a priori obvious why asbestos fibres—enormously larger than the diameter of DNA—should be so toxic. The mystery was solved when it was realised that asbestos does in fact contain and bind iron, that the sharp surface of the iron-loaded fibres can catalyse hydroxyl radical formation and that it is this that provides the toxic mechanism (Aljandali et al. 2001; Burrows and Muller 1998; Chao et al. 1996; Chen et al. 1996; Dai and Churg 2001; Fubini and Arean 1999; Gilmour et al. 1997; Governa et al. 1999; Gulumian and van Wyk 1987; Hardy and Aust 1995a, b; Hippeli and Elstner 1999; Jaurand 1997; Kamp et al. 1992; Kamp and Weitzman 1997, 1999; Kelleher et al. 2000; Levresse et al. 2000; Lund and Aust 1991, 1992; Mossman and Churg 1998; Mossman et al. 1987; Park and Aust 1998; Quinlan et al. 1994; Shatos et al. 1987; Shen et al. 1995; Weinberg 1989; Weitzman and Graceffa 1984; Xu et al. 1999; Azad et al. 2008; Bhattacharya et al. 2005; Daghino et al. 2006; Fantauzzi et al. 2010; Fattman et al. 2006; Gazzano et al. 2007; Ghio et al. 2004, 2009; Heintz et al. 2010; Kamp 2009; Knaapen et al. 2004; Liu et al. 2010; Nymark et al. 2008; Panduri et al. 2006; Poser et al. 2004; Quinlan et al. 2002; Shannahan et al.; Shukla et al. 2003a, b; Sørensen et al. 2007; Toyokuni 2009a, b; Upadhyay and Kamp 2003; Valavanidis et al. 2009).
Dioxins
Dioxins are formed typically via the thermal combustion of halogen-containing compounds, and a chief product, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, often referred to simply as ‘dioxin’ (ChEBI 28119), notoriously produced during the 1976 Seveso chemical plant explosion and as an impurity in Agent Orange (a roughly 1:1 mixture of the plant hormone (auxin—ChEBI 22676) analogues) 2,4-dichlorophenoxyacetic acid (2,4-D; ChEBI 28854) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T; ChEBI 27903), is both a cause of chloracne and is a hepato-carcinogen. It was found that mice low in iron are resistant to this hepatotoxicity however (Poland and Knutson 1982; Sweeny et al. 1979), strongly implicating iron. In addition, iron enhances the toxicity of dioxin (Al-Turk et al. 1988; Smith et al. 1998) while iron chelators reduce it (Al-Bayati and Stohs 1987; Wahba et al. 1990). While the exact details have still not been worked out, there is little doubt that iron is important in the toxicity of a wide variety of polyhalogenated aromatics (Smith and Chernova 2009; Smith and Elder 2010), including hexachlorobenzene (ChEBI 5692) (Franklin et al. 2005).
Hydrazines
Phenylhydrazine (ChEBI 27924) (like other hydrazines) is a reactive substance that is commonly used in toxicity studies. It is known (Agrawal et al. 2001; Atanasova et al. 2004; Ferrali et al. 1997a, b; Fraenkel et al. 2009; Karbownik et al. 2000; Kim et al. 2009; Latunde-Dada et al. 2004; Rokushima et al.2007a, b; Sharma and Haldar 2009; Yamamoto and Kawanishi 1992) to induce haemolytic anaemia and iron release and thence cytotoxicity. Melatonin can be protective (Sharma and Haldar 2009).
Methyl mercury
Originally highlighted via ‘Minamata disease’ (a prominent symptom of which, as with many neurological disease, is ataxia), methylmercury (ChEBI 30785) is a well-known neurotoxicant (Clarkson et al. 2003). Like most if not all other xenobiotics (Dobson et al. 2009a; Dobson and Kell 2008; Dobson et al. 2009b; Kell and Dobson 2009), it requires one or more carriers for cellular uptake—in this case, when complexed to L-cysteine, the ‘large amino acid’ transporter (Kanai and Endou 2003; Simmons-Willis et al. 2002; Yin et al. 2008). There is certainly evidence that exposure to this agent causes oxidative stress (Ali et al. 1992; Aschner et al. 2007; Atchison and Hare 1994; Franco et al. 2009; Johansson et al. 2007; Sanfeliu et al. 2003; Sarafian et al. 1994; Yee and Choi 1994, 1996; Yin et al. 2007) and raises cellular iron levels (Ilbäck et al. 2000) and that anti-oxidants (Daré et al. 2000; Kaur et al. 2006; Sarafian and Verity 1991; Shanker and Aschner 2003; Usuki et al. 2001; Watanabe et al. 2009; Yamashita et al. 2004; Zurich and Monnet-Tschudi 2009), metallothioneins (West et al. 2008) and/or iron chelators (Castoldi et al. 2001; LeBel et al. 1992; Sarafian and Verity 1991; Stohs and Bagchi 1995) can be protective. Taken together, this does imply strongly a role for iron-mediated cytotoxicity of the type highlighted herein in the pathogenesis of methylmercury poisoning.
Paraquat
Paraquat (N,N′-dimethyl-4,4′-bipyridinium dichloride; ChEBI 34905) is a well-known herbicide and toxicant, whose mode of action relies on the generation of superoxide radicals. As such, its toxicity may be expected to be exacerbated by iron, and this is the case (Andersen 2003; Ayaki et al. 2005; Fernandez et al. 2000; Peng et al. 2007, 2009). In addition, again as expected from the above (and consistent with its structural similarity to the Parkinson-inducing agent MPTP-1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, ChEBI 17963), it can induce Parkinson-like symptoms (Kaur et al. 2009; Miller et al. 2009; Ossowska et al. 2006; Peng et al. 2007, 2009). The protection by molecules—such as ferritin (Kaur et al. 2009) and polyphenols (Jimenez-Del-Rio et al. 2010)—capable of chelating iron harmlessly is thus of considerable interest.
Other toxicants in which iron release has been implicated as part of the mechanisms of their toxicity
The list of known and potential toxicants is limitless, and part of the role of this review is to alert domain experts to the likelihood that iron, and dysregulation in iron metabolism, may mediate the toxicity of such molecules (and thus that its chelation may be important, under both acute and chronic conditions, in assisting resolution of the toxicity). To this end, I note the role of iron-induced oxidative stress in the toxicity of anthracyclines (including adriamycin/doxorubicin ChEBI 28748) (Ceyssens et al. 2004; Kaiserová et al. 2007; Kotamraju et al. 2002, 2004; Luft 2006; Othman et al. 2008; Šimůnek et al. 2009; Thougaard et al. 2010; Xu et al. 2008), arsenic (De Vizcaya-Ruiz et al. 2009; Fujihara et al. 2009), beer brewed in cast iron pots (Kew and Asare 2007), bleomycin (ChEBI 3139) (Wu et al. 2004; Yang et al. 2010), C60 fullerenes (Usenko et al. 2008), carbon nanotubes (Murray et al. 2009) (and see Guo et al. 2007; Jurkschat et al. 2007), carbon tetrachloride (Mansour 2000; Ogeturk et al. 2004), organophosphates (Goel et al. 2006), sulphur mustard agents (Korkmaz et al. 2008; Korkmaz et al. 2006; Naghii 2002) and talc nanoparticles (Akhtar et al. 2010). Welding fumes are toxic and contain both iron and manganese (Flynn and Susi 2010; Sriram et al. 2010), and the toxicity of nano-iron itself has also been studied (Li et al. 2009).
Apoptosis or necrosis?
As is well known, cells can die by both necrosis and by a more regulated form, apoptosis. While this—nor ‘normal’ ageing (summarised e.g. by Bishop et al. 2010b; Finkel 2005; Franco et al. 2007; Gems 2009; Kenyon 2010; Kirkwood 2008; Muller et al. 2007; Vaupel 2010), in which various degradative processes including iron dysregulation can be implicated (e.g. Brewer 2010; Cass et al. 2007; Cook and Yu 1998; Facchini 2002; Galaris et al. 2008; Kell 2009a; Kujoth et al. 2005; Kurz et al. 2008b; Massie et al. 1983; Napoli et al. 2006; Pfefferbaum et al. 2009; Terman et al. 2010; Xu et al. 2010)—is not the focus of this review, it is reasonable to enquire as to what kind of evidence exists for the mechanism(s) of cell death contingent upon iron dysregulation. As it turns out, while there is a little evidence that iron overload can cause necrosis (Babatasi et al. 1996; Traoré and Meyer 2007; Vanden Berghe et al. 2009; Vanlangenakker et al. 2008), there is abundant evidence for the ability of iron to cause apoptosis (Kotamraju et al. 2004a, b; Kurz et al. 2007, 2008b; Vanden Berghe et al. 2009), and—depending presumably on the exact liganding of the iron (Kell 2009a)—this can be effected when iron is ostensibly depleted (e.g. Bergman et al. 2005; Bidle and Bender 2008; Brard et al. 2006; Buss et al. 2003; Fukuchi et al. 1994; Greene et al. 2002; Haq et al. 1995; Hileti et al. 1995; Jiang et al. 2002; Jin et al. 2007; Koc et al. 2006; Kovář et al. 1997, 2001; Leardi et al. 1998; Lee et al. 2006b, c; Pan et al. 2004; Simonart et al. 2000; Sun et al. 2009; Truksa et al. 2003; Zhang et al. 2003; Zhao et al. 2004) or in excess (e.g. Aljandali et al. 2001; Cozzi et al. 2003, 2010; Devireddy et al. 2005; Franke et al. 2010; Jacob et al. 1997, 2000; Javadi et al. 2004; Jímenez Del Río and Vélez-Pardo 2004; Kamp et al. 2002; Kawabata et al. 1997; Kermer et al. 2004; Kooncumchoo et al. 2006; Kruman et al. 1997; Kummer et al. 2008; Kurz et al. 2007, 2008b; Lakshmi Devi and Anuradha 2009; Lee et al. 2006d; Levenson 2005; Lu et al. 2008; Ma et al. 2009; Messer et al. 2009; Murakami et al. 2006; Natoli et al. 2009; Persson 2005; Polla et al. 2003; Schlawe et al. 2004; Shin and Kim 2009; Shukla et al. 2003; Tenopoulou et al. 2005; Upadhyay and Kamp 2003; Vélez-Pardo et al. 1997; Zhao et al. 1997).
Inflammation and NF-κB
As reviewed in the previous survey (Kell 2009a) and elsewhere (e.g. Shoelson and Goldfine 2009; Shoelson et al. 2007), inflammation lies at the heart of many of these processes, and pathways involving NF-κB (see e.g. http://www.nf-kb.org/) are especially core here (e.g. Ali and Mann 2004; Circu and Aw 2010; Granic et al. 2009; Karin 2008; Karin and Greten 2005; Li et al. 2005; Maeda and Omata 2008; Simmonds and Foxwell 2008; Zhang and Rigas 2006). Of particular relevance in ROS/iron metabolism are the facts that NF-κB is redox-sensitive (e.g. Aggarwal and Sung 2009; Baeuerle and Henkel 1994; Chandrasekaran and Taylor 2008; Cho et al. 1998; Dröge 2002; Fulda et al. 2010; Galaris et al. 2008; Gloire et al. 2006; Gloire and Piette 2009; Ha et al. 2010; Haddad et al. 2000; Hayashi et al. 1993; Janssen-Heininger et al. 2008; Lagan et al. 2008; Lukosz et al. 2010; Nishi et al. 2002; Phillips et al. 2010; Schreck et al. 1991, 1992; Schreck and Baeuerle 1994; Sen and Packer 1996; She et al. 2002; Surh et al. 2005; Surh and Na 2008; Thannickal and Fanburg 2000; Trachootham et al. 2008, 2009; Valko et al. 2007; Weinberg and Chandel 2009; Wondrak 2009; Xiong et al. 2003) and that there are links between NF-κB and NGAL/lcn2 (Bu et al. 2006; Cowland et al. 2003, 2006, 2008; Karlsen et al. 2010) and/or hepcidin (Sow et al. 2009; Tacchini et al. 2008). Specificity is likely to come, in part, via the frequency encoding of NF-κB activity (Ashall et al. 2009; Nelson et al. 2004).
Catalytic behaviour of polypeptides and proteins
I have mentioned several times the idea that different proteins can bind unliganded iron to catalyse the production of hydroxyl radical, and it is certainly the case that the degree of liganding can affect these kinds of activities (Kell 2009a). Here it is appropriate to highlight the abilities of polypeptides and even poly(aminoacids)—whether binding metals (or not)—to exhibit catalytic activities, e.g. poly(leucine) (Carrea et al. 2005; Porter et al. 1999), and how adding metals to polypeptides to make metalloenzymes can have huge effects on their catalytic activities (Pierron et al. 2008; Pordea and Ward 2008; Turner 2009). The recent development of orthogonal ribosomes capable of incorporating novel amino acids into polypeptides (Neumann et al. 2010) opens up considerable possibilities here.
Dietary and/or pharmacological treatment of iron-mediated toxicity
Pharmaceutical
Developing novel pharmaceutical agents is a costly enterprise, and many candidate drugs fall by the wayside during the development process (see e.g. Dobson et al. 2009b; Kola and Landis 2004; Leeson and Springthorpe 2007). As most recently reviewed (Agarwal 2010; Berdoukas et al. 2009; Bolognin et al. 2009a; Cappellini and Pattoneri 2009; Cappellini et al. 2010; Clifton et al. 2009; Hider et al. 2008; Kell 2009a; Kontoghiorghes et al. 2010; Kwiatkowski 2010; Mladĕnka et al. 2009; Pennell et al. 2010; Perez and Franz 2010; Richardson et al. 2009b and references therein), a number of (pharmacological drug) approved iron chelators do exist and thus clearly have the potential to prove beneficial in treating diseases and the sequelae of toxin exposure whose aetiology involves poorly liganded iron via the Fenton reaction. The benefits of administration of desferrioxamine in Alzheimer’s, for instance, have been known for nearly 20 years (Crapper McLachlan et al. 1991), benefits since observed with other iron chelators (e.g. Amit et al. 2008; Bolognin et al. 2009a; Bush 2008; Liu et al. 2009b, 2010b; Perez and Franz 2010; Smith et al. 2010; Zhang et al. 2009). The implications for this and other neurodegenerative diseases—and potentially for the more acute and even chronic effects of other toxins—are obvious. Those approved clinically include Deferoxamine (Desferal; ChEBI 31460), Deferiprone (Ferriprox; PubChem CID 2972) and Deferasirox (Exjade; ChEBI 49005) (Fig. 11). Note that—unlike the other two—Deferoxamine is not orally active and must be injected, and even then may fail to cross cellular membranes to exert its action appropriately (Warkentin et al. 2010).
Dietary or nutritional/nutraceutical
In addition, many organisms make so-called ‘secondary’ or natural products, whose functions are often unclear but that clearly have evolutionary benefits for the host (Hadacek et al. 2010; Kell et al. 1995). As I also reviewed (Kell 2009a), many iron-chelating natural products exist in (or may be added to, Pócsi et al. 2008) foodstuffs for which no full pharmaceutical regulatory controls are required and which are classed as nutritional substances. Many are polyphenols (ChEBI 26195) (Amic et al. 2007; Arts and Hollman 2005; Auclair et al. 2009; Cemeli et al. 2009; Cheynier 2005; Geronikaki and Gavalas 2006; Halliwell 2007; Jovanovic and Simic 2000; Kandaswami and Middleton 1994; Kiokias et al. 2008; Korkina and Afanas’ev 1997; Linseisen and Rohrmann 2008; Loke et al. 2009; Manach et al. 2004, 2005a, b; Pérez-Jiménez et al. 2010; Perron and Brumaghim 2009; Perron et al. 2008; Petti and Scully 2009; Pietta 2000; Rice-Evans and Packer 2003; Scalbert et al. 2005; Scalbert and Williamson 2000; Seifried et al. 2007; Slemmer et al. 2008; Spencer et al. 2008; Yadav and Bhatnagar 2007a, b, 2010), including the anthocyanins that proved chemoprotective (Butelli et al. 2008) in a mouse p53 cancer model. Since the previous review (Kell 2009a), I would draw particular attention to a comprehensive overview of the subject (Perron and Brumaghim 2009), as well as new reviews and papers on the chief polyphenol in green tea, (−)epigallocatechin-3-gallate (ChEBI 4806) (Adhami et al. 2009; Butt and Sultan 2009; Hsieh et al. 2009; Jimenez-Del-Rio et al. 2010; Johnson et al. 2010; Pandey and Gupta 2009; Paterniti et al. 2009; Ramesh et al. 2009; Reznichenko et al. 2010; Suh et al. 2009; Weinreb et al. 2009a), on curcumin (ChEBI 3962) (a constituent of turmeric) (Aggarwal and Harikumar 2009; Aggarwal and Sung 2009; Anand et al. 2010; Basile et al. 2009; Epstein et al. 2009; Hegde et al. 2009; Iqbal et al. 2009; Jiao et al. 2009; Jurenka 2009; Kalpravidh et al. 2010; Majumdar et al. 2009; Messner et al. 2009; Patil et al. 2009; Ravindran et al. 2009; Rivera-Espinoza and Muriel 2009; Thephinlap et al. 2009; Thomas et al. 2009; Tuntipopipat et al. 2009; Wang et al. 2009a), on eugenol (ChEBI 4917) (Nagababu et al. 2010) (and cf. Allen and Cornforth 2009), on nepetoidin caffeic acid esters (Maioli et al. 2010), on quercetin (ChEBI 16243) (Chobot 2010; Kitagawa et al. 2009; Krukoski et al. 2009; Lorrain et al. 2010; Park et al. 2010; Pavlica and Gebhardt 2010; Smirnova et al. 2009; Terao 2009; Vlachodimitropoulou et al. 2010) and on melatonin (CHEBI 16796) (Borah and Mohanakumar 2009; Korkmaz et al. 2008, 2009; Reiter et al. 2009; Sener et al. 2009; Sharma and Haldar 2009; Signorini et al. 2009). Many of the protective effects observed are likely due to the iron chelating, as well as directly antioxidative (redox) properties of these molecules (e.g. Ferrali et al. 1997a; Hague et al. 2006; Halliwell 2009; Halliwell et al. 2005; Hider et al. 2001; Končić et al. 2010; Lopes et al. 1999; Mandel et al. 2004; Mandel and Youdim 2004; Morel et al. 1994; Nijveldt et al. 2001; Rice-Evans et al. 1997; Rice-Evans and Packer 2003; Weinreb et al. 2007, 2009a; Yoshino and Murakami 1998). Other molecules that are not themselves directly antioxidant, but have iron-binding characteristics, may also be of benefit. This is certainly true of phytic acid (phytate; myo-inositol-1,2,3,4,5,6-hexakis phosphate; CHEBI 17401) (Aljandali et al. 2001; Allen and Cornforth 2009; Brune et al. 1989; Davidsson et al. 1994; Doria et al. 2009; Glahn et al. 2002; Graf and Eaton 1990, 1993; Graf et al. 1987; Grases et al. 2001; Hallberg et al. 1989; Han et al. 1994; Hanson et al. 2006; Hawkins et al. 1993; Kamp et al. 1995a, b, 2002; Kim et al. 2008; Kumar et al. 2010; Minihane and Rimbach 2002; Miyamoto et al. 2000; Panduri et al. 2006; Poser et al. 2004; Rao et al. 1991; Rimbach et al. 2008; Sandberg et al. 1999; Schlemmer et al. 2009; Shamsuddin 1995; Vucenik and Shamsuddin 2006; Xu et al. 2008).
Phlebotomy
In addition, as follows implicitly from Sullivan’s ‘iron hypothesis’ (Sullivan 1981, 2001, 2003, 2004) (and see Kell 2009a), one way to decrease the amount of iron in the body is to remove it by blood-letting or phlebotomy. While phlebotomy is a very traditional nostrum, often assumed or considered to have rather dubious or at best modest scientific support, there is in fact increasing literature implying its benefits in a variety of conditions (e.g. Aigner et al. 2008; Beutler 2007; Broedbaek et al. 2009; Brudevold et al. 2008; Busca et al. 2010; DePalma et al. 2007, 2010; Dereure et al. 2008; Desai et al. 2008; Dwyer et al. 2009; Equitani et al. 2008; Facchini et al. 2002; Fargion et al. 2002; Fernández-Real et al. 2002; Fujita et al. 2009; Fujita and Takei 2007; Hayashi et al. 2006; Hayashi and Yano 2002; Heathcote 2004; Horwitz and Rosenthal 1999; Hua et al. 2001; Kaito 2007; Kaito et al. 2006; Kato et al. 2001, 2007; Kom et al. 2006; Rajpathak et al. 2009; Sullivan 2009; Sumida et al. 2009; Tanaka et al. 2007, 2009; Toyokuni 2009b; Zacharski et al. 2008). Plausibly such benefits are due to its role in decreasing iron stores.
Discussion: toxicology as systems biology
“When sorrows come, they come not single spies but in battalions.” (Shakespeare, Hamlet.)
It is certainly the case that reasons for the co-occurrence of two or more observables are that one causes the other or they have the same, separate cause. Since I have argued strongly here for the essentiality of iron dysregulation in a variety of syndromes (and have omitted many others, such as non-alcoholic (Baptista-Gonzalez et al. 2008; Kirsch et al. 2006; Kowdley 2010) or alcoholic liver disease (Breitkopf et al. 2009; Lecube et al. 2009)), and indeed cancers of various kinds (see Kell 2009a), the implication is that iron dysregulation can manifest in multiple different ways and that these might co-occur. There is considerable and increasing evidence for that (see e.g. Adibhatla and Hatcher 2010; Baker et al. 2009; Beeri et al. 2008; Cole et al. 2007; Franco et al. 2009; Janson et al. 2004; Kell 2009a; Li and Hölscher 2007; Mascitelli et al. 2009; Neumann et al. 2008; Pandolfo 2009; Roriz-Filho et al. 2009; Schnaider Beeri et al. 2004; Starley et al. 2010; Utzschneider and Kowdley 2010; Verduzco and Nathan 2009; Xu et al. 2009), with the exact outcomes being determined by different levels of expression (see for instance http://www.proteinatlas.org/) of different proteins in different tissues.
In a similar vein, it is not news (Attwood et al. 2009; Evans 2008; Hull et al. 2008; Kostoff 2002) that the large floods of literature have led to something of a ‘balkanisation’ of the literature into increasingly specialised segments, and that this makes it hard to seek to bring together such disparate parts of the literature so as to draw out general truths (Dobson and Kell 2008; Kell 2009a), via the methods of inductive reasoning (Kell and Oliver 2004; King et al. 2004, 2009). As previously discussed, it is well established on theoretical grounds alone that the activity of any step in a biochemical network can influence the concentration of any variable (such as—fleetingly—the amount of hydroxyl radicals produced). Thus, while it is better to derive quantitative models (e.g. Coveney and Fowler 2005; Csikasz-Nágy 2009; Kell 2006a, b; Kell and Knowles 2006; Kell and Westerhoff 1986a, b; Milo et al. 2010; Mogilner et al. 2006; Noble 2002, 2006, 2010; Oates et al. 2009; Phillips and Milo 2009; Tomlin and Axelrod 2007), at the very minimum one should be looking qualitatively at those factors that should be increased or decreased in order to improve the organism’s ability to resist oxidative stress (Fig. 12).
Indeed, it is the very act of doing this that leads one to recognise these kinds of complex problems as problems of systems or network biology (Alon 2006; Klipp et al. 2005; Palsson 2006). What do I mean by this?
A systems biology view of toxicology: toxicology as systems biology
In the present case, I have adduced evidence that a range of distinct aetologies, which manifest as a variety of different syndromes or diseases, converge at some point on an autocatalytic series of reactions, involving imperfectly liganded iron, in the production of the very damaging hydroxyl radical, that by causing the destruction of appropriate cellular structures and proteins liberates further ‘free’ iron and other reactive oxygen species, thereby effecting positive feedback (also known as ‘vicious cycles’, Lin and Beal 2006; Proctor and Gray 2010; Proctor et al. 2007) that leads to the production of yet further damage. The implication of this is that to be more specific we need suitable network models of the biological systems of interest (Kell 2007), and of how toxicants of any kind can interact with their components.
Of course in the steady state, the amount of a substance is governed both by the processes that create it and those that decompose it (Fig. 13), and individual effects of particular substances can vary qualitatively depending on concentrations, as in hormesis (e.g. Calabrese 2008a, b, c; Calabrese and Baldwin 1997, 2001, 2003; Calabrese and Blain 2005, 2009; Cook and Calabrese 2006; Cox 2009; Gems and Partridge 2008; Gómez-Pinilla 2008; Hadacek et al. 2010; Hayes 2009; Hoffmann 2009; Mattson 2008a, b; Radak et al. 2008; Rattan 2004, 2008; Rattan and Demirovic 2009; Ristow and Zarse 2010; Sørensen et al. 2009), so any analysis should be a systems analysis. The simple fact that, for instance, iron chelators improve prospects in Alzheimer’s implies that there is a background level of natural repair (otherwise chelators would only slow the neurodegeneration and not reverse it) and that is not necessarily apparent from measuring only net effects. Hence, the need for accurate systems biology models.
The very fact that multiple reactions typically lead to the creation (and destruction) of a given metabolite (such as the hydroxyl radical) means that to minimise its concentration it is best to modify many reactions simultaneously. This exceptionally important point from systems biology should underpin any scientific and therapeutic programs.
Systems biology methods
Systems biology methods are typically classified into ‘forward’ and inverse’ (Fig. 14). For the purposes of this discussion, I shall concentrate on metabolic networks (in contrast to signalling networks they transfer both matter and information, Kell 2004). Metabolic and other networks are ‘graphs’ in that they consist of entities (‘nodes’, such as metabolites) that interact with each other via ‘edges’ (enzymatic reactions). There may of course be other interactions such those that do not involve substrates or products but are mediated via molecules (effectors) that modify the rates of reactions. The distinction between forward and inverse models is based on whether one starts with the parameters or variables of the system. In metabolic networks, the parameters are typically the starting concentrations of substances present and the kinetic constants (K m, K i, V max, etc.) of the enzymes. The variables are typically the concentrations of metabolic intermediates, and the metabolic fluxes that change over time until a steady state (if such exists) is attained. It is important to note that only the parameters control the system. In forward systems biology, we thus seek to create a model of the network based on its parameters, and this is done in four phases, the first two of which are qualitative and the second quantitative (Herrgård et al. 2008; Kell and Knowles 2006; Thiele and Palsson 2010).
Forward modelling
The first qualitative stage is one in which are listed all the reactions that are known to occur in the system or organism of interest; in the modern era, and especially for metabolic networks, these reaction lists are often derived in part from genomic annotations (Arakawa et al. 2006; Palsson 2006) with curation based on literature (‘bibliomic’) data (Duarte et al. 2007; Thiele and Palsson 2010). A second stage, again qualitative, adds known effectors (e.g. those involved in forming feedback loops), while the third and fourth stages—essentially amounting to molecular enzymology—include the known kinetic rate equations and the values of their parameters. Armed with such information, it is then possible to provide stochastic and ordinary differential equation models of the entire metabolic network of interest. In some cases, it is possible to make simplifying assumptions that require minimal knowledge of the kinetic parameters of the system and allow one to generate a time series of the system variables (Smallbone et al. 2007). An attractive feature of metabolism, for the purposes of modelling, is that, in contrast to signalling pathways, metabolism is subject to direct thermodynamic and (in particular) stoichiometric constraints (Palsson 2006). These constraints are extremely important in reducing the search space for the values of the kinetic parameters of the model, and indeed constrain significantly the states that the system can adopt (Kell 2004; Palsson 2006).
The modelling community has access to many tools, which are interoperable because they can be exchanged using community standards (Kell 2008) of which the most popular (Klipp et al. 2007) is SBML, the Systems Biology Markup Language (Hucka et al. 2003) (http://www.sbml.org/). Models are best made, and made available, in SBML. In addition, there is recognition that it is desirable to be clear about the meaning (semantics) of the elements in the model (Ananiadou et al. 2006; Attwood et al. 2009; Kell and Mendes 2008), since SBML puts no constraints on the names given to entities, and we have pioneered (Herrgård et al. 2008; Pettifer et al. 2009) the use of semantic annotation of all the entities, whether by reference to persistent databases or via database-independent means (e.g. the use of SMILES (Weininger 1988) and InChI (Coles et al. 2005; Stein et al. 2003) strings for small molecules). This is consistent with the MIRIAM proposals for describing the contents of a model accurately (Le Novère et al. 2005), and we have also used (Herrgård et al. 2008) the Systems Biology Ontology (http://www.ebi.ac.uk/sbo/) and the Gene Ontology (http://www.geneontology.org/) as especially pertinent and useful ontologies. Tools also exist for creating such networks using CellDesigner (http://www.celldesigner.org/) and SBMLMerge (Schulz et al. 2006) for merging submodels, including visualising them (via the Systems Biology Graphical Notation (Le Novère et al. 2009) (http://www.sbgn.org/) e.g. via Arcadia (Villéger et al. 2010) (http://www.sourceforge.net/projects/arcadiapathways/). Armed with such networks, it is possible to analyse their dynamics and other properties using tools/ modelling environments such as COPASI (Hoops et al. 2006) (http://www.copasi.org/) and to compare model and experimental data, conveniently stored using SBRML (Dada et al. 2010). Models and attendant metadata are best stored directly in databases, such as that set down in the B-Net database model (see Li et al. 2003; Mehrotra et al. 2010) as used in our yeast model (Herrgård et al. 2008) (http://www.comp-sys-bio.org/yeastnet).
Interrogating the model
Sensitivity analysis
Armed with a model, there are a number of important studies that are usefully performed. The first is the study of the contribution of individual steps to the control of the variables of interest. This is the domain of local and global sensitivity analysis (Saltelli et al. 2004, 2008), to which we have made significant contributions (e.g. Lüdtke et al. 2008; Sahle et al. 2008; Yue et al. 2006, 2007, 2008). Sensitivity analysis studies the quantitative contribution of individual steps to the overall control of a process. The ‘local’ version has been popularised in biochemistry as ‘metabolic control analysis’ (e.g. Fell 1996; Heinrich and Schuster 1996; Kell and Westerhoff 1986a), and, for instance COPASI (Hoops et al. 2006) calculates the control or sensitivity coefficients directly from a model. The results of these kinds of analyses are represented by a rank order of the most important reactions or parameters, which provides an important starting point for the combinatorial analysis of these complex networks. Formalisms such as Metabolic Control Analysis also explain why the modulation of individual steps singly rarely has much of an effect on an overall pathway (see e.g. Pérez et al. 2009a, b), reviewed by Gems (Gems and Doonan 2009), for a typical example.
Combinatorial analysis of parameter sensitivity
As indicated above, it is commonly the case in complex biochemical networks that while changes in individual parameters have little effect on the behaviour of biochemical pathways—indeed evolution is thought to have selected for robustness to individual parameter changes (Bornholdt and Sneppen 2000; Kitano 2007; Lehár et al. 2008; Morohashi et al. 2002; Stelling et al. 2004; Wagner 2005)—combinations of parameter changes can and do have major effects. A straightforward corollary of this is that in order to cause or inhibit the progression of diseases—including inflammatory diseases such as asthma (Biswas and Rahman 2009; Bowler 2004; Bowler and Crapo 2002; Boyer and Liu 2004; Chodaczek et al. 2007; Ciencewicki et al. 2008; Comhair and Erzurum 2010; Galli et al. 2008; Greene 1995; Han et al. 2001; Horváth et al. 1998; Jarvis et al. 2005; Kocyigit et al. 2004; Muñoz et al. 2009; Narula et al. 2007; Nemery 1990; Pae et al. 2008; Riedl and Nel 2008; Shore 2008; Vlašić et al. 2009)—it is necessary to affect multiple steps simultaneously. The problem is to find which ones, and that is a combinatorial problem that scales poorly. It is exactly here where the ability to interrogate a suitable model of the pathway comes to the fore, since computations to determine which combinations of parameters to affect can be done enormously more rapidly than can ‘wet’ experiments. To be explicit, if manipulating (let us say knocking out) just three genes out of 1,000 is necessary to secure a desired effect (as is often the case) and the question is to choose which three, the answer involves recognizing that this is a combinatorial optimization problem involving ca 100 million combinations—a trivial number computationally given a model and an easy number (i.e. 3) to work with post hoc, but an implausible number to interrogate with present experimental methods. See (Park et al. 2007, 2008) for a nice example of the approach.
At all events, the results of running a systems biology model in the forward direction are the time-varying concentrations of variables that result from the network topology and parameters of the system, and these can then be compared with measured variables to see how accurate the model is.
Of course it is typically the case (i) that the variables predicted by the model differ from those that one has measured, and (ii) one has only measurements of the variables and not the parameters anyway. Since it is the parameters that determine the system behaviour, it is then necessary to try and estimate or infer the parameters of the systems from measurements of the variables, the domain of inverse modelling (Liu and Wang 2008; Mendes and Kell 1996) or system identification (Bongard and Lipson 2005; Ljung 1987).
Inverse modelling
Finding the sets of parameters that can reflect a set (or sets) of variables is a much harder problem. If there are just 20 parameters, each of which can take just 10 values, there are at once 1020 combinations, although there may be multiple combinations that give a reasonable solution (the problem or system is then said to be under-determined). At all events, finding these combinations requires methods that can effectively search these combinatorial search spaces to find good solutions (they are not provably optimal solutions as these kinds of problems are NP-hard, Garey and Johnson 1979).
There are many kinds of approaches to attacking these kinds of combinatorial optimisation problems, and I give two examples. The first uses evolutionary methods to modify the search for these parameters, while the second is Bayesian in character.
Evolutionary computing methods (e.g. Bäck et al. 1997; Goldberg 1989, 2002) use ideas from Darwinian evolution to select and modify parameters to achieve a desired objective (or objectives—this is known as multi-objective optimisation, Handl et al. 2007). In an evolutionary algorithm, one selects a population of models encoding a different candidate set of parameters, assesses their performance, rejects those that perform poorly and modifies the parameters of the better-performing models using methods akin to recombination and mutation to create a second generation population and so on. Finally, one finds a candidate set of solutions that perform well and can indeed interrogate these solutions to determine the parameters that differ most frequently in the selected solutions from those in the starting pathway. These kinds of methods are recognised as highly effective (Banga 2008; Banga and Balsa-Canto 2008), and they have proved so for both modelling (Dada and Mendes 2009; Mendes and Kell 1998; Moles et al. 2003; Rodriguez-Fernandez et al. 2006) and other optimisation problems (e.g. Broadhurst et al. 1997; Kell et al. 2001; Knight et al. 2009; O’Hagan et al. 2005, 2007; Platt et al. 2009; Rowe et al. 2010). A related and novel approach that we have developed (Wilkinson et al. 2008) (and see Brown et al. 2010) also evolves estimates of parameters, but in a somewhat different way. Evolutionary computing methods are amenable to (‘coarse-grained’) parallelisation that allows them to run on many (hundreds or even thousands of) processors simultaneously, and we have used this approach with great effectiveness (Wedge and Kell 2008; Wedge et al. 2009). Overall, the result of this kind of approach is the effective determination of the multiple parameters that are required to explain the system variables.
A conceptually quite different approach, which may of course be expected to lead to the same result (and which in consequence strengthens further any conclusions from e.g. an evolutionary computing approach), is an approach based on Bayesian inference (Jayawardhana et al. 2008; Liebermeister and Klipp 2006; Vyshemirsky and Girolami 2008) and known as Markov Chain Monte Carlo or MCMC. In this approach (which we do not rehearse in detail as those details have recently been published by us, Jayawardhana et al. 2008), we have knowledge of the variables of interest and sample candidate models that can explain those variables using various estimates of their parameters from a distribution. The MCMC algorithm then slowly builds up the likeliest values of the parameters and their possible distribution given the demonstrable uncertainty.
Detecting the modes or sites of action of a toxin
Clearly, if we have measurements of the variables of a system before and after the addition of a toxin, we can estimate the parameters in the two cases and see which have most plausibly changed by applying these inferencing methods to the two cases. This automatically gives the mode(s) or site(s) of action of the intervention, whether of a toxic inhibitor or by any other means. In other words these methods can detect the modes or sites of action of anything that causes a system to change its state. Taking the Bayesian approach (see also e.g. Nidhi et al. 2006; O’Hagan 2006; Vyshemirsky and Girolami 2008; Wilkinson 2007), in the work described above (Jayawardhana et al. 2008) we ‘calibrated’ our approach using purely simulated data in which we knew exactly which parameters had been changed (and in which direction) and showed that we could detect this accurately. We then applied our approach to an experimental system of metabolic engineering in Lactococcus lactis (Hoefnagel et al. 2002) where the investigators had manipulated the activity of a specific enzyme to achieve a desired metabolic effect. Our approach detected both the enzyme whose activity had been modified and suggested the site of another significant pleiotropic effect (these always occur, Featherstone and Broadie 2002). Thus, the power of this general approach is that it is free of specific hypotheses and thus guides us to the hypotheses that can best explain the data. These are classic data mining methods (e.g. Breiman 2001; Kell and King 2000; Kell and Oliver 2004; Wu et al. 2008) that allow us to discover mechanisms of action that are unknown and/or have not previously been contemplated. Note, however, that such methods need careful handling from a statistical point of view (e.g. Altman and Royston 2000; Broadhurst and Kell 2006; Hastie et al. 2001; Ioannidis 2005, 2008). A variety of other network inferencing methods also exist (e.g. Lorenz et al. 2009; Managbanag et al. 2008; Stolovitzky and Califano 2006; Stolovitzky et al. 2009).
Why most toxins will have multiple sites of action
A variety of means are available for testing these mechanisms further, including the use of modulation of the expression of constituent parts of the proposed pathways by genetic means (e.g. Jones et al. 2007), and in particular the use of appropriate iron chelators and antioxidants, alone or (preferably) in combination. The combinatorial aspect is important, since there are theoretical grounds, as well as abundant evidence, to the effect that several steps in a network must be modulated simultaneously to exhibit substantial effects (see e.g. Ágoston et al. 2005; Ambesi-Impiombato and di Bernardo 2006; Bender et al. 2006; Borisy et al. 2003; Csermely et al. 2005; Dancey and Chen 2006; Fan et al. 2003, 2006; Haggarty et al. 2003; Hopkins et al. 2006; Kaelin 2005; Keith et al. 2005; Kostoff and Delafuente 2006; Millan 2006; Perlstein et al. 2006; Sharom et al. 2004; Shiomi et al. 1999; Wermuth 2004) (Brötz-Oesterhelt and Brunner 2008; Chong and Sullivan 2007; Costanzo et al. 2010; Gerber et al. 2007; Goltsov et al. 2009; Hegreness et al. 2008; Huang et al. 2008b; Jia et al. 2009; Koufaki et al. 2009; Kuhn et al. 2010; Lee et al. 2007; Lehár et al. 2007, 2008a, b; Li et al. 2007; Liby et al. 2007; Morphy and Rankovic 2007; Pachauri et al. 2009; Reznichenko et al. 2010; Sarkar and Li 2009; Sarkar et al. 2009; Sawyers 2007; Schrattenholz and Šoškić 2008; Silver et al. 2009; Silver 2007; Sporn et al. 2007; Stommel et al. 2007; Suchy et al. 2009; Ulrich-Merzenich et al. 2009; Watterson et al. 2008; Weinreb et al. 2009b; Whitehurst et al. 2007; Wong et al. 2008; Yeh et al. 2006, 2009; Ying et al. 2008; Zamin et al. 2009; Zhang 2006; Zhang et al. 2010; Zimmermann et al. 2007). The same holds true for biotechnology (Kell and Westerhoff 1986a), in which typically we seek to maximise fluxes (Park et al. 2007, 2008). Of course some molecules—both nutrients/ natural products and pharmaceutical agents—can act as both antioxidants and iron chelators; these are likely to be especially useful in minimising the damage from toxicants of many kinds.
Concluding remarks
Science consists of both analysis and synthesis, and thus a substantial amount of Science involves finding regularities (‘Laws’) that can be extracted from observables, even when some of those observables or their presumed causes seem to have little in common (Kell and Oliver 2004; Oldroyd 1986; Westerhoff and Kell 2007). Thus, there are many kinds of toxins that can cause death or injury, but it is not necessarily obvious what they share in terms of their modes of action. What I have therefore sought to do here is to bring together the evidence for the autocatalytic involvement of poorly liganded iron (leading, inter alia, to hydroxyl radical formation) in a considerable number of these. Certainly the exact molecular mechanisms, cascades and networks involved in each case depend on many other factors, and appropriate systems biology methods can tell us what they are, but I would argue that the extensive evidence for iron’s involvement that I cite here and elsewhere (Kell 2009a) is very hard to ignore. Hence, the view that iron is the nexus.
Abbreviations
- Aβ:
-
β-amyloid
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis (Lou Gehrig’s disease)
- BSE:
-
Bovine spongiform encephalopathy
- CJD:
-
Creutzfeldt-Jakob disease
- CNS:
-
Central nervous system
- EAE:
-
Experimental autoimmune encephalomyelitis
- GBA:
-
Glucocerebrosidase
- HD:
-
Huntington’s disease
- Htt:
-
Huntingtin protein
- MCMC:
-
Markov chain Monte Carlo
- MIRIAM:
-
Minimum information requested in the annotation of biochemical models
- MPTP:
-
1-methyl-4-phenyl-1,2,3,6-tetrahydro-pyridine
- MS:
-
Multiple sclerosis
- PD:
-
Parkinson’s disease
- RNS(s):
-
Reactive nitrogen species
- ROS(s):
-
Reactive oxygen species
- SBML:
-
Systems biology markup language
- SBRML:
-
Systems biology results markup language
- SIRS:
-
Systemic inflammatory response syndrome
- TCDD, ‘dioxin’:
-
2,3,7,8-tetrachlorodibenzo-p-dioxin
- TSEs:
-
Transmissible spongiform encephalopathies
- 2,4-D:
-
2,4-dichlorophenoxyacetic acid
- 2,4,5-T:
-
2,4,5-trichlorophenoxyacetic acid
References
Aamodt AH, Stovner LJ, Thorstensen K, Lydersen S, White LR, Aasly JO (2007) Prevalence of haemochromatosis gene mutations in Parkinson’s disease. J Neurol Neurosurg Psychiatry 78(3):315–317
Abdelrahman M, Mazzon E, Bauer M, Bauer I, Delbosc S, Cristol JP et al (2005) Inhibitors of NADPH oxidase reduce the organ injury in hemorrhagic shock. Shock 23(2):107–114
Abdelrahman M, Sharples EJ, McDonald MC, Collin M, Patel NS, Yaqoob MM et al (2004) Erythropoietin attenuates the tissue injury associated with hemorrhagic shock and myocardial ischemia. Shock 22(1):63–69
Abrous DN, Koehl M, Le Moal M (2005) Adult neurogenesis: from precursors to network and physiology. Physiol Rev 85(2):523–569
Adamkiewicz TV, Abboud MR, Paley C, Olivieri N, Kirby-Allen M, Vichinsky E et al (2009) Serum ferritin level changes in children with sickle cell disease on chronic blood transfusion are nonlinear and are associated with iron load and liver injury. Blood 114(21):4632–4638
Adams CWM (1988) Perivascular iron deposition and other vascular damage in multiple sclerosis. J Neurol Neurosurg Psychiatry 51(2):260–265
Adams RJ (2007) Big strokes in small persons. Arch Neurol 64(11):1567–1574
Adhami VM, Siddiqui IA, Sarfaraz S, Khwaja SI, Hafeez BB, Ahmad N et al (2009) Effective prostate cancer chemopreventive intervention with green tea polyphenols in the TRAMP model depends on the stage of the disease. Clin Cancer Res 15(6):1947–1953
Adibhatla RM, Hatcher JF (2010) Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal 12:125–169
Adlard PA, Bush AI (2006) Metals and Alzheimer’s disease. J Alzheimers Dis 10(2–3):145–163
Agarwal MB (2010) Deferasirox: oral, once daily iron chelator–an expert opinion. Indian J Pediatr 77(2):185–191
Agarwal R, Vasavada N, Sachs NG, Chase S (2004) Oxidative stress and renal injury with intravenous iron in patients with chronic kidney disease. Kidney Int 65(6):2279–2289
Aggarwal BB, Harikumar KB (2009) Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int J Biochem Cell Biol 41(1):40–59
Aggarwal BB, Sung B (2009) Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci 30(2):85–94
Ágoston V, Csermely P, Pongor S (2005) Multiple weak hits confuse complex systems: a transcriptional regulatory network as an example. Phys Rev E 71(5):051909
Agrawal R, Sharma PK, Rao GS (2001) Release of iron from ferritin by metabolites of benzene and superoxide radical generating agents. Toxicology 168(3):223–230
Aguzzi A (2006) Prion diseases of humans and farm animals: epidemiology, genetics, and pathogenesis. J Neurochem 97(6):1726–1739
Aguzzi A, Heikenwälder M (2006) Pathogenesis of prion diseases: current status and future outlook. Nat Rev Microbiol 4(10):765–775
Aguzzi A, Heikenwalder M, Polymenidou M (2007) Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol 8(7):552–561
Aguzzi A, Baumann F, Bremer J (2008a) The prion’s elusive reason for being. Annu Rev Neurosci 31:439–477
Aguzzi A, Sigurdson C, Heikenwaelder M (2008b) Molecular mechanisms of prion pathogenesis. Annu Rev Pathol 3:11–40
Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R (2004) Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 351(19):1972–1977
Ahluwalia N, Genoux A, Ferrieres J, Perret B, Carayol M, Drouet L et al (2010) Iron status is associated with carotid atherosclerotic plaques in middle-aged adults. J Nutr 140(4):812–816
Aigner E, Theurl I, Theurl M, Lederer D, Haufe H, Dietze O et al (2008) Pathways underlying iron accumulation in human nonalcoholic fatty liver disease. Am J Clin Nutr 87(5):1374–1383
Akaike T, Noguchi Y, Ijiri S, Setoguchi K, Suga M, Zheng YM et al (1996) Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proc Natl Acad Sci USA 93(6):2448–2453
Akhtar MJ, Kumar S, Murthy RC, Ashquin M, Khan MI, Patil G et al. (2010) The primary role of iron-mediated lipid peroxidation in the differential cytotoxicity caused by two varieties of talc nanoparticles on A(549) cells and lipid peroxidation inhibitory effect exerted by ascorbic acid. Toxicol In Vitro
Al-Bayati ZA, Stohs SJ (1987) The role of iron in 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced lipid peroxidation by rat liver microsomes. Toxicol Lett 38(1–2):115–121
Al-Turk WA, Shara MA, Mohammadpour H, Stohs SJ (1988) Dietary iron and 2,3,7,8-tetrachlorodibenzo-p-dioxin induced alterations in hepatic lipid peroxidation, glutathione content and body weight. Drug Chem Toxicol 11(1):55–70
Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD et al (1997) Oxidative DNA damage in the parkinsonian brain: An apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem 69(3):1196–1203
Alavian SM, Tabatabaei SV (2009) Therapy of hepatitis C in thalassemia: the influence of iron on achieving sustained viral response. Ann Hematol
Ali S, Mann DA (2004) Signal transduction via the NF-kappaB pathway: a targeted treatment modality for infection, inflammation and repair. Cell Biochem Funct 22(2):67–79
Ali SF, LeBel CP, Bondy SC (1992) Reactive oxygen species formation as a biomarker of methylmercury and trimethyltin neurotoxicity. Neurotoxicology 13:637–648
Alissa EM, Ahmed WH, Al-Ama N, Ferns GA (2007) Relationship between indices of iron status and coronary risk factors including diabetes and the metabolic syndrome in Saudi subjects without overt coronary disease. J Trace Elem Med Biol 21(4):242–254
Aljandali A, Pollack H, Yeldandi A, Li Y, Weitzman SA, Kamp DW (2001) Asbestos causes apoptosis in alveolar epithelial cells: role of iron-induced free radicals. J Lab Clin Med 137(5):330–339
Alla V, Bonkovsky HL (2005) Iron in nonhemochromatotic liver disorders. Semin Liver Dis 25(4):461–472
Allen K, Cornforth D (2009) Effect of chelating agents and spice-derived antioxidants on myoglobin oxidation in a lipid-free model system. J Food Sci 74(5):C375–379
Alon U (2006) An introduction to systems biology: design principles of biological circuits. Chapman and Hall/CRC, London
Altamura C, Squitti R, Pasqualetti P, Gaudino C, Palazzo P, Tibuzzi F et al (2009) Ceruloplasmin/Transferrin system is related to clinical status in acute stroke. Stroke 40(4):1282–1288
Altamura S, Muckenthaler MU (2009) Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. J Alzheimers Dis 16(4):879–895
Altman DG, Royston P (2000) What do we mean by validating a prognostic model? Stat Med 19(4):453–473
Altman R (2003) Risk factors in coronary atherosclerosis athero-inflammation: the meeting point. Thromb J 1(1):4
Ambesi-Impiombato A, di Bernardo D (2006) Computational biology and drug discovery: from single-target to network drugs. Curr Bioinform 1(1):3–13
Ameli M, Besharati S, Nemati K, Zamani F (2008) Relationship between elevated liver enzyme with iron overload and viral hepatitis in thalassemia major patients in Northern Iran. Saudi Med J 29(11):1611–1615
Amic D, Davidovic-Amic D, Beslo D, Rastija V, Lucic B, Trinajstic N (2007) SAR and QSAR of the antioxidant activity of flavonoids. Curr Med Chem 14(7):827–845
Amit T, Avramovich-Tirosh Y, Youdim MB, Mandel S (2008) Targeting multiple Alzheimer’s disease etiologies with multimodal neuroprotective and neurorestorative iron chelators. FASEB J 22(5):1296–1305
Anand P, Nair HB, Sung B, Kunnumakkara AB, Yadav VR, Tekmal RR et al (2010) Design of curcumin-loaded PLGA nanoparticles formulation with enhanced cellular uptake, and increased bioactivity in vitro and superior bioavailability in vivo. Biochem Pharmacol 79(3):330–338
Ananiadou S, Kell DB, Tsujii J-i (2006) Text mining and its potential applications in systems biology. Trends Biotechnol 24:571–579
Andersen JK (2003) Paraquat and iron exposure as possible synergistic environmental risk factors in Parkinson’s disease. Neurotox Res 5(5):307–313
Andersen JK (2004) Iron dysregulation and Parkinson’s disease. J Alzheimers Dis 6(6 Suppl):S47–S52
Anderson PR, Kirby K, Hilliker AJ, Phillips JP (2005) RNAi-mediated suppression of the mitochondrial iron chaperone, frataxin, in Drosophila. Hum Mol Genet 14(22):3397–3405
Anderson PR, Kirby K, Orr WC, Hilliker AJ, Phillips JP (2008) Hydrogen peroxide scavenging rescues frataxin deficiency in a Drosophila model of Friedreich’s ataxia. Proc Natl Acad Sci USA 105(2):611–616
Andrades M, Ritter C, Moreira JC, Dal-Pizzol F (2005) Oxidative parameters differences during non-lethal and lethal sepsis development. J Surg Res 125(1):68–72
Andreini C, Bertini I, Cavallaro G, Holliday GL, Thornton JM (2008) Metal ions in biological catalysis: from enzyme databases to general principles. J Biol Inorg Chem 13(8):1205–1218
Andreini C, Bertini I, Cavallaro G, Najmanovich RJ, Thornton JM (2009) Structural analysis of metal sites in proteins: non-heme iron sites as a case study. J Mol Biol 388(2):356–380
Andrews SC, Robinson AK, Rodriguez-Quinones F (2003) Bacterial iron homeostasis. FEMS Microbiol Rev 27(2–3):215–237
Arakawa K, Yamada Y, Shinoda K, Nakayama Y, Tomita M (2006) GEM System: automatic prototyping of cell-wide metabolic pathway models from genomes. BMC Bioinform 7:168
Armand P, Kim HT, Cutler CS, Ho VT, Koreth J, Alyea EP et al (2007) Prognostic impact of elevated pretransplantation serum ferritin in patients undergoing myeloablative stem cell transplantation. Blood 109(10):4586–4588
Armengou A, Davalos A (2002) A review of the state of research into the role of iron in stroke. J Nutr Health Aging 6(3):207–208
Armstrong RA, Lantos PL, Cairns NJ (2001) Spatial correlations between the vacuolation, prion protein deposits, and surviving neurons in the cerebral cortex in sporadic Creutzfeldt-Jakob disease. Neuropathology 21(4):266–271
Arosio P, Ingrassia R, Cavadini P (2009) Ferritins: a family of molecules for iron storage, antioxidation and more. Biochim Biophys Acta 1790:589–599
Arreguin S, Nelson P, Padway S, Shirazi M, Pierpont C (2009) Dopamine complexes of iron in the etiology and pathogenesis of Parkinson’s disease. J Inorg Biochem 103(1):87–93
Arts ICW, Hollman PCH (2005) Polyphenols and disease risk in epidemiologic studies. Am J Clin Nutr 81(1):317S–325S
Aschner M, Syversen T, Souza DO, Rocha JB, Farina M (2007) Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Braz J Med Biol Res 40(3):285–291
Ashall L, Horton CA, Nelson DE, Paszek P, Ryan S, Sillitoe K et al (2009) Pulsatile stimulation determines timing and specificity of NFkappa-B-dependent transcription. Science 324:242–246
Aslan M, Ryan TM, Adler B, Townes TM, Parks DA, Thompson JA et al (2001) Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc Natl Acad Sci 98(26):15215–15220
Assenza G, Zappasodi F, Squitti R, Altamura C, Ventriglia M, Ercolani M et al (2009) Neuronal functionality assessed by magnetoencephalography is related to oxidative stress system in acute ischemic stroke. Neuroimage 44(4):1267–1273
Atamna H (2009) Amino acids variations in amyloid-beta peptides, mitochondrial dysfunction, and new therapies for Alzheimer’s disease. J Bioenerg Biomembr 41:457–464
Atamna H, Frey W H 2nd, Ko N (2009) Human and rodent amyloid-beta peptides differentially bind heme: relevance to the human susceptibility to Alzheimer’s disease. Arch Biochem Biophys 487(1):59–65
Atanasova B, Mudway IS, Laftah AH, Latunde-Dada GO, McKie AT, Peters TJ et al (2004) Duodenal ascorbate levels are changed in mice with altered iron metabolism. J Nutr 134(3):501–505
Atchison WD, Hare MF (1994) Mechanisms of methylmercury-induced neurotoxicity. FASEB J 8(9):622–629
Atkinson A, Winge DR (2009) Metal acquisition and availability in the mitochondria. Chem Rev 109(10):4708–4721
Attwood TK, Kell DB, McDermott P, Marsh J, Pettifer SR, Thorne D (2009) Calling international rescue: knowledge lost in literature and data landslide!. Biochem J 424:317–333
Auclair S, Milenkovic D, Besson C, Chauvet S, Gueux E, Morand C et al (2009) Catechin reduces atherosclerotic lesion development in apo E-deficient mice: a transcriptomic study. Atherosclerosis 204(2):e21–e27
Avramovich-Tirosh Y, Amit T, Bar-Am O, Zheng HL, Fridkin M, Youdim MBH (2007a) Therapeutic targets and potential of the novel brain- permeable multifunctional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer’s disease. J Neurochem 100(2):490–502
Avramovich-Tirosh Y, Reznichenko L, Mit T, Zheng H, Fridkin M, Weinreb O et al (2007b) Neurorescue activity, APP regulation and amyloid-beta peptide reduction by novel multi-functional brain permeable iron- chelating- antioxidants, M-30 and green tea polyphenol, EGCG. Curr Alzheimer Res 4(4):403–411
Ayaki H, Lee MJ, Sumino K, Nishio H (2005) Different cytoprotective effect of antioxidants and change in the iron regulatory system in rodent cells exposed to paraquat or formaldehyde. Toxicology 208(1):73–79
Ayala-Castro C, Saini A, Outten FW (2008) Fe-S cluster assembly pathways in bacteria. Microbiol Mol Biol Rev 72(1): 110–125. Table of contents
Azad N, Rojanasakul Y, Vallyathan V (2008) Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J Toxicol Environ Health B Crit Rev 11(1):1–15
Ba LA, Doering M, Burkholz T, Jacob C (2009) Metal trafficking: from maintaining the metal homeostasis to future drug design. Metallomics 1(4):292–311
Babady NE, Carelle N, Wells RD, Rouault TA, Hirano M, Lynch DR et al (2007) Advancements in the pathophysiology of Friedreich’s Ataxia and new prospects for treatments. Mol Genet Metab 92(1–2):23–35
Babatasi G, Massetti M, Galateau F, Mosquet B, Khayat A, Evrard C (1996) Bronchial necrosis induced by inhalation of an iron tablet. J Thorac Cardiovasc Surg 112(5):1397–1399
Babior BM (2000) Phagocytes and oxidative stress. Am J Med 109(1):33–44
Bäck T, Fogel DB, Michalewicz Z (eds) (1997) Handbook of evolutionary computation. IOP Publishing/Oxford University Press, Oxford
Baeuerle PA, Henkel T (1994) Function and activation of NF-kappa B in the immune system. Annu Rev Immunol 12:141–179
Bailey DM, Robach P, Thomsen JJ, Lundby C (2006) Erythropoietin depletes iron stores: antioxidant neuroprotection for ischemic stroke? Stroke 37(10):2453
Baker ML, Wang JJ, Rogers S, Klein R, Kuller LH, Larsen EK et al (2009) Early age-related macular degeneration, cognitive function, and dementia: the cardiovascular health study. Arch Ophthalmol 127(5):667–673
Bakshi R, Benedict RH, Bermel RA, Caruthers SD, Puli SR, Tjoa CW et al (2002) T2 hypointensity in the deep gray matter of patients with multiple sclerosis: a quantitative magnetic resonance imaging study. Arch Neurol 59(1):62–68
Bakshi R, Dmochowski J, Shaikh ZA, Jacobs L (2001) Gray matter T2 hypointensity is related to plaques and atrophy in the brains of multiple sclerosis patients. J Neurol Sci 185(1):19–26
Bakshi R, Shaikh ZA, Janardhan V (2000) MRI T2 shortening (‘black T2’) in multiple sclerosis: frequency, location, and clinical correlation. Neuroreport 11(1):15–21
Bakshi R, Thompson AJ, Rocca MA, Pelletier D, Dousset V, Barkhof F et al (2008) MRI in multiple sclerosis: current status and future prospects. Lancet Neurol 7(7):615–625
Bal W, Kasprzak KS (2002) Induction of oxidative DNA damage by carcinogenic metals. Toxicol Lett 127(1–3):55–62
Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F et al (1992) Ferritin: a cytoprotective antioxidant strategem of endothelium. J Biol Chem 267(25):18148–18153
Bandyopadhyay S, Huang X, Cho H, Greig NH, Youdim MB, Rogers JT (2006) Metal specificity of an iron-responsive element in Alzheimer’s APP mRNA 5′ untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator. J Neural Trans Suppl 71:237–247
Banga JR (2008) Optimization in computational systems biology. BMC Syst Biol 2(1):47
Banga JR, Balsa-Canto E (2008) Parameter estimation and optimal experimental design. Essays Biochem 45:195–209
Baptista-Gonzalez H, Chavez-Tapia NC, Zamora-Valdes D, Uribe M, Mendez-Sanchez N (2008) Importance of iron and iron metabolism in nonalcoholic fatty liver disease. Mini Rev Med Chem 8(2):171–174
Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA et al (2010) Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature 464(7293):1351–1356
Barapatre N, Morawski M, Butz T, Reinert T (2010) Trace element mapping in Parkinsonian brain by quantitative ion beam microscopy. Nucl Instrum Methods Phys Res B 268:2156–2159
Bareggi SR, Braida D, Pollera C, Bondiolotti G, Formentin E, Puricelli M et al (2009) Effects of clioquinol on memory impairment and the neurochemical modifications induced by scrapie infection in golden hamsters. Brain Res 1280:195–200
Barichello T, Machado RA, Constantino L, Valvassori SS, Reus GZ, Martins MR et al (2007) Antioxidant treatment prevented late memory impairment in an animal model of sepsis. Crit Care Med 35(9):2186–2190
Barker JD Jr, de Carle DJ, Anuras S (1977) Chronic excessive acetaminophen use and liver damage. Ann Intern Med 87(3):299–301
Barnham KJ, Cappai R, Beyreuther K, Masters CL, Hill AF (2006) Delineating common molecular mechanisms in Alzheimer’s and prion diseases. Trends Biochem Sci 31(8):465–472
Barnham KJ, Masters CL, Bush AI (2004) Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 3(3):205–214
Barry SM, Challis GL (2009) Recent advances in siderophore biosynthesis. Curr Opin Chem Biol 13(2):205–215
Bartzokis G, Lu PH, Tishler TA, Peters DG, Kosenko A, Barrall KA et al (2010) Prevalent iron metabolism gene variants associated with increased brain ferritin iron in healthy older men. J Alzheimers Dis 20(1):333–341
Bartzokis G, Tishler TA, Lu PH, Villablanca P, Altshuler LL, Carter M et al (2007) Brain ferritin iron may influence age- and gender-related risks of neurodegeneration. Neurobiol Aging 28(3):414–423
Basak R, Chowdhury A, Fatmi L, Saha N, Mollah A, Yasmin S (2008) Stroke in the young: relationship with iron deficiency anemia and thrombocytosis. Mymensingh Med J 17(1):74–77
Basile V, Ferrari E, Lazzari S, Belluti S, Pignedoli F, Imbriano C (2009) Curcumin derivatives: molecular basis of their anti-cancer activity. Biochem Pharmacol 78(10):1305–1315
Bassett ML (2007) Iron and hepatitis C: beginning to make sense. J Gastroenterol Hepatol 22(11):1703–1704
Basu S, Mohan ML, Luo X, Kundu B, Kong Q, Singh N (2007) Modulation of proteinase K-resistant prion protein in cells and infectious brain homogenate by redox iron: implications for prion replication and disease pathogenesis. Mol Biol Cell 18(9):3302–3312
Batts KP (2007) Iron overload syndromes and the liver. Mod Pathol 20(Suppl 1):S31–S39
Baudrexel S, Nurnberger L, Rub U, Seifried C, Klein JC, Deller T et al. (2010) Quantitative mapping of T1 and T2* discloses nigral and brainstem pathology in early Parkinson’s disease. Neuroimage
Baue AE, Durham R, Faist E (1998) Systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), multiple organ failure (MOF): are we winning the battle? Shock 10(2):79–89
Bauer PO, Nukina N (2009) The pathogenic mechanisms of polyglutamine diseases and current therapeutic strategies. J Neurochem 110(6):1737–1765
Baune BT, Neuhauser H, Ellert U, Berger K (2010) The role of the inflammatory markers ferritin, transferrin and fibrinogen in the relationship between major depression and cardiovascular disorders—the German health interview and examination survey. Acta Psychiatr Scand 121(2):135–142
Beal AL, Cerra FB (1994) Multiple organ failure syndrome in the 1990s—systemic inflammatory response and organ dysfunction. JAMA 271(3):226–233
Beck MA, Handy J, Levander OA (2004) Host nutritional status: the neglected virulence factor. Trends Microbiol 12(9):417–423
Becker E, Richardson DR (2001) Frataxin: its role in iron metabolism and the pathogenesis of Friedreich’s ataxia. Int J Biochem Cell Biol 33(1):1–10
Becker JS (2010) Bioimaging of metals in brain tissue from micrometre to nanometre scale by laser ablation inductively coupled plasma mass spectrometry: state of the art and perspectives. Int J Mass Spectrom 289(2–3):65–75
Becker JS, Matusch A, Palm C, Salber D, Morton KA, Becker S (2010a) Bioimaging of metals in brain tissue by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) and metallomics. Metallomics 2(2):104–111
Becker JS, Zoriy M, Matusch A, Wu B, Salber D, Palm C et al (2010b) Bioimaging of metals by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS). Mass Spectrom Rev 29(1):156–175
Beckett GJ, Foster GR, Hussey AJ, Oliveira DB, Donovan JW, Prescott LF et al (1989) Plasma glutathione S-transferase and F protein are more sensitive than alanine aminotransferase as markers of paracetamol (acetaminophen)-induced liver damage. Clin Chem 35(11):2186–2189
Beckman JS (1996) Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol 9(5):836–844
Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA (1990) Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA 87(4):1620–1624
Beckman JS, Ischiropoulos H, Zhu L, van der Woerd M, Smith C, Chen J et al (1992) Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch Biochem Biophys 298(2):438–445
Beckman JS, Koppenol WH (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271(5 Pt 1):C1424–C1437
Bedard K, Krause KH (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87(1):245–313
Beeri MS, Schmeidler J, Silverman JM, Gandy S, Wysocki M, Hannigan CM et al (2008) Insulin in combination with other diabetes medication is associated with less Alzheimer neuropathology. Neurology 71(10):750–757
Bekyarova G, Tancheva S, Hristova M (2009) Protective effect of melatonin against oxidative hepatic injury after experimental thermal trauma. Methods Find Exp Clin Pharmacol 31(1):11–14
Ben-Shachar D, Riederer P, Youdim MB (1991) Iron-melanin interaction and lipid peroxidation: implications for Parkinson’s disease. J Neurochem 57(5):1609–1614
Benarroch EE (2009) Brain iron homeostasis and neurodegenerative disease. Neurology 72(16):1436–1440
Bencze KZ, Kondapalli KC, Cook JD, McMahon S, Millán-Pacheco C, Pastor N et al (2006) The structure and function of frataxin. Crit Rev Biochem Mol Biol 41(5):269–291
Bencze KZ, Yoon T, Millán-Pacheco C, Bradley PB, Pastor N, Cowan JA et al (2007) Human frataxin: iron and ferrochelatase binding surface. Chem Commun (Camb) 18:1798–1800
Bender A, Jenkins JL, Glick M, Deng Z, Nettles JH, Davies JW (2006) “Bayes affinity fingerprints” improve retrieval rates in virtual screening and define orthogonal bioactivity space: when are multitarget drugs a feasible concept? J Chem Inf Model 46(6):2445–2456
Berdoukas V, Chouliaras G, Moraitis P, Zannikos K, Berdoussi E, Ladis V (2009) The efficacy of iron chelator regimes in reducing cardiac and hepatic iron in patients with thalassaemia major: a clinical observational study. J Cardiovasc Magn Reson 11(1):20
Berg D (2006) In vivo detection of iron and neuromelanin by transcranial sonography—a new approach for early detection of substantia nigra damage. J Neural Transm 113(6):775–780
Berg D (2007) Disturbance of iron metabolism as a contributing factor to SN hyperechogenicity in Parkinson’s disease: implications for idiopathic and monogenetic forms. Neurochem Res 32(10):1646–1654
Berg D, Gerlach M, Youdim MBH, Double KL, Zecca L, Riederer P et al (2001) Brain iron pathways and their relevance to Parkinson’s disease. J Neurochem 79(2):225–236
Berg D, Hochstrasser H (2006) Iron metabolism in parkinsonian syndromes. Mov Disord 21(9):1299–1310
Berg D, Hochstrasser H, Schweitzer KJ, Riess O (2006) Disturbance of iron metabolism in Parkinson’s disease ultrasonography as a biomarker. Neurotox Res 9(1):1–13
Berg D, Riederer P, Gerlach M (2008) Contribution of disturbed iron metabolism to the pathogenesis of Parkinson’s disease. Future Med 3(4):447–461
Bergman M, Salman H, Pinchasi R, Straussberg R, Djaldetti M, Bessler H (2005) Phagocytic capacity and apoptosis of peripheral blood cells from patients with iron deficiency anemia. Biomed Pharmacother 59(6):307–311
Bermel RA, Puli SR, Rudick RA, Weinstock-Guttman B, Fisher E, Munschauer F E 3rd et al (2005) Prediction of longitudinal brain atrophy in multiple sclerosis by gray matter magnetic resonance imaging T2 hypointensity. Arch Neurol 62(9):1371–1376
Beutler E (2007) Iron storage disease: facts, fiction and progress. Blood Cells Mol Dis 39(2):140–147
Bharath S, Hsu M, Kaur D, Rajagopalan S, Andersen JK (2002) Glutathione, iron and Parkinson’s disease. Biochem Pharmacol 64(5–6):1037–1048
Bhattacharya K, Dopp E, Kakkar P, Jaffery FN, Schiffmann D, Jaurand MC et al (2005) Biomarkers in risk assessment of asbestos exposure. Mutat Res 579(1–2):6–21
Bhidayasiri R, Truong DD (2004) Chorea and related disorders. Postgrad Med J 80(947):527–534
Biasiotto G, Goldwurm S, Finazzi D, Tunesi S, Zecchinelli A, Sironi F et al (2008) HFE gene mutations in a population of Italian Parkinson’s disease patients. Parkinsonism Relat Disord 14(5):426–430
Bidle KD, Bender SJ (2008) Iron starvation and culture age activate metacaspases and programmed cell death in the marine diatom Thalassiosira pseudonana. Eukaryot Cell 7(2):223–236
Binder CJ, Chang MK, Shaw PX, Miller YI, Hartvigsen K, Dewan A et al (2002) Innate and acquired immunity in atherogenesis. Nat Med 8(11):1218–1226
Biran Y, Masters CL, Barnham KJ, Bush AI, Adlard PA (2009) Pharmacotherapeutic targets in Alzheimer’s disease. J Cell Mol Med 13(1):61–86
Bishop GM, Robinson SR (2001) Quantitative analysis of cell death and ferritin expression in response to cortical iron: implications for hypoxia-ischemia and stroke. Brain Res 907(1–2):175–187
Bishop GM, Robinson SR, Liu Q, Perry G, Atwood CS, Smith MA (2002) Iron: a pathological mediator of Alzheimer disease? Dev Neurosci 24(2–3):184–187
Bishop GM, Dang TN, Dringen R, Robinson SR (2010a) Accumulation of non-transferrin-bound iron by neurons, astrocytes, and microglia. Neurotox Res (in press)
Bishop NA, Lu T, Yankner BA (2010b) Neural mechanisms of ageing and cognitive decline. Nature 464(7288):529–535
Biswas SK, Rahman I (2009) Environmental toxicity, redox signaling and lung inflammation: the role of glutathione. Mol Aspects Med 30(1–2):60–76
Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C (2008) Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database of Systematic Reviews 2008, CD007176.
Blake GJ, Ridker PM (2001) Novel clinical markers of vascular wall inflammation. Circ Res 89(9):763–771
Blasiak J, Sklodowska A, Ulinska M, Szaflik JP (2009) Iron and age-related macular degeneration. Klin Oczna 111(4–6):174–177
Blázquez L, De Juan D, Ruiz-Martinez J, Emparanza JI, Saenz A, Otaegui D et al (2007) Genes related to iron metabolism and susceptibility to Alzheimer’s disease in Basque population. Neurobiol Aging 28(12):1941–1943
Blum D, Torch S, Lambeng N, Nissou MF, Benabid AL, Sadoul R et al (2001) Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson’s disease. Prog Neurobiol 65(2):135–172
Boddaert N, Le Quan Sang KH, Rotig A, Leroy-Willig A, Gallet S, Brunelle F et al (2007) Selective iron chelation in Friedreich ataxia: biologic and clinical implications. Blood 110(1):401–408
Boelaert JR, Weinberg GA, Weinberg ED (1996) Altered iron metabolism in HIV infection: mechanisms, possible consequences, and proposals for management. Infect Agents Dis 5(1):36–46
Bolognin S, Drago D, Messori L, Zatta P (2009a) Chelation therapy for neurodegenerative diseases. Med Res Rev 29(4):547–570
Bolognin S, Messori L, Zatta P (2009b) Metal ion physiopathology in neurodegenerative disorders. Neuromol Med 11(4):223–238
Bolton CF (1996) Sepsis and the systemic inflammatory response syndrome: neuromuscular manifestations. Crit Care Med 24(8):1408–1416
Bomhard EM, Herbold BA (2005) Genotoxic activities of aniline and its metabolites and their relationship to the carcinogenicity of aniline in the spleen of rats. Crit Rev Toxicol 35(10):783–835
Bone RC (1996) Toward a theory regarding the pathogenesis of the systemic inflammatory response syndrome: what we do and do not know about cytokine regulation. Crit Care Med 24(1):163–172
Bongard JC, Lipson H (2005) Nonlinear system identification using coevolution of models and tests. IEEE Trans Evol Comput 9(4):361–384
Bonkovsky HL (2002) Iron as a comorbid factor in chronic viral hepatitis. Am J Gastroenterol 97(1):1–4
Bonkovsky HL, Naishadham D, Lambrecht RW, Chung RT, Hoefs JC, Nash SR et al (2006) Roles of iron and HFE mutations on severity and response to therapy during retreatment of advanced chronic hepatitis C. Gastroenterology 131(5):1440–1451
Borah A, Mohanakumar KP (2009) Melatonin inhibits 6-hydroxydopamine production in the brain to protect against experimental parkinsonism in rodents. J Pineal Res 47(4):293–300
Borgna-Pignatti C, Cappellini MD, De Stefano P, Del Vecchio GC, Forni GL, Gamberini MR et al (2005) Survival and complications in thalassemia. Ann NY Acad Sci 1054:40–47
Borisy AA, Elliott PJ, Hurst NW, Lee MS, Lehar J, Price ER et al (2003) Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci USA 100(13):7977–7982
Bornholdt S, Sneppen K (2000) Robustness as an evolutionary principle. Proc R Soc B Biol Sci 267(1459):2281–2286
Bosomtwi A, Jiang Q, Ding GL, Zhang L, Zhang ZG, Lu M et al (2008) Quantitative evaluation of microvascular density after stroke in rats using MRI. J Cereb Blood Flow Metab 28(12):1978–1987
Bowern N, Ramshaw IA, Clark IA, Doherty PC (1984) Inhibition of autoimmune neuropathological process by treatment with an iron-chelating agent. J Exp Med 160(5):1532–1543
Bowler RP (2004) Oxidative stress in the pathogenesis of asthma. Curr Allergy Asthma Rep 4(2):116–122
Bowler RP, Crapo JD (2002) Oxidative stress in allergic respiratory diseases. J Allergy Clin Immunol 110(3):349–356
Boyer J, Liu RH (2004) Apple phytochemicals and their health benefits. Nutr J 3:5
Brar S, Henderson D, Schenck J, Zimmerman EA (2009) Iron accumulation in the substantia nigra of patients with Alzheimer disease and parkinsonism. Arch Neurol 66(3):371–374
Brard L, Granai CO, Swamy N (2006) Iron chelators deferoxamine and diethylenetriamine pentaacetic acid induce apoptosis in ovarian carcinoma. Gynecol Oncol 100(1):116–127
Brass SD, Benedict RH, Weinstock-Guttman B, Munschauer F, Bakshi R (2006a) Cognitive impairment is associated with subcortical magnetic resonance imaging grey matter T2 hypointensity in multiple sclerosis. Mult Scler 12(4):437–444
Brass SD, Chen NK, Mulkern RV, Bakshi R (2006b) Magnetic resonance imaging of iron deposition in neurological disorders. Top Magn Reson Imaging 17(1):31–40
Braun S, Ndrepepa G, von Beckerath N, Vogt W, Schomig A, Kastrati A (2004) Value of serum ferritin and soluble transferrin receptor for prediction of coronary artery disease and its clinical presentations. Atherosclerosis 174(1):105–110
Breiman L (2001) Statistical modeling: the two cultures. Stat Sci 16(3):199–215
Breitkopf K, Nagy LE, Beier JI, Mueller S, Weng H, Dooley S (2009) Current experimental perspectives on the clinical progression of alcoholic liver disease. Alcohol Clin Exp Res 33(10):1647–1655
Brennan LA, Kantorow M (2009) Mitochondrial function and redox control in the aging eye: role of MsrA and other repair systems in cataract and macular degenerations. Exp Eye Res 88(2):195–203
Brewer GJ (2007) Iron and copper toxicity in diseases of aging, particularly atherosclerosis and Alzheimer’s disease. Exp Biol Med 232(2):323–335
Brewer GJ (2010) Risks of copper and iron toxicity during aging in humans. Chem Res Toxicol 23(2):319–326
Broadhurst D, Goodacre R, Jones A, Rowland JJ, Kell DB (1997) Genetic algorithms as a method for variable selection in multiple linear regression and partial least squares regression, with applications to pyrolysis mass spectrometry. Anal Chim Acta 348(1–3):71–86
Broadhurst D, Kell DB (2006) Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics 2(4):171–196. doi:10.1007/s11306-006-0037-z
Broedbaek K, Poulsen HE, Weimann A, Kom GD, Schwedhelm E, Nielsen P et al (2009) Urinary excretion of biomarkers of oxidatively damaged DNA and RNA in hereditary hemochromatosis. Free Radic Biol Med 47:1230–1233
Brötz-Oesterhelt H, Brunner NA (2008) How many modes of action should an antibiotic have? Curr Opin Pharmacol 8:567–573
Brown DR (2009a) Brain proteins that mind metals: a neurodegenerative perspective. Dalton Trans 21:4069–4076
Brown DR (2009b) Role of microglia in age-related changes to the nervous system. ScientificWorldJournal 9:1061–1071
Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R et al (1997) The cellular prion protein binds copper in vivo. Nature 390(6661):684–687
Brown M, He F, Wilkinson SJ (2010) Properties of the proximate parameter tuning regularization algorithm. Bull Math Biol 72(3):697–718
Brudevold R, Hole T, Hammerstrøm J (2008) Hyperferritinemia is associated with insulin resistance and fatty liver in patients without iron overload. PLoS One 3(10):e3547
Brune M, Rossander L, Hallberg L (1989) Iron absorption and phenolic compounds: importance of different phenolic structures. Eur J Clin Nutr 43(8):547–557
Bu DX, Hemdahl AL, Gabrielsen A, Fuxe J, Zhu C, Eriksson P et al (2006) Induction of neutrophil gelatinase-associated lipocalin in vascular injury via activation of nuclear factor-kappaB. Am J Pathol 169(6):2245–2253
Buchanan DD, Silburn PA, Chalk JB, Le Couteur DG, Mellick GD (2002) The Cys282Tyr polymorphism in the HFE gene in Australian Parkinson’s disease patients. Neurosci Lett 327(2):91–94
Büeler H (2009) Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp Neurol 218(2):235–246
Bugianesi E, Manzini P, D’Antico S, Vanni E, Longo F, Leone N et al (2004) Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology 39(1):179–187
Bulger EM, Maier RV (2001) Antioxidants in critical illness. Arch Surg 136(10):1201–1207
Bulgin MS, Melson SS (2007) What veterinary practitioners should know about scrapie. J Am Vet Med Assoc 230(8):1158–1164, 1120.
Bulteau AL, Dancis A, Gareil M, Montagne JJ, Camadro JM, Lesuisse E (2007) Oxidative stress and protease dysfunction in the yeast model of Friedreich ataxia. Free Radic Biol Med 42(10):1561–1570
Bulucu F, Ocal R, Karadurmus N, Sahin M, Kenar L, Aydin A et al (2009) Effects of N-acetylcysteine, deferoxamine and selenium on doxorubicin-induced hepatotoxicity. Biol Trace Elem Res 132:184–196
Burrows CJ, Muller JG (1998) Oxidative nucleobase modifications leading to strand scission. Chem Rev 98(3):1109–1152
Busca A, Falda M, Manzini P, D’Antico S, Valfre A, Locatelli F et al (2010) Iron overload in patients receiving allogeneic hematopoietic stem cell transplantation: quantification of iron burden by a superconducting quantum interference device (SQUID) and therapeutic effectiveness of phlebotomy. Biol Blood Marrow Transpl 16(1):115–122
Bush AI (2000) Metals and neuroscience. Curr Opin Chem Biol 4(2):184–191
Bush AI (2003) The metallobiology of Alzheimer’s disease. Trends Neurosci 26(4):207–214
Bush AI (2008) Drug development based on the metals hypothesis of Alzheimer’s disease. J Alzheimers Dis 15(2):223–240
Buss JL, Neuzil J, Gellert N, Weber C, Ponka P (2003) Pyridoxal isonicotinoyl hydrazone analogs induce apoptosis in hematopoietic cells due to their iron-chelating properties. Biochem Pharmacol 65(2):161–172
Butelli E, Titta L, Giorgio M, Mock HP, Matros A, Peterek S et al (2008) Enrichment of tomato fruit with health-promoting anthocyanins by expression of select transcription factors. Nat Biotechnol 26(11):1301–1308
Butensky James E, Harmatz P, Lee M, Kennedy C, Petru A, Wara D et al (2009) Altered iron metabolism in children with human immunodeficiency virus disease. Pediatr Hematol Oncol 26(2):69–84
Butt MS, Sultan MT (2009) Green tea: nature’s defense against malignancies. Crit Rev Food Sci Nutr 49(5):463–473
Butterfield DA, Reed T, Newman SF, Sultana R (2007) Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic Biol Med 43(5):658–677
Buzan T (2002) How to mind map. Thorsons, London
Cadenas S, Cadenas AM (2002) Fighting the stranger-antioxidant protection against endotoxin toxicity. Toxicology 180(1):45–63
Cai J, Nelson KC, Wu M, Sternberg P Jr, Jones DP (2000) Oxidative damage and protection of the RPE. Prog Retin Eye Res 19(2):205–221
Calabrese E (2008a) Hormesis: why it is important to toxicology and toxicologists. Environ Toxicol Chem 27:1451–1474
Calabrese EJ (2008b) Converging concepts: adaptive response, preconditioning, and the Yerkes-Dodson Law are manifestations of hormesis. Ageing Res Rev 7(1):8–20
Calabrese EJ (2008c) Hormesis and medicine. Br J Clin Pharmacol
Calabrese EJ, Baldwin LA (1997) The dose determines the stimulation (and poison): development of a chemical hormesis database. Int J Toxicol 16(6):545–559
Calabrese EJ, Baldwin LA (2001) Hormesis: U-shaped dose responses and their centrality in toxicology. Trends Pharmacol Sci 22(6):285–291
Calabrese EJ, Baldwin LA (2003) Hormesis: the dose-response revolution. Annu Rev Pharmacol Toxicol 43:175–197
Calabrese EJ, Blain R (2005) The occurrence of hormetic dose responses in the toxicological literature, the hormesis database: an overview. Toxicol Appl Pharmacol 202(3):289–301
Calabrese EJ, Blain RB (2009) Hormesis and plant biology. Environ Pollut 157(1):42–48
Camaschella C, Merlini R (2005) Inherited hemochromatosis: from genetics to clinics. Minerva Med 96(3):207–222
Campanella A, Rovelli E, Santambrogio P, Cozzi A, Taroni F, Levi S (2009) Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: hypothesis for a protective role in Friedreich ataxia. Hum Mol Genet 18(1):1–11
Candore G, Licastro F, Chiappelli M, Franceschi C, Lio D, Rita Balistreri C et al (2003) Association between the HFE mutations and unsuccessful ageing: a study in Alzheimer’s disease patients from Northern Italy. Mech Ageing Dev 124(4):525–528
Cao A, Galanello R (2010) Beta-thalassemia. Genet Med 12(2):61–76
Cappellini MD, Pattoneri P (2009) Oral iron chelators. Annu Rev Med 60:25–38
Cappellini MD, Porter J, El-Beshlawy A, Li CK, Seymour JF, Elalfy M et al (2010) Tailoring iron chelation by iron intake and serum ferritin: the prospective EPIC study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematologica 95(4):557–566
Carbonell T, Rama R (2007) Iron, oxidative stress and early neurological deterioration in ischemic stroke. Curr Med Chem 14(8):857–874
Carrea G, Colonna S, Kelly DR, Lazcano A, Ottolina G, Roberts SM (2005) Polyamino acids as synthetic enzymes: mechanism, applications and relevance to prebiotic catalysis. Trends Biotechnol 23(10):507–513
Carri MT, Ferri A, Cozzolino M, Calabrese L, Rotilio G (2003) Neurodegeneration in amyotrophic lateral sclerosis: the role of oxidative stress and altered homeostasis of metals. Brain Res Bull 61(4):365–374
Carrillo-Vico A, Lardone PJ, Naji L, Fernández-Santos JM, Martín-Lacave I, Guerrero JM et al (2005) Beneficial pleiotropic actions of melatonin in an experimental model of septic shock in mice: regulation of pro-/anti-inflammatory cytokine network, protection against oxidative damage and anti-apoptotic effects. J Pineal Res 39(4):400–408
Casadesus G, Smith MA, Zhu X, Aliev G, Cash AD, Honda K et al (2004) Alzheimer disease: evidence for a central pathogenic role of iron-mediated reactive oxygen species. J Alzheimers Dis 6(2):165–169
Cass WA, Grondin R, Andersen AH, Zhang Z, Hardy PA, Hussey-Andersen LK et al (2007) Iron accumulation in the striatum predicts aging-related decline in motor function in rhesus monkeys. Neurobiol Aging 28(2):258–271
Cassol OJ Jr, Rezin GT, Petronilho FC, Scaini G, Goncalves CL, Ferreira GK et al. (2009) Effects of N-acetylcysteine/deferoxamine, taurine and RC-3095 on respiratory chain complexes and creatine kinase activities in rat brain after sepsis. Neurochem Res
Castellani RJ, Moreira PI, Liu G, Dobson J, Perry G, Smith MA et al (2007) Iron: the redox-active center of oxidative stress in Alzheimer disease. Neurochem Res 32(10):1640–1645
Castellani RJ, Perry G, Siedlak SL, Nunomura A, Shimohama S, Zhang J et al (2002) Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem lewy bodies in humans. Neurosci Lett 319(1):25–28
Castoldi AF, Coccini T, Ceccatelli S, Manzo L (2001) Neurotoxicity and molecular effects of methylmercury. Brain Res Bull 55(2):197–203
Caughey B, Baron GS (2006) Prions and their partners in crime. Nature 443(7113):803–810
Caughey B, Lansbury PT (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26:267–298
Cavadini P, Gellera C, Patel PI, Isaya G (2000) Human frataxin maintains mitochondrial iron homeostasis in Saccharomyces cerevisiae. Hum Mol Genet 9(17):2523–2530
Cavadini P, O’Neill HA, Benada O, Isaya G (2002) Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum Mol Genet 11(3):217–227
Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S et al (2006) NADPH oxidases in cardiovascular health and disease. [Review]. Antioxid Redox Signal 8(5–6):691–728
Cazzola M, Della Porta MG, Malcovati L (2008) Clinical relevance of anemia and transfusion iron overload in myelodysplastic syndromes. Hematol Am Soc Hematol Educ Program 2008:166–175
Ceccarelli A, Filippi M, Neema M, Arora A, Valsasina P, Rocca MA et al (2009) T2 hypointensity in the deep gray matter of patients with benign multiple sclerosis. Mult Scler 15(6):678–686
Ceccarelli A, Rocca MA, Neema M, Martinelli V, Arora A, Tauhid S et al (2010) Deep gray matter T2 hypointensity is present in patients with clinically isolated syndromes suggestive of multiple sclerosis. Mult Scler 16(1):39–44
Cemeli E, Baumgartner A, Anderson D (2009) Antioxidants and the comet assay. Mutat Res 681(1):51–67
Ceyssens B, Pauwels M, Meulemans B, Verbeelen D, Van den Branden C (2004) Increased oxidative stress in the mouse adriamycin model of glomerulosclerosis is accompanied by deposition of ferric iron and altered GGT activity in renal cortex. Ren Fail 26(1):21–27
Chakravarty A (2003) Friedreich’s ataxia—yesterday, today and tomorrow. Neurol India 51(2):176–182
Chandrasekaran V, Taylor EW (2008) Molecular modeling of the oxidized form of nuclear factor-kappa B suggests a mechanism for redox regulation of DNA binding and transcriptional activation. J Mol Graph Model 26(5):861–867
Chandrasekaran VR, Hsu DZ, Liu MY (2009) The protective effect of sesamol against mitochondrial oxidative stress and hepatic injury in acetaminophen-overdosed rats. Shock 32(1):89–93
Chang EF, Claus CP, Vreman HJ, Wong RJ, Noble-Haeusslein LJ (2005) Heme regulation in traumatic brain injury: relevance to the adult and developing brain. J Cereb Blood Flow Metab 25(11):1401–1417
Chao CC, Park SH, Aust AE (1996) Participation of nitric oxide and iron in the oxidation of DNA in asbestos-treated human lung epithelial cells. Arch Biochem Biophys 326(1):152–157
Chard DT, Miller DH (2009) What you see depends on how you look: gray matter lesions in multiple sclerosis. Neurology 73(12):918–919
Charkoudian LK, Dentchev T, Lukinova N, Wolkow N, Dunaief JL, Franz KJ (2008) Iron prochelator BSIH protects retinal pigment epithelial cells against cell death induced by hydrogen peroxide. J Inorg Biochem 102(12):2130–2135
Chau LY (2000) Iron and atherosclerosis. Proc Natl Sci Counc Repub China B 24(4):151–155
Chen-Roetling J, Chen L, Regan RF (2009) Minocycline attenuates iron neurotoxicity in cortical cell cultures. Biochem Biophys Res Commun
Chen H, Lukas TJ, Du N, Suyeoka G, Neufeld AH (2009) Dysfunction of the retinal pigment epithelium with age: increased iron decreases phagocytosis and lysosomal activity. Invest Ophthalmol Vis Sci 50(4):1895–1902
Chen LW, Wang YQ, Wei LC, Shi M, Chan YS (2007) Chinese herbs and herbal extracts for neuroprotection of dopaminergic neurons and potential therapeutic treatment of Parkinson’s disease. CNS Neurol Disord Drug Targets 6(4):273–281
Chen Q, Marsh J, Ames B, Mossman B (1996) Detection of 8-oxo-2’-deoxyguanosine, a marker of oxidative DNA damage, in culture medium from human mesothelial cells exposed to crocidolite asbestos. Carcinogenesis 17(11):2525–2527
Chen W, Stambolian D, Edwards AO, Branham KE, Othman M, Jakobsdottir J et al (2010) Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc Natl Acad Sci USA
Chen X, Scholl TO, Stein TP (2006) Association of elevated serum ferritin levels and the risk of gestational diabetes mellitus in pregnant women: the Camden study. Diabetes Care 29(5):1077–1082
Cheynier V (2005) Polyphenols in foods are more complex than often thought. Am J Clin Nutr 81(1 Suppl):223S–229S
Chiueh CC, Andoh T, Lai AR, Lai E, Krishna G (2000) Neuroprotective strategies in Parkinson’s disease: protection against progressive nigral damage induced by free radicals. Neurotox Res 2(2–3):293–310
Cho S, Urata Y, Iida T, Goto S, Yamaguchi M, Sumikawa K et al (1998) Glutathione downregulates the phosphorylation of I kappa B: autoloop regulation of the NF-kappa B-mediated expression of NF-kappa B subunits by TNF-alpha in mouse vascular endothelial cells. Biochem Biophys Res Commun 253(1):104–108
Cho TH, Nighoghossian N, Wiart M, Desestret V, Cakmak S, Berthezene Y et al (2007) USPIO-enhanced MRI of neuroinflammation at the sub-acute stage of ischemic stroke: preliminary data. Cerebrovasc Dis 24(6):544–546
Chobot V (2010) Simultaneous detection of pro- and antioxidative effects in the variants of the deoxyribose degradation assay. J Agric Food Chem 58(4):2088–2094
Chodaczek G, Saavedra-Molina A, Bacsi A, Kruzel ML, Sur S, Boldogh I (2007) Iron-mediated dismutation of superoxide anion augments antigen-induced allergic inflammation: effect of lactoferrin. Postepy Hig Med Dosw (Online) 61:268–276
Choi CJ, Kanthasamy A, Anantharam V, Kanthasamy AG (2006) Interaction of metals with prion protein: possible role of divalent cations in the pathogenesis of prion diseases. Neurotoxicology 27(5):777–787
Choi KM, Lee KW, Kim HY, Seo JA, Kim SG, Kim NH et al (2005) Association among serum ferritin, alanine aminotransferase levels, and metabolic syndrome in Korean postmenopausal women. Metabolism 54(11):1510–1514
Choi YG, Park JH, Lim S (2009) Acupuncture inhibits ferric iron deposition and ferritin-heavy chain reduction in an MPTP-induced parkinsonism model. Neurosci Lett 450(2):92–96
Chong CR, Sullivan D J Jr (2007) New uses for old drugs. Nature 448(7154):645–646
Chopra V, Fox JH, Lieberman G, Dorsey K, Matson W, Waldmeier P et al (2007) A small-molecule therapeutic lead for Huntington’s disease: preclinical pharmacology and efficacy of C2–8 in the R6/2 transgenic mouse. Proc Natl Acad Sci USA 104(42):16685–16689
Chowers I, Wong R, Dentchev T, Farkas RH, Iacovelli J, Gunatilaka TL et al (2006) The iron carrier transferrin is upregulated in retinas from patients with age-related macular degeneration. Invest Ophthalmol Vis Sci 47(5):2135–2140
Christen Y (2000) Oxidative stress and Alzheimer disease. Am J Clin Nutr 71(2):621S–629S
Christie BR, Cameron HA (2006) Neurogenesis in the adult hippocampus. Hippocampus 16(3):199–207
Chu J, Zhou CC, Lu N, Zhang X, Dong FT (2008) Genetic variants in three genes and smoking show strong associations with susceptibility to exudative age-related macular degeneration in a Chinese population. Chin Med J (Engl) 121(24):2525–2533
Chung YL, Williams A, Ritchie D, Williams SC, Changani KK, Hope J et al (1999) Conflicting MRI signals from gliosis and neuronal vacuolation in prion diseases. Neuroreport 10(17):3471–3477
Ciencewicki J, Trivedi S, Kleeberger SR (2008) Oxidants and the pathogenesis of lung diseases. J Allergy Clin Immunol 122(3):456–468 (quiz 469–470)
Circu ML, Aw TY (2010) Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med 48(6):749–762
Clark LN, Ross BM, Wang Y, Mejia-Santana H, Harris J, Louis ED et al (2007) Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 69(12):1270–1277
Clarkson TW, Magos L, Myers GJ (2003) The toxicology of mercury—current exposures and clinical manifestations. N Engl J Med 349(18):1731–1737
Clifton MC, Corrent C, Strong RK (2009) Siderocalins: siderophore-binding proteins of the innate immune system. Biometals 22(4):557–564
Closa D, Folch-Puy E (2004) Oxygen free radicals and the systemic inflammatory response. IUBMB Life 56(4):185–191
Cohen AR, Galanello R, Pennell DJ, Cunningham MJ, Vichinsky E (2004) Thalassemia. Hematology 14–34
Cole AR, Astell A, Green C, Sutherland C (2007) Molecular connexions between dementia and diabetes. Neurosci Biobehav Rev 31(7):1046–1063
Coles SJ, Day NE, Murray-Rust P, Rzepa HS, Zhang Y (2005) Enhancement of the chemical semantic web through the use of InChI identifiers. Organic & Biomolecular Chemistry 3(10):1832–1834
Collingwood J, Dobson J (2006) Mapping and characterization of iron compounds in Alzheimer’s tissue. J Alzheimers Dis 10(2–3):215–222
Collingwood JF, Chong RK, Kasama T, Cervera-Gontard L, Dunin-Borkowski RE, Perry G et al (2008) Three-dimensional tomographic imaging and characterization of iron compounds within Alzheimer’s plaque core material. J Alzheimers Dis 14(2):235–245
Collins FS (2010) The langage of life. Profile Books, London
Combarros O, Garcia-Roman M, Fontalba A, Fernandez-Luna JL, Llorca J, Infante J et al (2003) Interaction of the H63D mutation in the hemochromatosis gene with the apolipoprotein E epsilon 4 allele modulates age at onset of Alzheimer’s disease. Dement Geriatr Cogn Disord 15(3):151–154
Comhair SA, Erzurum SC (2010) Redox control of asthma: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal 12(1):93–124
Connor JR, Lee SY (2006) HFE mutations and Alzheimer’s disease. J Alzheimers Dis 10(2–3):267–276
Connor JR, Menzies SL, St Martin SM, Mufson EJ (1992) A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. J Neurosci Res 31(1):75–83
Cook CI, Yu BP (1998) Iron accumulation in aging: modulation by dietary restriction. Mech Ageing Devel 102(1):1–13
Cook JD, Bencze KZ, Jankovic AD, Crater AK, Busch CN, Bradley PB et al (2006) Monomeric yeast frataxin is an iron-binding protein. Biochemistry 45(25):7767–7777
Cook R, Calabrese EJ (2006) The importance of hormesis to public health. Environ Health Perspect 114(11):1631–1635
Cooke MS, Evans MD, Dizdaroglu M, Lunec J (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17(10):1195–1214
Cooke MS, Olinski R, Loft S (2008) Measurement and meaning of oxidatively modified DNA lesions in urine. Cancer Epidemiol Biomarkers Prev 17(1):3–14
Cornelis P, Andrews SC (eds) (2010) Iron uptake and homeostasis in microorganisms. Horizon Press, New York
Correia AR, Pastore C, Adinolfi S, Pastore A, Gomes CM (2008) Dynamics, stability and iron-binding activity of frataxin clinical mutants. FEBS J 275(14):3680–3690
Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS et al (2010) The genetic landscape of a cell. Science 327(5964):425–431
Coveney PV, Fowler PW (2005) Modelling biological complexity: a physical scientist’s perspective. J R Soc Interface 2(4):267–280
Cowan KJ, Diamond MI, Welch WJ (2003) Polyglutamine protein aggregation and toxicity are linked to the cellular stress response. Hum Mol Genet 12(12):1377–1391
Cowland JB, Muta T, Borregaard N (2006) IL-1beta-specific up-regulation of neutrophil gelatinase-associated lipocalin is controlled by IkappaB-zeta. J Immunol 176(9):5559–5566
Cowland JB, Sorensen OE, Sehested M, Borregaard N (2003) Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1 beta, but not by TNF-alpha. J Immunol 171(12):6630–6639
Cox LA Jr (2009) Hormesis without cell killing. Risk Anal 29(3):393–400
Cozzi A, Levi S, Corsi B, Santambrogio P, Campanella A, Gerardi G et al (2003) Role of iron and ferritin in TNFalpha-induced apoptosis in HeLa cells. FEBS Lett 537(1–3):187–192
Cozzi A, Rovelli E, Frizzale G, Campanella A, Amendola M, Arosio P et al (2010) Oxidative stress and cell death in cells expressing L-ferritin variants causing neuroferritinopathy. Neurobiol Dis 37(1):77–85
Cozzolino M, Ferri A, Carri MT (2008) Amyotrophic lateral sclerosis: from current developments in the laboratory to clinical implications. Antioxid Redox Signal 10(3):405–443
Craelius W, Migdal MW, Luessenhop CP, Sugar A, Mihalakis I (1982) Iron deposits surrounding multiple sclerosis plaques. Arch Pathol Lab Med 106(8):397–399
Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W et al (1991) Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337(8753):1304–1308
Crichton RR, Ward RJ (2006) Metal-based neurodegeneration. Wiley/Blackwell, Chichester
Crimi E, Sica V, Slutsky AS, Zhang HB, Williams-Ignarro S, Ignarro LJ et al (2006a) Role of oxidative stress in experimental sepsis and multisystem organ dysfunction. Free Radic Res 40(7):665–672
Crimi E, Sica V, Williams-Ignarro S, Zhang H, Slutsky AS, Ignarro LJ et al (2006b) The role of oxidative stress in adult critical care. Free Radic Biol Med 40(3):398–406
Crozet C, Beranger F, Lehmann S (2008) Cellular pathogenesis in prion diseases. Vet Res 39(4):44
Csermely P, Agoston V, Pongor S (2005) The efficiency of multi-target drugs: the network approach might help drug design. Trends Pharmacol Sci 26(4):178–182
Csete M, Doyle J (2004) Bow ties, metabolism and disease. Trends Biotechnol 22(9):446–450
Csikasz-Nágy A (2009) Computational systems biology of the cell cycle. Brief Bioinform 10(4):424–434
Cuajungco MP, Fagét KY, Huang X, Tanzi RE, Bush AI (2000) Metal chelation as a potential therapy for Alzheimer’s disease. Ann NY Acad Sci 920:292–304
Cuijpers ML, Raymakers RA, Mackenzie MA, de Witte TJ, Swinkels DW (2010) Recent advances in the understanding of iron overload in sideroblastic myelodysplastic syndrome. Br J Haematol 149:322–333
Cuzzocrea S, Di Paola R, Mazzon E, Patel NS, Genovese T, Muia C et al (2006) Erythropoietin reduces the development of nonseptic shock induced by zymosan in mice. Crit Care Med 34(4):1168–1177
Cuzzocrea S, Riley DP, Caputi AP, Salvemini D (2001) Antioxidant therapy: a new pharmacological approach in shock, inflammation, and ischemia/reperfusion injury. Pharmacol Rev 53(1):135–159
Cuzzocrea S, Thiemermann C, Salvemini D (2004) Potential therapeutic effect of antioxidant therapy in shock and inflammation. Curr Med Chem 11(9):1147–1162
Dada JO, Mendes P (2009) Design and architecture of web services for simulation of biochemical systems. Data Integr Life Sci Proc 5647:182–195
Dada JO, Spasic I, Paton NW, Mendes P (2010) SBRML: a markup language for associating systems biology data with models. Bioinformatics
Daghino S, Turci F, Tomatis M, Favier A, Perotto S, Douki T et al (2006) Soil fungi reduce the iron content and the DNA damaging effects of asbestos fibers. Environ Sci Technol 40(18):5793–5798
Dai J, Churg A (2001) Relationship of fiber surface iron and active oxygen species to expression of procollagen, PDGF-A, and TGF-beta(1) in tracheal explants exposed to amosite asbestos. Am J Respir Cell Mol Biol 24(4):427–435
Dalle-Donne I (2007) Familial amyotrophic lateral sclerosis (FALS): emerging hints from redox proteomics. Highlight commentary on: “Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice—a model of familial amyotrophic lateral sclerosis”. Free Radic Biol Med 43(2):157–159
Dancey JE, Chen HX (2006) Strategies for optimizing combinations of molecularly targeted anticancer agents. Nat Rev Drug Discov 5(8):649–659
Daré E, Götz ME, Zhivotovsky B, Manzo L, Ceccatelli S (2000) Antioxidants J811 and 17beta-estradiol protect cerebellar granule cells from methylmercury-induced apoptotic cell death. J Neurosci Res 62(4):557–565
Dávalos A, Fernandezreal JM, Ricart W, Soler S, Molins A, Planas E et al (1994) Iron-related damage in acute ischemic stroke. Stroke 25(8):1543–1546
Davidsson L, Galan P, Kastenmayer P, Cherouvrier F, Juillerat MA, Hercberg S et al (1994) Iron bioavailability studied in infants: the influence of phytic acid and ascorbic acid in infant formulas based on soy isolate. Pediatr Res 36(6):816–822
Davies BW, Kohanski MA, Simmons LA, Winkler JA, Collins JJ, Walker GC (2009) Hydroxyurea induces hydroxyl radical-mediated cell death in Escherichia coli. Mol Cell 36(5):845–860
Davies MG, Hagen PO (1997) Systemic inflammatory response syndrome. Br J Surg 84(7):920–935
Davies MJ (2005) The oxidative environment and protein damage. Biochim Biophys Acta 1703(2):93–109
Day SM, Duquaine D, Mundada LV, Menon RG, Khan BV, Rajagopalan S et al (2003) Chronic iron administration increases vascular oxidative stress and accelerates arterial thrombosis. Circulation 107(20):2601–2606
de Carvalho CC, Fernandes P (2010) Production of metabolites as bacterial responses to the marine environment. Mar Drugs 8(3):705–727
De Freitas J, Wintz H, Kim JH, Poynton H, Fox T, Vulpe C (2003) Yeast, a model organism for iron and copper metabolism studies. Biometals 16(1):185–197
de Monyé C, Karcher DS, Boelaert JR, Gordeuk VR (1999) Bone marrow macrophage iron grade and survival of HIV-seropositive patients. Aids 13(3):375–380
de Valk B, Marx JJM (1999) Iron, atherosclerosis, and ischemic heart disease. Arch Internal Med 159(14):1542–1548
De Vizcaya-Ruiz A, Barbier O, Ruiz-Ramos R, Cebrian ME (2009) Biomarkers of oxidative stress and damage in human populations exposed to arsenic. Mutat Res 674(1–2):85–92
Debebe Z, Ammosova T, Jerebtsova M, Kurantsin-Mills J, Niu X, Charles S et al (2007) Iron chelators ICL670 and 311 inhibit HIV-1 transcription. Virology 367(2):324–333
Dekker MC, Giesbergen PC, Njajou OT, van Swieten JC, Hofman A, Breteler MM et al (2003) Mutations in the hemochromatosis gene (HFE), Parkinson’s disease and parkinsonism. Neurosci Lett 348(2):117–119
Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R et al (2008) Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med 36(1):296–327
Demir O, Singh S, Klimaschewski L, Kurnaz IA (2009) From birth till death: neurogenesis, cell cycle, and neurodegeneration. Anat Rec (Hoboken) 292(12):1953–1961
Demougeot C, Van Hoecke M, Bertrand N, Prigent-Tessier A, Mossiat C, Beley A et al (2004) Cytoprotective efficacy and mechanisms of the liposoluble iron chelator 2, 2 ‘-dipyridyl in the rat photothrombotic ischemic stroke model. J Pharm Exp Therapeut 311(3):1080–1087
Dentchev T, Hahn P, Dunaief JL (2005) Strong labeling for iron and the iron-handling proteins ferritin and ferroportin in the photoreceptor layer in age-related macular degeneration. Arch Ophthalmol 123(12):1745–1746
DePalma RG, Hayes VW, Chow BK, Shamayeva G, May PE, Zacharski LR (2010) Ferritin levels, inflammatory biomarkers, and mortality in peripheral arterial disease: a substudy of the iron (Fe) and atherosclerosis study (FeAST) Trial. J Vasc Surg
DePalma RG, Hayes VW, Zacharski LR (2007) Bloodletting: past and present. J Am Coll Surg 205(1):132–144
Dereure O, Jumez N, Bessis D, Gallix B, Guillot B (2008) Measurement of liver iron content by magnetic resonance imaging in 20 patients with overt porphyria cutanea tarda before phlebotomy therapy: a prospective study. Acta Derm Venereol 88(4):341–345
Desai TK, Jamil LH, Balasubramaniam M, Koff R, Bonkovsky HL (2008) Phlebotomy improves therapeutic response to interferon in patients with chronic hepatitis C: a meta-analysis of six prospective randomized controlled trials. Dig Dis Sci 53(3):815–822
Dever JB, Mallory MA, Mallory JE, Wallace D, Kowdley KV (2010) Phenotypic characteristics and diagnoses of patients referred to an iron overload clinic. Dig Dis Sci 55:803–807
Devireddy LR, Gazin C, Zhu X, Green MR (2005) A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 123(7):1293–1305
Di Marco V, Capra M, Gagliardotto F, Borsellino Z, Cabibi D, Barbaria F et al (2008) Liver disease in chelated transfusion-dependent thalassemics: the role of iron overload and chronic hepatitis C. Haematologica 93(8):1243–1246
Di Paola R, Mazzon E, Muia C, Crisafulli C, Genovese T, Di Bella P et al (2006) Green tea polyphenol extract attenuates zymosan-induced non-septic shock in mice. Shock 26(4):402–409
Dickens MG, Franz KJ (2010) A prochelator activated by hydrogen peroxide prevents metal-induced amyloid beta aggregation. Chembiochem 11(1):59–62
Diedrich JF, Bendheim PE, Kim YS, Carp RI, Haase AT (1991) Scrapie-associated prion protein accumulates in astrocytes during scrapie infection. Proc Natl Acad Sci USA 88(2):375–379
DiMauro S, Schon EA (2008) Mitochondrial disorders in the nervous system. Annu Rev Neurosci 31:91–123
Ding B, Chen KM, Ling HW, Sun F, Li X, Wan T et al (2009) Correlation of iron in the hippocampus with MMSE in patients with Alzheimer’s disease. J Magn Reson Imaging 29(4):793–798
Dobson P, Lanthaler K, Oliver SG, Kell DB (2009a) Implications of the dominant role of cellular transporters in drug uptake. Curr Top Med Chem 9:163–184
Dobson PD, Patel Y, Kell DB (2009b) “Metabolite-likeness” as a criterion in the design and selection of pharmaceutical drug libraries. Drug Disc Today 14:31–40
Dobson PD, Kell DB (2008) Carrier-mediated cellular uptake of pharmaceutical drugs: an exception or the rule? Nat Rev Drug Discov 7:205–220
Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G et al (2003) Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 42(10):2768–2773
Doraiswamy PM, Finefrock AE (2004) Metals in our minds: therapeutic implications for neurodegenerative disorders. Lancet Neurol 3(7):431–434
Doria E, Galleschi L, Calucci L, Pinzino C, Pilu R, Cassani E et al (2009) Phytic acid prevents oxidative stress in seeds: evidence from a maize (Zea mays L.) low phytic acid mutant. J Exp Bot 60(3):967–978
Double KL, Dedov VN, Fedorow H, Kettle E, Halliday GM, Garner B et al (2008) The comparative biology of neuromelanin and lipofuscin in the human brain. Cell Mol Life Sci 65(11):1669–1682
Double KL, Gerlach M, Schunemann V, Trautwein AX, Zecca L, Gallorini M et al (2003a) Iron-binding characteristics of neuromelanin of the human substantia nigra. Biochem Pharmacol 66(3):489–494
Double KL, Halliday GM, Henderson J, Griffiths FM, Heinemann T, Riederer P et al (2003b) The dopamine receptor agonist lisuride attenuates iron-mediated dopaminergic neurodegeneration. Exp Neurol 184(1):530–535
Double KL, Gerlach M, Youdim MB, Riederer P (2000) Impaired iron homeostasis in Parkinson’s disease. J Neural Transm Suppl 60: 37–58
Drakesmith H, Prentice A (2008) Viral infection and iron metabolism. Nat Rev Microbiol 6(7):541–552
Drayer B, Burger P, Hurwitz B, Dawson D, Cain J (1987) Reduced signal intensity on MR images of thalamus and putamen in multiple sclerosis—increased iron content. Am J Neuroradiol 8(3):413–419
Dreyfus F (2008) The deleterious effects of iron overload in patients with myelodysplastic syndromes. Blood Rev 22(Suppl 2):S29–S34
Dröge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82(1):47–95
Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD et al (2007) Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci 104(6):1777–1782
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464(7293):1357–1361
Duffy SJ, Biegelsen ES, Holbrook M, Russell JD, Gokce N, Keaney J F Jr et al (2001) Iron chelation improves endothelial function in patients with coronary artery disease. Circulation 103(23):2799–2804
Dunaief JL (2006) Iron induced oxidative damage as a potential factor in age-related macular degeneration: The Cogan Lecture. Invest Ophthalmol Vis Sci 47(11):4660–4664
Durant R, Klouche K, Delbosc S, Morena M, Amigues L, Beraud JJ et al (2004) Superoxide anion overproduction in sepsis: effects of vitamin E and simvastatin. Shock 22(1):34–39
Duvigneau JC, Piskernik C, Haindl S, Kloesch B, Hartl RT, Huttemann M et al (2008) A novel endotoxin-induced pathway: upregulation of heme oxygenase 1, accumulation of free iron, and free iron-mediated mitochondrial dysfunction. Lab Invest 88(1):70–77
Dwyer BE, Zacharski LR, Balestra DJ, Lerner AJ, Perry G, Zhu X et al (2009a) Getting the iron out: phlebotomy for Alzheimer’s disease? Med Hypotheses 72(5):504–509
Dwyer DJ, Kohanski MA, Collins JJ (2009b) Role of reactive oxygen species in antibiotic action and resistance. Curr Opin Microbiol 12(5):482–489
Dwyer DJ, Kohanski MA, Hayete B, Collins JJ (2007) Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol Syst Biol 3:91
Dwyer JH, Allayee H, Dwyer KM, Fan J, Wu H, Mar R et al (2004) Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N Engl J Med 350(1):29–37
Eissa A, Lebel RM, Korzan JR, Zavodni AE, Warren KG, Catz I et al (2009) Detecting lesions in multiple sclerosis at 4.7 tesla using phase susceptibility-weighting and T2-weighting. J Magn Reson Imaging 30(4):737–742
Ekblom K, Hultdin J, Stegmayr B, Johansson I, Van Guelpen B, Hallmans G et al (2007) Iron stores and HFE genotypes are not related to increased risk of ischemic stroke. A prospective nested case-referent study. Cerebrovasc Dis 24(5):405–411
Ekshyyan O, Aw TY (2004) Apoptosis: a key in neurodegenerative disorders. Curr Neurovasc Res 1(4):355–371
El Tannir El Tayara N, Delatour B, Le Cudennec C, Guegan M, Volk A, Dhenain M (2006) Age-related evolution of amyloid burden, iron load, and MR relaxation times in a transgenic mouse model of Alzheimer’s disease. Neurobiol Dis 22(1):199–208
Ellervik C, Birgens H, Tybjaerg-Hansen A, Nordestgaard BG (2007) Hemochromatosis genotypes and risk of 31 disease endpoints: meta-analyses including 66, 000 cases and 226, 000 controls. Hepatology 46(4):1071–1080
Epstein J, Sanderson IR, MacDonald TT (2009) Curcumin as a therapeutic agent: the evidence from in vitro, animal and human studies. Br J Nutr. doi:10.1017/S0007114509993667
Equitani F, Fernandez-Real JM, Menichella G, Koch M, Calvani M, Nobili V et al (2008) Bloodletting ameliorates insulin sensitivity and secretion in parallel to reducing liver iron in carriers of HFE gene mutations. Diabetes Care 31(1):3–8
Erkinjuntti T, Benavente O, Eliasziw M, Munoz DG, Sulkava R, Haltia M et al (1996) Diffuse vacuolization (spongiosis) and arteriolosclerosis in the frontal white matter occurs in vascular dementia. Arch Neurol 53(4):325–332
Escames G, Acuna-Castroviejo D, López LC, Tan DX, Maldonado MD, Sanchez-Hidalgo M et al (2006) Pharmacological utility of melatonin in the treatment of septic shock: experimental and clinical evidence. J Pharm Pharmacol 58(9):1153–1165
Evans JA (2008) Electronic publication and the narrowing of science and scholarship. Science 321(5887):395–399
Exley C (2006) Aluminium and iron, but neither copper nor zinc, are key to the precipitation of beta-sheets of Abeta_{42} in senile plaque cores in Alzheimer’s disease. J Alzheimers Dis 10(2–3):173–177
Exley C, Mamutse G, Korchazhkina O, Pye E, Strekopytov S, Polwart A et al (2006) Elevated urinary excretion of aluminium and iron in multiple sclerosis. Mult Scler 12(5):533–540
Fábián I, Csordás V (2003) Metal ion catalyzed autoxidation reactions: Kinetics and mechanisms. Adv Inorg Chem 54:395–461
Facchini FS (2002) The iron factor of aging: why do Americans age faster?. Fenestra Books, Tucson, AZ
Facchini FS, Hua NW, Stoohs RA (2002) Effect of iron depletion in carbohydrate-intolerant patients with clinical evidence of nonalcoholic fatty liver disease. Gastroenterology 122(4):931–939
Fakih S, Podinovskaia M, Kong X, Collins HL, Schaible UE, Hider RC (2008) Targeting the lysosome: fluorescent iron(III) chelators to selectively monitor endosomal/lysosomal labile iron pools. J Med Chem 51(15):4539–4552
Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D et al (2006) A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 9(5):341–349
Fan QW, Specht KM, Zhang C, Goldenberg DD, Shokat KM, Weiss WA (2003) Combinatorial efficacy achieved through two-point blockade within a signaling pathway-a chemical genetic approach. Cancer Res 63(24):8930–8938
Fantauzzi M, Pacella A, Atzei D, Gianfagna A, Andreozzi GB, Rossi A (2010) Combined use of X-ray photoelectron and Mössbauer spectroscopic techniques in the analytical characterization of iron oxidation state in amphibole asbestos. Anal Bioanal Chem 396(8):2889–2898
Fargion S, Fracanzani AL, Rossini A, Borzio M, Riggio O, Belloni G et al (2002) Iron reduction and sustained response to interferon-alpha therapy in patients with chronic hepatitis C: results of an Italian multicenter randomized study. Am J Gastroenterol 97(5):1204–1210
Fargion S, Mattioli M, Fracanzani AL, Sampietro M, Tavazzi D, Fociani P et al (2001) Hyperferritinemia, iron overload, and multiple metabolic alterations identify patients at risk for nonalcoholic steatohepatitis. Am J Gastroenterol 96(8):2448–2455
Fasano M, Bergamasco B, Lopiano L (2006) Modifications of the iron-neuromelanin system in Parkinson’s disease. J Neurochem 96(4):909–916
Fato R, Bergamini C, Leoni S, Strocchi P, Lenaz G (2008) Generation of reactive oxygen species by mitochondrial complex I: implications in neurodegeneration. Neurochem Res 33(12):2487–2501
Fattman CL, Tan RJ, Tobolewski JM, Oury TD (2006) Increased sensitivity to asbestos-induced lung injury in mice lacking extracellular superoxide dismutase. Free Radic Biol Med 40(4):601–607
Faucheux BA, Martin ME, Beaumont C, Hauw JJ, Agid Y, Hirsch EC (2003) Neuromelanin associated redox-active iron is increased in the substantia nigra of patients with Parkinson’s disease. J Neurochem 86(5):1142–1148
Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J et al (2010) PBT2 rapidly improves cognition in Alzheimer’s disease: additional phase II analyses. J Alzheimers Dis
Featherstone DE, Broadie K (2002) Wrestling with pleiotropy: genomic and topological analysis of the yeast gene expression network. Bioessays 24(3):267–274
Fedorow H, Tribl F, Halliday G, Gerlach M, Riederer P, Double KL (2005) Neuromelanin in human dopamine neurons: comparison with peripheral melanins and relevance to Parkinson’s disease. Prog Neurobiol 75(2):109–124
Fell DA (1996) Understanding the control of metabolism. Portland Press, London
Fernaeus S, Halldin J, Bedecs K, Land T (2005a) Changed iron regulation in scrapie-infected neuroblastoma cells. Brain Res Mol Brain Res 133(2):266–273
Fernaeus S, Land T (2005) Increased iron-induced oxidative stress and toxicity in scrapie-infected neuroblastoma cells. Neurosci Lett 382(3):217–220
Fernaeus S, Reis K, Bedecs K, Land T (2005b) Increased susceptibility to oxidative stress in scrapie-infected neuroblastoma cells is associated with intracellular iron status. Neurosci Lett 389(3):133–136
Fernandes de Godoy M, Takakura IT, Machado RD, Grassi LV, Nogueira PR (2007) Serum ferritin and obstructive coronary artery disease: angiographic correlation. Arquivos Brasileiros De Cardiologia 88(4):430–433
Fernández-Real JM, Peñarroja G, Castro A, García-Bragado F, López-Bermejo A, Ricart W (2002) Blood letting in high-ferritin type 2 diabetes: effects on vascular reactivity. Diabetes Care 25(12):2249–2255
Fernandez Y, Anglade F, Mitjavila S (2000) Paraquat and iron-dependent lipid peroxidation. NADPH versus NADPH-generating systems. Biol Trace Elem Res 74(3):191–201
Ferrali M, Signorini C, Caciotti B, Sugherini L, Ciccoli L, Giachetti D et al (1997a) Protection against oxidative damage of erythrocyte membrane by the flavonoid quercetin and its relation to iron chelating activity. FEBS Lett 416(2):123–129
Ferrali M, Signorini C, Sugherini L, Pompella A, Lodovici M, Caciotti B et al (1997b) Release of free, redox-active iron in the liver and DNA oxidative damage following phenylhydrazine intoxication. Biochem Pharmacol 53(11):1743–1751
Ferrara DE, Taylor WR (2005) Iron chelation and vascular function: in search of the mechanisms. Arterioscler Thromb Vasc Biol 25(11):2235–2237
Ferrara F, Ventura P, Vegetti A, Guido M, Abbati G, Corradini E et al (2009) Serum ferritin as a predictor of treatment outcome in patients with chronic hepatitis C. Am J Gastroenterol 104(3):605–616
Ferro JM, Dávalos A (2006) Other neuroprotective therapies on trial in acute stroke. Cerebrovasc Dis 21(Suppl 2):127–130
Ferrucci L, Semba RD, Guralnik JM, Ershler WB, Bandinelli S, Patel KV et al (2010) Proinflammatory state, hepcidin, and anemia in older persons. Blood 115(18):3810–3816
Filippi M, Agosta F (2009) Closing the clinical-imaging gap in multiple sclerosis? Imaging iron deposition in deep gray matter. J Neuroimaging 19(1):1–2
Finch CA, Bellotti V, Stray S, Lipschitz DA, Cook JD, Pippard MJ et al (1986) Plasma ferritin determination as a diagnostic tool. West J Med 145(5):657–663
Fine SL (2005) Age-related macular degeneration 1969–2004: a 35-year personal perspective. Am J Ophthalmol 139(3):405–420
Finefrock AE, Bush AI, Doraiswamy PM (2003) Current status of metals as therapeutic targets in Alzheimer’s disease. J Am Geriatr Soc 51(8):1143–1148
Finkel T (2005) Radical medicine: treating ageing to cure disease. Nat Rev Mol Cell Biol 6(12):971–976
Firdaus WJ, Wyttenbach A, Giuliano P, Kretz-Remy C, Currie RW, Arrigo AP (2006) Huntingtin inclusion bodies are iron-dependent centers of oxidative events. FEBS J 273(23):5428–5441
Flynn MR, Susi P (2010) Manganese, iron, and total particulate exposures to welders. J Occup Environ Hyg 7(2):115–126
Ford ES, Cogswell ME (1999) Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care 22(12):1978–1983
Forouhi NG, Harding AH, Allison M, Sandhu MS, Welch A, Luben R et al (2007) Elevated serum ferritin levels predict new-onset type 2 diabetes: results from the EPIC-Norfolk prospective study. Diabetologia 50(5):949–956
Forrester JS (2002) Prevention of plaque rupture: a new paradigm of therapy. Ann Intern Med 137(10):823–833
Forrester JS (2004) Common ancestors: chronic progressive diseases have the same pathogenesis. Clin Cardiol 27(4):186–190
Foster J, Goldmann W, Parnham D, Chong A, Hunter N (2001) Partial dissociation of PrP(Sc) deposition and vacuolation in the brains of scrapie and BSE experimentally affected goats. J Gen Virol 82(Pt 1):267–273
Foury F (1997) Human genetic diseases: a cross-talk between man and yeast. Gene 195:1–10
Foury F, Cazzalini O (1997) Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett 411:373–377
Foury F, Talibi D (2001) Mitochondrial control of iron homeostasis. A genome wide analysis of gene expression in a yeast frataxin-deficient strain. J Biol Chem 276(11):7762–7768
Fraenkel PG, Gibert Y, Holzheimer JL, Lattanzi VJ, Burnett SF, Dooley KA et al (2009) Transferrin-a modulates hepcidin expression in zebrafish embryos. Blood 113(12):2843–2850
Franchini M, Targher G, Capra F, Montagnana M, Lippi G (2008) The effect of iron depletion on chronic hepatitis C virus infection. Hepatol Internat 2(3):335–340
Franco JL, Posser T, Dunkley PR, Dickson PW, Mattos JJ, Martins R et al (2009a) Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free Radic Biol Med 47(4):449–457
Franco R, Sánchez-Olea R, Reyes-Reyes EM, Panayiotidis MI (2009b) Environmental toxicity, oxidative stress and apoptosis: menage a trois. Mutat Res 674(1–2):3–22
Franco OH, Kirkwood TB, Powell JR, Catt M, Goodwin J, Ordovas JM et al (2007) Ten commandments for the future of ageing research in the UK: a vision for action. BMC Geriatr 7:10
Franke JC, Plotz M, Prokop A, Geilen CC, Schmalz HG, Eberle J (2010) New caspase-independent but ROS-dependent apoptosis pathways are targeted in melanoma cells by an iron-containing cytosine analogue. Biochem Pharmacol 79(4):575–586
Franklin MR, Phillips JD, Kushner JP (2005) Attenuation of polychlorinated biphenyl induced uroporphyria by iron deprivation. Environ Toxicol Pharmacol 20(3):417–423
Franks NP (2006) Molecular targets underlying general anaesthesia. Br J Pharmacol 147(Suppl 1):S72–S81
Franks NP (2008) General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci 9(5):370–386
Freixes M, Rodríguez A, Dalfó E, Ferrer I (2006) Oxidation, glycoxidation, lipoxidation, nitration, and responses to oxidative stress in the cerebral cortex in Creutzfeldt-Jakob disease. Neurobiol Aging 27(12):1807–1815
Friedman A, Gałązka-Friedman J, Bauminger ER (2007) Iron as a trigger of neurodegeneration in Parkinson’s disease. Handb Clin Neurol 83:493–505
Friedman A, Galazka-Friedman J, Koziorowski D (2009) Iron as a cause of Parkinson disease—a myth or a well established hypothesis? Parkinsonism Rel Disord 15S3:S212–S214
Fubini B, Arean CO (1999) Chemical aspects of the toxicity of inhaled mineral dusts. Chem Soc Rev 28(6):373–381
Fuchs E, Gould E (2000) Mini-review: in vivo neurogenesis in the adult brain: regulation and functional implications. Eur J Neurosci 12(7):2211–2214
Fujihara J, Agusa T, Tanaka J, Fujii Y, Moritani T, Hasegawa M et al (2009) 8-Hydroxy-2’-deoxyguanosine (8-OHdG) as a possible marker of arsenic poisoning: a clinical case study on the relationship between concentrations of 8-OHdG and each arsenic compound in urine of an acute promyelocytic leukemia patient being treated with arsenic trioxide. Forensic Toxicol 27(1):41–44
Fujita N, Horiike S, Sugimoto R, Tanaka H, Iwasa M, Kobayashi Y et al (2007a) Hepatic oxidative DNA damage correlates with iron overload in chronic hepatitis C patients. Free Radic Biol Med 42(3):353–362
Fujita N, Sugimoto R, Takeo M, Urawa N, Mifuji R, Tanaka H et al (2007b) Hepcidin expression in the liver: relatively low level in patients with chronic hepatitis C. Mol Med 13(1–2):97–104
Fujita N, Sugimoto R, Urawa N, Tanaka H, Konishi M, Kobayashi Y et al (2007c) Influence of phlebotomy on iron-related gene expression levels in the livers of patients with chronic hepatitis C. J Gastroenterol 42(4):326–327
Fujita N, Miyachi H, Tanaka H, Takeo M, Nakagawa N, Kobayashi Y et al (2009) Iron overload is associated with hepatic oxidative damage to DNA in nonalcoholic steatohepatitis. Cancer Epidemiol Biomarkers Prev 18(2):424–432
Fujita N, Takei Y (2007) Iron, hepatitis C virus, and hepatocellular carcinoma: iron reduction preaches the gospel for chronic hepatitis C. J Gastroenterol 42(11):923–926
Fukuchi K, Tomoyasu S, Tsuruoka N, Gomi K (1994) Iron deprivation-induced apoptosis in HL-60 cells. FEBS Lett 350(1):139–142
Fulda S, Gorman AM, Hori O, Samali A (2010) Cellular stress responses: cell survival and cell death. Int J Cell Biol 2010:214074
Gackowski D, Kruszewski M, Bartlomiejczyk T, Jawien A, Ciecierski M, Olinski R (2002) The level of 8-oxo-7, 8-dihydro-2’-deoxyguanosine is positively correlated with the size of the labile iron pool in human lymphocytes. J Biol Inorg Chem 7(4–5):548–550
Gajda M, Banas K, Banas A, Jawien J, Mateuszuk L, Chlopicki S et al (2008) Distribution of selected elements in atherosclerotic plaques of apoE/LDLR-double knockout mice assessed by synchrotron radiation-induced micro-XRF spectrometry. X-Ray Spectrom 37(5):495–502
Gajdusek DC (2008) Review. Kuru and its contribution to medicine. Philos Trans R Soc Lond B Biol Sci 363(1510):3697–3700
Gakh O, Park S, Liu G, Macomber L, Imlay JA, Ferreira GC et al (2006) Mitochondrial iron detoxification is a primary function of frataxin that limits oxidative damage and preserves cell longevity. Human Mol Genet 15(3):467–479
Gal S, Fridkin M, Amit T, Zheng H, Youdim MBH (2006) M30, a novel multifunctional neuroprotective drug with potent iron chelating and brain selective monoamine oxidase-ab inhibitory activity for Parkinson’s disease. J Neural Transm Suppl 70:447–456
Gal S, Zheng H, Fridkin M, Youdim MB (2009) Restoration of nigrostriatal dopamine neurons in post-MPTP treatment by the novel multifunctional brain-permeable iron chelator-monoamine oxidase inhibitor drug, M30. Neurotox Res
Galaris D, Mantzaris M, Amorgianiotis C (2008a) Oxidative stress and aging: the potential role of iron. Hormones (Athens) 7(2):114–122
Galaris D, Skiada V, Barbouti A (2008b) Redox signaling and cancer: the role of “labile” iron. Cancer Lett 266(1):21–29
Galley HF, Davies MJ, Webster NR (1996) Ascorbyl radical formation in patients with sepsis: effect of ascorbate loading. Free Radic Biol Med 20(1):139–143
Galley HF, Howdle PD, Walker BE, Webster NR (1997) The effects of intravenous antioxidants in patients with septic shock. Free Radic Biol Med 23(5):768–774
Galley HF, Webster NR (1996) Elevated serum bleomycin-detectable iron concentrations in patients with sepsis syndrome. Intensive Care Med 22(3):226–229
Galli SJ, Tsai M, Piliponsky AM (2008) The development of allergic inflammation. Nature 454(7203):445–454
Gamberini MR, De Sanctis V, Gilli G (2008) Hypogonadism, diabetes mellitus, hypothyroidism, hypoparathyroidism: incidence and prevalence related to iron overload and chelation therapy in patients with thalassaemia major followed from 1980 to 2007 in the Ferrara Centre. Pediatr Endocrinol Rev 6(Suppl 1):158–169
Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T et al (2008) Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 70(24):2277–2283
Gan EK, Ayonrinde OT, Trinder D, Olynyk JK (2010) Phenotypic expression of hereditary hemochromatosis: what have we learned from the population studies? Curr Gastroenterol Rep 12(1):7–12
Gao F, Linhartova L, Johnston AM, Thickett DR (2008) Statins and sepsis. Br J Anaesth 100(3):288–298
García-Castiñeiras S (2010) Iron, the retina and the lens: A focused review. Exp Eye Res 90:664–678
Garey M, Johnson D (1979) Computers and intractability: a guide to the theory of NP-completeness. Freeman, San Francisco
Garoufi AJ, Prassouli AA, Attilakos AV, Voudris KA, Katsarou ES (2006) Homozygous MTHFR C677T gene mutation and recurrent stroke in an infant. Pediatr Neurol 35(1):49–51
Gattermann N (2008) Overview of guidelines on iron chelation therapy in patients with myelodysplastic syndromes and transfusional iron overload. Int J Hematol 88(1):24–29
Gattermann N (2009) The treatment of secondary hemochromatosis. Dtsch Ärztebl Int 106(30): 499–504, I
Gazzano E, Turci F, Foresti E, Putzu MG, Aldieri E, Silvagno F et al (2007) Iron-loaded synthetic chrysotile: a new model solid for studying the role of iron in asbestos toxicity. Chem Res Toxicol 20(3):380–387
Ge Y, Jensen JH, Lu H, Helpern JA, Miles L, Inglese M et al (2007) Quantitative assessment of iron accumulation in the deep gray matter of multiple sclerosis by magnetic field correlation imaging. AJNR Am J Neuroradiol 28(9):1639–1644
Gems D (2009) Ageing and oxidants in the nematode Caenorhabditis elegans. SEB Exp Biol Ser 62:31–56
Gems D, Doonan R (2009) Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong? Cell Cycle 8(11):1681–1687
Gems D, Partridge L (2008) Stress-response hormesis and aging: “that which does not kill us makes us stronger”. Cell Metab 7(3):200–203
Gennero L, Roos MA, D’Amelio P, Denysenko T, Morra E, Sperber K et al (2010) Iron metabolism markers and haptoglobin phenotypes in susceptibility to HSV-1 or/and HSV-2 lesion relapses. Cell Biochem Funct 28(2):142–148
Georgiou NA, van der Bruggen T, Oudshoorn M, Nottet HH, Marx JJ, van Asbeck BS (2000) Combining iron chelators with the nucleoside analog didanosine in anti-HIV therapy. Transfus Sci 23(3):249–250
Gerber S, Assmus H, Bakker B, Klipp E (2007) Drug-efficacy depends on the inhibitor type and the target position in a metabolic network—a systematic study. J Theor Biol 255:442–455
Gerlach M, Benshachar D, Riederer P, Youdim MBH (1994) Altered brain metabolism of iron as a cause of neurodegenerative diseases. J Neurochem 63(3):793–807
Gerlach M, Double KL, Ben-Shachar D, Zecca L, Youdim MB, Riederer P (2003) Neuromelanin and its interaction with iron as a potential risk factor for dopaminergic neurodegeneration underlying Parkinson’s disease. Neurotox Res 5(1–2):35–44
Gerlach M, Riederer P, Double KL (2008) Neuromelanin-bound ferric iron as an experimental model of dopaminergic neurodegeneration in Parkinson’s disease. Parkinsonism Relat Disord 14(Suppl 2):S185–S188
Geronikaki AA, Gavalas AM (2006) Antioxidants and inflammatory disease: synthetic and natural antioxidants with anti-inflammatory activity. Comb Chem High Throughput Screen 9(6):425–442
Ghio A, Tan RJ, Ghio K, Fattman CL, Oury TD (2009) Iron accumulation and expression of iron-related proteins following murine exposure to crocidolite. J Environ Pathol Toxicol Oncol 28(2):153–162
Ghio AJ, Carter JD, Richards JH, Richer LD, Grissom CK, Elstad MR (2003) Iron and iron-related proteins in the lower respiratory tract of patients with acute respiratory distress syndrome. Crit Care Med 31(2):395–400
Ghio AJ, Churg A, Roggli VL (2004) Ferruginous bodies: implications in the mechanism of fiber and particle toxicity. Toxicol Pathol 32(6):643–649
Ghosh B, Antonio T, Reith ME, Dutta AK (2010) Discovery of 4-(4-(2-((5-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)ethyl) piperazin-1-yl)quinolin-8-ol and its analogues as highly potent dopamine D2/D3 agonists and as iron chelator: in vivo activity indicates potential application in symptomatic and neuroprotective therapy for Parkinson’s disease. J Med Chem 53(5):2114–2125
Ghoshal N, Cali I, Perrin RJ, Josephson SA, Sun N, Gambetti P et al (2009) Codistribution of amyloid beta plaques and spongiform degeneration in familial Creutzfeldt-Jakob disease with the E200K–129M haplotype. Arch Neurol 66(10):1240–1246
Ghribi O, Golovko MY, Larsen B, Schrag M, Murphy EJ (2006) Deposition of iron and beta-amyloid plaques is associated with cortical cellular damage in rabbits fed with long-term cholesterol-enriched diets. J Neurochem 99(2):438–449
Gieseg SP, Leake DS, Flavall EM, Amit Z, Reid L, Yang YT (2009) Macrophage antioxidant protection within atherosclerotic plaques. Frontiers Biosci 14:1230–1246
Gillum RF, Sempos CT, Makuc DM, Looker AC, Chien CY, Ingram DD (1996) Serum transferrin saturation, stroke incidence, and mortality in women and men. The NHANES I epidemiologic followup study. National health and nutrition examination survey. Am J Epidemiol 144(1):59–68
Gilmour PS, Brown DM, Beswick PH, MacNee W, Rahman I, Donaldson K (1997) Free radical activity of industrial fibers: role of iron in oxidative stress and activation of transcription factors. Environ Health Perspect 105(Suppl 5):1313–1317
Girelli D, Pasino M, Goodnough JB, Nemeth E, Guido M, Castagna A et al (2009) Reduced serum hepcidin levels in patients with chronic hepatitis C. J Hepatol 51(5):845–852
Giustarini D, Dalle-Donne I, Tsikas D, Rossi R (2009) Oxidative stress and human diseases: origin, link, measurement, mechanisms, and biomarkers. Crit Rev Clin Lab Sci 46(5–6):241–281
Glahn RP, Wortley GM, South PK, Miller DD (2002) Inhibition of iron uptake by phytic acid, tannic acid, and ZnCl2: studies using an in vitro digestion/Caco-2 cell model. J Agric Food Chem 50(2):390–395
Gloire G, Legrand-Poels S, Piette J (2006) NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 72(11):1493–1505
Gloire G, Piette J (2009) Redox regulation of nuclear post-translational modifications during NF-kappaB activation. Antioxid Redox Signal 11(9):2209–2222
Goel A, Dani V, Dhawan DK (2006) Role of zinc in mitigating the toxic effects of chlorpyrifos on hematological alterations and electron microscopic observations in rat blood. Biometals 19(5):483–492
Goldberg DE (1989) Genetic algorithms in search, optimization and machine learning. Addison-Wesley, Reading, MA
Goldberg DE (2002) The design of innovation: lessons from and for competent genetic algorithms. Kluwer, Boston
Goldstein S, Merényi G (2008) The chemistry of peroxynitrite: implications for biological activity. Methods Enzymol 436:49–61
Goldstein S, Meyerstein D, Czapski G (1993) The Fenton reagents. Free Radic Biol Med 15(4):435–445
Goltsov A, Maryashkin A, Swat M, Kosinsky Y, Humphery-Smith I, Demin O et al (2009) Kinetic modelling of NSAID action on COX-1: focus on in vitro/in vivo aspects and drug combinations. Eur J Pharm Sci 36(1):122–136
Gómez-Pinilla F (2008) The influences of diet and exercise on mental health through hormesis. Ageing Res Rev 7(1):49–62
Gómez Ravetti M, Rosso OA, Berretta R, Moscato P (2010) Uncovering molecular biomarkers that correlate cognitive decline with the changes of hippocampus’ gene expression profiles in Alzheimer’s disease. PLoSOne 5(4):e10153
Goncalves S, Paupe V, Dassa EP, Rustin P (2008) Deferiprone targets aconitase: implication for Friedreich’s ataxia treatment. BMC Neurol 8:20
González-Cabo P, Llorens JV, Palau F, Moltó MD (2009) Friedreich ataxia: an update on animal models, frataxin function and therapies. Adv Exp Med Biol 652:247–261
González-Cabo P, Vázquez-Manrique RP, García-Gimeno MA, Sanz P, Palau F (2005) Frataxin interacts functionally with mitochondrial electron transport chain proteins. Hum Mol Genet 14(15):2091–2098
Good PF, Hsu A, Werner P, Perl DP, Olanow CW (1998) Protein nitration in Parkinson’s disease. J Neuropathol Exp Neurol 57(4):338–342
Good PF, Perl DP, Bierer LM, Schmeidler J (1992) Selective accumulation of aluminum and iron in the neurofibrillary tangles of Alzheimer’s disease: a laser microprobe (LAMMA) study. Ann Neurol 31(3):286–292
Good PF, Werner P, Hsu A, Olanow CW, Perl DP (1996) Evidence of neuronal oxidative damage in Alzheimer’s disease. Am J Pathol 149(1):21–28
Goodall EF, Haque MS, Morrison KE (2008) Increased serum ferritin levels in amyotrophic lateral sclerosis (ALS) patients. J Neurol 255(11):1652–1656
Goode HF, Webster NR (1993) Free radicals and antioxidants in sepsis. Crit Care Med 21(11):1770–1776
Goralska M, Ferrell J, Harned J, Lall M, Nagar S, Fleisher LN et al (2009a) Iron metabolism in the eye: a review. Exp Eye Res 88(2):204–215
Goralska M, Nagar S, Colitz CM, Fleisher LN, McGahan MC (2009b) Changes in ferritin H- and L-chains in canine lenses with age-related nuclear cataract. Invest Ophthalmol Vis Sci 50(1):305–310
Goralska M, Nagar S, Fleisher LN, McGahan MC (2009c) Distribution of ferritin chains in canine lenses with and without age-related nuclear cataracts. Mol Vis 15:2404–2410
Gordeuk VR, Delanghe JR, Langlois MR, Boelaert JR (2001) Iron status and the outcome of HIV infection: an overview. J Clin Virol 20(3):111–115
Gordeuk VR, Onojobi G, Schneider MF, Dawkins FW, Delapenha R, Voloshin Y et al (2006) The association of serum ferritin and transferrin receptor concentrations with mortality in women with human immunodeficiency virus infection. Haematologica 91(6):739–743
Goris RJ (1990) Mediators of multiple organ failure. Intensive Care Med 16(Suppl 3):S192–196
Gorria M, Tekpli X, Rissel M, Sergent O, Huc L, Landvik N et al (2008) A new lactoferrin- and iron-dependent lysosomal death pathway is induced by benzo[a]pyrene in hepatic epithelial cells. Toxicol Appl Pharmacol 228(2):212–224
Götz M, Huttner WB (2005) The cell biology of neurogenesis. Nat Rev Mol Cell Biol 6(10):777–788
Gotz ME, Double K, Gerlach M, Youdim MBH, Riederer P (2004) The relevance of iron in the pathogenesis of Parkinson’s disease. Redox-Active Metals Neurol Disord 1012:193–208
Gould E (2007) How widespread is adult neurogenesis in mammals? Nat Rev Neurosci 8(6):481–488
Governa M, Amati M, Fontana S, Visona I, Botta GC, Mollo F et al (1999) Role of iron in asbestos-body-induced oxidant radical generation. J Toxicol Environ Health A 58(5):279–287
Gozzelino R, Jeney V, Soares MP (2010) Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol 50:323–354
Graf E, Eaton JW (1990) Antioxidant functions of phytic acid. Free Radic Biol Med 8(1):61–69
Graf E, Eaton JW (1993) Suppression of colonic cancer by dietary phytic acid. Nutr Cancer 19(1):11–19
Graf E, Empson KL, Eaton JW (1987) Phytic acid. A natural antioxidant. J Biol Chem 262(24):11647–11650
Graf E, Mahoney JR, Bryant RG, Eaton JW (1984) Iron-catalyzed hydroxyl radical formation. Stringent requirement for free iron coordination site. J Biol Chem 259(6):3620–3624
Grainger DJ (2007) TGF-beta and atherosclerosis in man. Cardiovasc Res 74(2):213–222
Granic I, Dolga AM, Nijholt IM, van Dijk G, Eisel ULM (2009) Inflammation and NF-kappaB in Alzheimer’s disease and diabetes. J Alzheimers Dis 16(4):809–821
Grases F, Simonet BM, Prieto RM, March JG (2001) Dietary phytate and mineral bioavailability. J Trace Elem Med Biol 15(4):221–228
Grasshoff C, Drexler B, Rudolph U, Antkowiak B (2006) Anaesthetic drugs: linking molecular actions to clinical effects. Curr Pharm Des 12(28):3665–3679
Greenberg PL (2006) Myelodysplastic syndromes: iron overload consequences and current chelating therapies. J Natl Compr Canc Netw 4(1):91–96
Greenberg PL, Attar E, Battiwalla M, Bennett JM, Bloomfield CD, DeCastro CM et al (2008) Myelodysplastic syndromes. J Natl Compr Canc Netw 6(9):902–926
Greene BT, Thorburn J, Willingham MC, Thorburn A, Planalp RP, Brechbiel MW et al (2002) Activation of caspase pathways during iron chelator-mediated apoptosis. J Biol Chem 277(28):25568–25575
Greene LS (1995) Asthma and oxidant stress—nutritional, environmental, and genetic risk factors. J Am Coll Nutr 14(4):317–324
Grimaud J, Millar J, Thorpe JW, Moseley IF, McDonald WI, Miller DH (1995) Signal intensity on MRI of basal ganglia in multiple sclerosis. J Neurol Neurosurg Psychiatry 59(3):306–308
Grootveld M, Halliwell B (1986) Aromatic hydroxylation as a potential measure of hydroxyl-radical formation in vivo. Identification of hydroxylated derivatives of salicylate in human body fluids. Biochem J 237(2):499–504
Grossi C, Francese S, Casini A, Rosi MC, Luccarini I, Fiorentini A et al (2009) Clioquinol decreases amyloid-beta burden and reduces working memory impairment in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis 96(3):423–440
Gu Y, Hua Y, Keep RF, Morgenstern LB, Xi G (2009) Deferoxamine reduces intracerebral hematoma-induced iron accumulation and neuronal death in piglets. Stroke 40(6):2241–2243
Gudmundsdóttir KB, Sigurdarson S, Kristinsson J, Eiríksson T, Jóhannesson T (2006) Iron and iron/manganese ratio in forage from Icelandic sheep farms: relation to scrapie. Acta Vet Scand 48:16
Gulumian M, van Wyk JA (1987) Hydroxyl radical production in the presence of fibres by a Fenton-type reaction. Chem Biol Interact 62(1):89–97
Guo L, Morris DG, Liu XY, Vaslet C, Hurt RH, Kane AB (2007) Iron bioavailability and redox activity in diverse carbon nanotube samples. Chem Mater 19(14):3472–3478
Gupta RC, Milatovic D (2009) Toxicants and neurodegenerative diseases special issue. Toxicol Appl Pharmacol 240(2):123
Gürkan E, Ergun Y, Zorludemir S, Başlamişli F, Koçak R (2005) Liver involvement in sickle cell disease. Turk J Gastroenterol 16(4):194–198
Gusarov I, Shatalin K, Starodubtseva M, Nudler E (2009) Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 325(5946):1380–1384
Gusella JF, MacDonald ME (2000) Unmasking polyglutamine triggers in neurodegenerative disease. Nat Rev Neurosci 1(2):109–115
Gutierrez JM, Rucavado A, Escalante T, Lomonte B, Angulo Y, Fox JW (2010) Tissue pathology induced by snake venoms: how to understand a complex pattern of alterations from a systems biology perspective. Toxicon 55(1):166–170
Gutteridge JMC, Mitchell J (1999) Redox imbalance in the critically ill. Br Med Bull 55(1):49–75
Guyader D, Thirouard AS, Erdtmann L, Rakba N, Jacquelinet S, Danielou H et al (2007) Liver iron is a surrogate marker of severe fibrosis in chronic hepatitis C. J Hepatol 46(4):587–595
Ha YM, Chung SW, Kim JM, Kim DH, Kim JY, Lee EK et al (2010) Molecular activation of NF-kappaB, pro-inflammatory mediators, and signal pathways in gamma-irradiated mice. Biotechnol Lett 32(3):373–378
Haacke EM, Cheng NY, House MJ, Liu Q, Neelavalli J, Ogg RJ et al (2005) Imaging iron stores in the brain using magnetic resonance imaging. Magn Reson Imaging 23(1):1–25
Haacke EM, Garbern J, Miao Y, Habib C, Liu M (2010) Iron stores and cerebral veins in MS studied by susceptibility weighted imaging. Int Angiol 29(2):149–157
Haacke EM, Makki M, Ge Y, Maheshwari M, Sehgal V, Hu J et al (2009) Characterizing iron deposition in multiple sclerosis lesions using susceptibility weighted imaging. J Magn Reson Imaging 29(3):537–544
Haas H, Eisendle M, Turgeon BG (2008) Siderophores in fungal physiology and virulence. Annu Rev Phytopathol 46:149–187
Hadacek F, Bachmann G, Engelmeier D, Chobot V (2010) Hormesis and a chemical raison d’être for secondary plant metabolites. Dose-response (in press)
Haddad JJ, Olver RE, Land SC (2000) Antioxidant/pro-oxidant equilibrium regulates HIF-1alpha and NF-kappa B redox sensitivity. Evidence for inhibition by glutathione oxidation in alveolar epithelial cells. J Biol Chem 275(28):21130–21139
Hadley D, Orlin A, Brown G, Brucker AJ, Ho AC, Regillo CD et al (2010) Analysis of six genetic risk factors highly associated with AMD in the region surrounding ARMS2 and HTRA1 on chromosome 10, region q26. Invest Ophthalmol Vis Sci 51(4):2191–2196
Hagen J (2006) Industrial Catalysis: A Practical Approach. Wiley VCH Chemie, Weinheim
Haggarty SJ, Clemons PA, Schreiber SL (2003) Chemical genomic profiling of biological networks using graph theory and combinations of small molecule perturbations. J Am Chem Soc 125(35):10543–10545
Hague T, Andrews PLR, Barker J, Naughton DP (2006) Dietary chelators as antioxidant enzyme mimetics: implications for dietary intervention in neurodegenerative diseases. Behav Pharmacol 17(5–6):425–430
Hahn P, Milam AH, Dunaief JL (2003) Maculas affected by age-related macular degeneration contain increased chelatable iron in the retinal pigment epithelium and Bruch’s membrane. Arch Ophthalmol 121(8):1099–1105
Hahn P, Qian Y, Dentchev T, Chen L, Beard J, Harris ZL et al (2004) Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc Natl Acad Sci U S A 101(38):13850–13855
Hallberg L, Brune M, Rossander L (1989) Iron absorption in man: ascorbic acid and dose-dependent inhibition by phytate. Am J Clin Nutr 49(1):140–144
Halliwell B (2007) Dietary polyphenols: good, bad, or indifferent for your health? Cardiovasc Res 73(2):341–347
Halliwell B (2009) The wanderings of a free radical. Free Radic Biol Med 46(5):531–542
Halliwell B, Gutteridge JMC (2006) Free radicals in biology and medicine, 4th edn. Oxford University Press, Oxford
Halliwell B, Rafter J, Jenner A (2005) Health promotion by flavonoids, tocopherols, tocotrienols, and other phenols: direct or indirect effects? Antioxidant or not? Am J Clin Nutr 81(1 Suppl):268S–276S
Hammond KE, Metcalf M, Carvajal L, Okuda DT, Srinivasan R, Vigneron D et al (2008) Quantitative in vivo magnetic resonance imaging of multiple sclerosis at 7 Tesla with sensitivity to iron. Ann Neurol 64(6):707–713
Han JY, Takeshita K, Utsumi H (2001) Noninvasive detection of hydroxyl radical generation in lung by diesel exhaust particles. Free Radic Biol Med 30(5):516–525
Han O, Failla ML, Hill AD, Morris ER, Smith J C Jr (1994) Inositol phosphates inhibit uptake and transport of iron and zinc by a human intestinal cell line. J Nutr 124(4):580–587
Handl J, Kell DB, Knowles J (2007) Multiobjective optimization in bioinformatics and computational biology. IEEE Trans Comput Biol Bioinform 4(2):279–292
Hanson L, Roeytenberg A, Martinez PM, Coppes VG, Sweet DC, Rao RJ et al (2009) Intranasal deferoxamine provides increased brain exposure and significant protection in rat ischemic stroke. J Pharmacol Exp Ther 330:679–686
Hanson LN, Engelman HM, Alekel DL, Schalinske KL, Kohut ML, Reddy MB (2006) Effects of soy isoflavones and phytate on homocysteine, C-reactive protein, and iron status in postmenopausal women. Am J Clin Nutr 84(4):774–780
Hansson GK (2001) Immune mechanisms in atherosclerosis. Arterioscler Thromb Vasc Biol 21(12):1876–1890
Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352(16):1685–1695
Haq RU, Wereley JP, Chitambar CR (1995) Induction of apoptosis by iron deprivation in human leukemic CCRF-CEM cells. Exp Hematol 23(5):428–432
Hardy JA, Aust AE (1995a) The effect of iron binding on the ability of crocidolite asbestos to catalyze DNA single-strand breaks. Carcinogenesis 16(2):319–325
Hardy JA, Aust AE (1995b) Iron in asbestos chemistry and carcinogenicity. Chem Rev 95(1):97–118
Harvey PT (2003) Common eye diseases of elderly people: identifying and treating causes of vision loss. Gerontology 49(1):1–11
Hasinoff BB, Rahimtula AD, Omar RF (1990) NADPH-cytochrome-P-450 reductase promoted hydroxyl radical production by the iron(III)-ochratoxin A complex. Biochim Biophys Acta 1036(1):78–81
Hassoun EA, Wang X (1999) Time- and concentration-dependent production of superoxide anion, nitric oxide, DNA damage and cellular death by ricin in the J774A.1 macrophage cells. J Biochem Mol Toxicol 13(3–4):179–185
Hastie T, Tibshirani R, Friedman J (2001) The elements of statistical learning: data mining, inference and prediction. Springer, Berlin
Hattori N, Oda S, Sadahiro T, Nakamura M, Abe R, Shinozaki K et al (2009) YKL-40 identified by proteomic analysis as a biomarker of sepsis. Shock 32(4):393–400
Haugen AC, Di Prospero NA, Parker JS, Fannin RD, Chou J, Meyer JN et al (2010) Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: cellular model of pathology. PLoS Genet 6(1):e1000812
Hawkins PT, Poyner DR, Jackson TR, Letcher AJ, Lander DA, Irvine RF (1993) Inhibition of iron-catalysed hydroxyl radical formation by inositol polyphosphates: a possible physiological function for myo-inositol hexakisphosphate. Biochem J 294(Pt 3):929–934
Hayashi H, Wakusawa S, Yano M (2006) Iron removal by phlebotomy for the prophylaxis of fulminant hepatitis in a Wilson disease model of Long-Evans Cinnamon Rats. Hepatol Res 35(4):276–280
Hayashi H, Yano M (2002) Iron cytotoxicity in chronic hepatitis C. J Health Sci 48(3):227–231
Hayashi T, Ueno Y, Okamoto T (1993) Oxidoreductive regulation of nuclear factor kappa B. Involvement of a cellular reducing catalyst thioredoxin. J Biol Chem 268(15):11380–11388
Hayes DP (2009) Nutritional hormesis and aging. Dose Response 8(1):10–15
He X, Hahn P, Iacovelli J, Wong R, King C, Bhisitkul R et al (2007) Iron homeostasis and toxicity in retinal degeneration. Prog Retin Eye Res 26(6):649–673
He X, Yablonskiy DA (2009) Biophysical mechanisms of phase contrast in gradient echo MRI. Proc Natl Acad Sci USA 106(32):13558–13563
Heathcote EJ (2004) Prevention of hepatitis C virus-related hepatocellular carcinoma. Gastroenterology 127(5 Suppl 1):S294–S302
Heckl S (2007) Future contrast agents for molecular imaging in stroke. Curr Med Chem 14(16):1713–1728
Hegde ML, Bharathi P, Suram A, Venugopal C, Jagannathan R, Poddar P et al (2009) Challenges associated with metal chelation therapy in Alzheimer’s disease. J Alzheimers Dis 17(3):457–468
Hegreness M, Shoresh N, Damian D, Hartl D, Kishony R (2008) Accelerated evolution of resistance in multidrug environments. Proc Natl Acad Sci USA 105(37):13977–13981
Heinrich R, Schuster S (1996) The regulation of cellular systems. Chapman & Hall, New York
Heintz NH, Janssen-Heininger YM, Mossman BT (2010) Asbestos, lung cancers, and mesotheliomas: from molecular approaches to targeting tumor survival pathways. Am J Respir Cell Mol Biol 42(2):133–139
Helal GK (2008) Systemic administration of Zn2+ during the reperfusion phase of transient cerebral ischaemia protects rat hippocampus against iron-catalysed postischaemic injury. Clin Exp Pharmacol Physiol 35(7):775–781
Henshall TL, Tucker B, Lumsden AL, Nornes S, Lardelli MT, Richards RI (2009) Selective neuronal requirement for huntingtin in the developing zebrafish. Hum Mol Genet 18(24):4830–4842
Herce-Pagliai C, Kotecha S, Shuker DEG (1998) Analytical methods for 3-nitrotyrosine as a marker of exposure to reactive nitrogen species: a review. Nitric Oxide-Biol Chem 2(5):324–336
Herrgård MJ, Swainston N, Dobson P, Dunn WB, Arga KY, Arvas M et al (2008) A consensus yeast metabolic network obtained from a community approach to systems biology. Nat Biotechnol 26(10):1155–1160
Hershko C, Weatherall DJ (1988) Iron-chelating therapy. Crit Rev Clin Lab Sci 26(4):303–345
Hider RC, Liu ZD, Khodr HH (2001) Metal chelation of polyphenols. Flavonoids Other Polyphenols 335:190–203
Hider RC, Ma Y, Molina-Holgado F, Gaeta A, Roy S (2008) Iron chelation as a potential therapy for neurodegenerative disease. Biochem Soc Trans 36(Pt 6):1304–1308
Hileti D, Panayiotidis P, Hoffbrand AV (1995) Iron chelators induce apoptosis in proliferating cells. Br J Haematol 89(1):181–187
Hill RH Jr, Todd GD, Kilbourne EM, Cline RE, McCraw J, Orti DL et al (1987) Gas chromatographic/mass spectrometric determination of aniline in food oils associated with the Spanish toxic oil syndrome. Bull Environ Contam Toxicol 39(3):511–515
Hillmer AS, Putcha P, Levin J, Hogen T, Hyman BT, Kretzschmar H et al (2009) Converse modulation of toxic alpha-synuclein oligomers in living cells by N’-benzylidene-benzohydrazide derivates and ferric iron. Biochem Biophys Res Commun
Himmelfarb J, Stenvinkel P, Ikizler TA, Hakim RM (2002) The elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int 62(5):1524–1538
Hininger I, Waters R, Osman M, Garrel C, Fernholz K, Roussel AM et al (2005) Acute prooxidant effects of vitamin C in EDTA chelation therapy and long-term antioxidant benefits of therapy. Free Rad Biol Med 38(12):1565–1570
Hinson JA, Reid AB, McCullough SS, James LP (2004) Acetaminophen-induced hepatotoxicity: role of metabolic activation, reactive oxygen/nitrogen species, and mitochondrial permeability transition. Drug Metab Rev 36(3–4):805–822
Hintze KJ, Theil EC (2006) Cellular regulation and molecular interactions of the ferritins. Cell Mol Life Sci 63(5):591–600
Hippeli S, Elstner EF (1999) Transition metal ion-catalyzed oxygen activation during pathogenic processes. FEBS Lett 443(1):1–7
Hirsch EC (2009) Iron transport in Parkinson’s disease. Parkinsonism Rel Disord 15:S209–S211
Hirsch EC, Faucheux BA (1998) Iron metabolism and Parkinson’s disease. Mov Disord 13(Suppl 1):39–45
Hoefnagel MH, Starrenburg MJ, Martens DE, Hugenholtz J, Kleerebezem M, Van S II et al (2002) Metabolic engineering of lactic acid bacteria, the combined approach: kinetic modelling, metabolic control and experimental analysis. Microbiology 148(Pt 4):1003–1013
Hoffmann GR (2009) A perspective on the scientific, philosophical, and policy dimensions of hormesis. Dose Response 7(1):1–51
Honda K, Casadesus G, Petersen RB, Perry G, Smith MA (2004) Oxidative stress and redox-active iron in Alzheimer’s disease. Ann NY Acad Sci 1012:179–182
Hoops S, Sahle S, Gauges R, Lee C, Pahle J, Simus N et al (2006) COPASI: a COmplex PAthway SImulator. Bioinformatics 22(24):3067–3074
Hopkins AL, Mason JS, Overington JP (2006) Can we rationally design promiscuous drugs? Curr Opin Struct Biol 16(1):127–136
Hori A, Mizoue T, Kasai H, Kawai K, Matsushita Y, Nanri A et al (2010) Body iron store as a predictor of oxidative DNA damage in healthy men and women. Cancer Sci 101(2):517–522
Horn KD (1998) Evolving strategies in the treatment of sepsis and systemic inflammatory response syndrome (SIRS). QJM 91(4):265–277
Horváth I, Donnelly LE, Kiss A, Paredi P, Kharitonov SA, Barnes PJ (1998) Raised levels of exhaled carbon monoxide are associated with an increased expression of heme oxygenase-1 in airway macrophages in asthma: a new marker of oxidative stress. Thorax 53(8):668–672
Horwitz LD, Rosenthal EA (1999) Iron-mediated cardiovascular injury. Vasc Med 4(2):93–99
Horwitz LD, Sherman NA, Kong YN, Pike AW, Gobin J, Fennessey PV et al (1998) Lipophilic siderophores Mycobacterium tuberculosis prevent cardiac reperfusion injury. Proc Natl Acad Sci 95(9):5263–5268
Hotchkiss RS, Karl IE (2003) The pathophysiology and treatment of sepsis. N Engl J Med 348(2):138–150
Howes PS, Zacharski LR, Sullivan J, Chow B (2000) Role of stored iron in atherosclerosis. J Vasc Nurs 18(4):109–114 (quiz 115–106)
Hsieh SR, Tsai DC, Chen JY, Tsai SW, Liou YM (2009) Green tea extract protects rats against myocardial infarction associated with left anterior descending coronary artery ligation. Pflugers Arch 458(4):631–642
Hua NW, Stoohs RA, Facchini FS (2001) Low iron status and enhanced insulin sensitivity in lacto-ovo vegetarians. Br J Nutr 86(4):515–519
Hua Y, Keep RF, Hoff JT, Xi G (2008) Deferoxamine therapy for intracerebral hemorrhage. Acta Neurochir Suppl 105:3–6
Huang J, Dizin E, Cowan JA (2008a) Mapping iron binding sites on human frataxin: implications for cluster assembly on the ISU Fe-S cluster scaffold protein. J Biol Inorg Chem 13(5):825–836
Huang XJ, Li CT, Zhang WP, Lu YB, Fang SH, Wei EQ (2008b) Dihydroartemisinin potentiates the cytotoxic effect of temozolomide in rat C6 glioma cells. Pharmacology 82(1):1–9
Huang KJ, Su IJ, Theron M, Wu YC, Lai SK, Liu CC et al (2005) An interferon-gamma-related cytokine storm in SARS patients. J Med Virol 75(2):185–194
Huang ML, Becker EM, Whitnall M, Rahmanto YS, Ponka P, Richardson DR (2009) Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc Natl Acad Sci USA 106(38):16381–16386
Hubel CA, Bodnar LM, Many A, Harger G, Ness RB, Roberts JM (2004) Nonglycosylated ferritin predominates in the circulation of women with preeclampsia but not intrauterine growth restriction. Clin Chem 50(5):948–951
Hucka M, Finney A, Sauro HM, Bolouri H, Doyle JC, Kitano H et al (2003) The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19(4):524–531
Hull D, Pettifer SR, Kell DB (2008) Defrosting the digital library: bibliographic tools for the next generation web. PLoS Comput Biol 4(10):e1000204. doi:1000210.1001371/journal.pcbi.1000204
Hur K, Kim JI, Choi SI, Choi EK, Carp RI, Kim YS (2002) The pathogenic mechanisms of prion diseases. Mech Ageing Dev 123(12):1637–1647
Hurn PD, Koehler RC, Blizzard KK, Traystman RJ (1995) Deferoxamine reduces early metabolic failure associated with severe cerebral ischemic acidosis in dogs. Stroke 26(4):688–694 (discussion 694–685)
Hwang D, Lee IY, Yoo H, Gehlenborg N, Cho JH, Petritis B et al (2009) A systems approach to prion disease. Mol Syst Biol 5:252
Iannetti A, Pacifico F, Acquaviva R, Lavorgna A, Crescenzi E, Vascotto C et al. (2008) The neutrophil gelatinase-associated lipocalin (NGAL), a NF-{kappa}B-regulated gene, is a survival factor for thyroid neoplastic cells. Proc Natl Acad Sci USA 105: 14058–14063
Ilbäck NG, Lindh U, Wesslén L, Fohlman J, Friman G (2000) Trace element distribution in heart tissue sections studied by nuclear microscopy is changed in Coxsackie virus B3 myocarditis in methyl mercury-exposed mice. Biol Trace Elem Res 78(1–3):131–147
Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C et al (2008) Huntington’s disease: from pathology and genetics to potential therapies. Biochem J 412(2):191–209
Imayoshi I, Sakamoto M, Ohtsuka T, Kageyama R (2009) Continuous neurogenesis in the adult brain. Dev Growth Differ 51(3):379–386
Imlay JA (2008) Cellular defenses against superoxide and hydrogen peroxide. Annu Rev Biochem 77:755–776
Imrie CE, Korre A, Munoz-Melendez G (2009) Spatial correlation between the prevalence of transmissible spongiform diseases and British soil geochemistry. Environ Geochem Health 31(1):133–145
Inati A, Musallam KM, Wood JC, Taher AT (2010) Iron overload indices rise linearly with transfusion rate in patients with sickle cell disease. Blood 115(14): 2980–2981; author reply 2981–2982
Ioannidis JP (2005) Why most published research findings are false. PLoS Med 2(8):e124
Ioannidis JP (2008) Why most discovered true associations are inflated. Epidemiology 19(5):640–648
Iqbal M, Okazaki Y, Okada S (2009) Curcumin attenuates oxidative damage in animals treated with a renal carcinogen, ferric nitrilotriacetate (Fe-NTA): implications for cancer prevention. Mol Cell Biochem 324(1–2):157–164
Irazusta V, Cabiscol E, Reverter-Branchat G, Ros J, Tamarit J (2006) Manganese is the link between frataxin and iron-sulfur deficiency in the yeast model of Friedreich ataxia. J Biol Chem 281(18):12227–12232
Irazusta V, Moreno-Cermeno A, Cabiscol E, Ros J, Tamarit J (2008) Major targets of iron-induced protein oxidative damage in frataxin-deficient yeasts are magnesium-binding proteins. Free Radic Biol Med
Ishizaka N, Saito K, Mori I, Matsuzaki G, Ohno M, Nagai R (2005) Iron chelation suppresses ferritin upregulation and attenuates vascular dysfunction in the aorta of angiotensin II-infused rats. Arterioscler Thromb Vasc Biol 25(11):2282–2288
Iwamaru Y, Shimizu Y, Imamura M, Murayama Y, Endo R, Tagawa Y et al (2008) Lactoferrin induces cell surface retention of prion protein and inhibits prion accumulation. J Neurochem 107(3):636–646
Jabbour E, Garcia-Manero G, Taher A, Kantarjian HM (2009) Managing iron overload in patients with myelodysplastic syndromes with oral deferasirox therapy. Oncologist 14(5):489–496
Jacob AK, Hotchkiss RS, DeMeester SL, Hiramatsu M, Karl IE, Swanson PE et al. (1997).Endothelial cell apoptosis is accelerated by inorganic iron and heat via an oxygen radical dependent mechanism. Surgery 122(2):243–253 (discussion 254)
Jacob AK, Hotchkiss RS, Swanson PE, Tinsley KW, Karl IE, Buchman TG (2000) Injection of iron compounds followed by induction of the stress response causes tissue injury and apoptosis. Shock 14(4):460–464
Jädersten M, Hellström-Lindberg E (2010) New clues to the molecular pathogenesis of myelodysplastic syndromes. Exp Cell Res
Jaeschke H, Bajt ML (2006) Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci 89(1):31–41
James LP, Mayeux PR, Hinson JA (2003) Acetaminophen-induced hepatotoxicity. Drug Metab Dispos 31(12):1499–1506
Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC (2004) Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53(2):474–481
Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T et al (2008) Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med 45(1):1–17
Jarvis J, Seed MJ, Elton R, Sawyer L, Agius R (2005) Relationship between chemical structure and the occupational asthma hazard of low molecular weight organic compounds. Occup Environ Med 62(4):243–250
Jaurand MC (1997) Mechanisms of fiber-induced genotoxicity. Environ Health Perspect 105(Suppl 5):1073–1084
Javadi P, Buchman TG, Stromberg PE, Husain KD, Dunne WM, Woolsey CA et al (2004) High-dose exogenous iron following cecal ligation and puncture increases mortality rate in mice and is associated with an increase in gut epithelial and splenic apoptosis. Crit Care Med 32(5):1178–1185
Javadi P, Buchman TG, Stromberg PE, Turnbull IR, Vyas D, Hotchkiss RS et al (2005) Iron dysregulation combined with aging prevents sepsis-induced apoptosis. J Surg Res 128(1):37–44
Jayawardhana B, Kell DB, Rattray M (2008) Bayesian inference of the sites of perturbations in metabolic pathways via Markov Chain Monte Carlo. Bioinformatics 24:1191–1197. doi:10.1093/bioinformatics/btn103
Jehn M, Clark JM, Guallar E (2004) Serum ferritin and risk of the metabolic syndrome in U.S. adults. Diabetes Care 27(10):2422–2428
Jehn ML, Guallar E, Clark JM, Couper D, Duncan BB, Ballantyne CM et al (2007) A prospective study of plasma ferritin level and incident diabetes: the atherosclerosis risk in communities (ARIC) study. Am J Epidemiol 165(9):1047–1054
Jellinger K, Paulus W, Grundke-Iqbal I, Riederer P, Youdim MB (1990) Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J Neural Transm Park Dis Dement Sect 2(4):327–340
Jellinger KA (1999) The role of iron in neurodegeneration—prospects for pharmacotherapy of Parkinson’s disease. Drugs Aging 14(2):115–140
Jellinger KA (2002) Recent developments in the pathology of Parkinson’s disease. J Neural Transm Suppl (62):347–376
Jellinger KA (2009) Recent advances in our understanding of neurodegeneration. J Neural Transm 116(9):1111–1162
Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53(Suppl 3):S26–S36 (discussion S28–S36)
Jenner P, Olanow CW (1996) Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology 47(6 Suppl 3):S161–S170
Jeong SY, Rathore KI, Schulz K, Ponka P, Arosio P, David S (2009) Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. J Neurosci 29(3):610–619
Jia J, Zhu F, Ma X, Cao ZW, Li YX, Chen YZ (2009) Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov 8(2):111–128
Jiang D, Li X, Williams R, Patel S, Men L, Wang Y et al (2009) Ternary complexes of iron, amyloid- and nitrilotriacetic acid: binding affinities, redox properties, and relevance to iron-induced oxidative stress in Alzheimers Disease. Biochemistry
Jiang XP, Wang F, Yang DC, Elliott RL, Head JF (2002) Induction of apoptosis by iron depletion in the human breast cancer MCF-7 cell line and the 13762NF rat mammary adenocarcinoma in vivo. Anticancer Res 22(5):2685–2692
Jiao Y, Wilkinson JT, Di X, Wang W, Hatcher H, Kock ND et al (2009) Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood 113(2):462–469
Jimenez-Del-Rio M, Guzman-Martinez C, Velez-Pardo C (2010) The effects of polyphenols on survival and locomotor activity in Drosophila melanogaster exposed to iron and paraquat. Neurochem Res 35(2):227–238
Jímenez Del Río M, Vélez-Pardo C (2004) Transition metal-induced apoptosis in lymphocytes via hydroxyl radical generation, mitochondria dysfunction, and caspase-3 activation: an in vitro model for neurodegeneration. Arch Med Res 35(3):185–193
Jin H, Terai S, Sakaida I (2007) The iron chelator deferoxamine causes activated hepatic stellate cells to become quiescent and to undergo apoptosis. J Gastroenterol 42(6):475–484
Johansson C, Castoldi AF, Onishchenko N, Manzo L, Vahter M, Ceccatelli S (2007) Neurobehavioural and molecular changes induced by methylmercury exposure during development. Neurotox Res 11(3–4):241–260
Johnson D, Mayers I (2001) Multiple organ dysfunction syndrome: a narrative review. Can J Anaesth 48(5):502–509
Johnson JJ, Bailey HH, Mukhtar H (2010) Green tea polyphenols for prostate cancer chemoprevention: a translational perspective. Phytomedicine 17(1):3–13
Johnson L (2008) Iron and siderophores in fungal-host interactions. Mycol Res 112(Pt 2):170–183
Johnson S (2000) Iron catalyzed oxidative damage, in spite of normal ferritin and transferrin saturation levels and its possible role in Werner’s syndrome, Parkinson’s disease, cancer, gout, rheumatoid arthritis, etc. Med Hypotheses 55(3):242–244
Jolkkonen J, Jokivarsi K, Laitinen T, Gröhn O (2007) Subacute hemorrhage and resolution of edema in Rose Bengal stroke model in rats coincides with improved sensorimotor functions. Neurosci Lett 428(2–3):99–102
Jones BC, Beard JL, Gibson JN, Unger EL, Allen RP, McCarthy KA et al (2007) Systems genetic analysis of peripheral iron parameters in the mouse. Am J Physiol Regul Integr Comp Physiol 293(1):R116–124
Jovanovic SV, Simic MG (2000) Antioxidants in nutrition. Ann NY Acad Sci 899:326–334
Julius C, Hutter G, Wagner U, Seeger H, Kana V, Kranich J et al (2008) Transcriptional stability of cultured cells upon prion infection. J Mol Biol 375(5):1222–1233
Jung T, Bader N, Grune T (2007) Lipofuscin: formation, distribution, and metabolic consequences. Ann NY Acad Sci 1119:97–111
Jurenka JS (2009) Anti-inflammatory properties of curcumin, a major constituent of Curcuma longa: a review of preclinical and clinical research. Altern Med Rev 14(2):141–153
Jurkschat K, Ji X, Crossley A, Compton RG, Banks CE (2007) Super-washing does not leave single walled carbon nanotubes iron-free. Analyst 132(1):21–23
Justicia C, Ramos-Cabrer P, Hoehn M (2008) MRI detection of secondary damage after stroke: chronic iron accumulation in the thalamus of the rat brain. Stroke 39(5):1541–1547
Kaelin WG Jr (2005) The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 5(9):689–698
Kagu MB, Khalil MI, Ahmed SG (2007) Bone marrow macrophage iron stores in patients with HIV infection and AIDS-associated Kaposi’s sarcoma. Afr J Med Med Sci 36(2):125–128
Kaiserová H, Šimůnek T, van der Vijgh WJ, Bast A, Kvasničková E (2007) Flavonoids as protectors against doxorubicin cardiotoxicity: role of iron chelation, antioxidant activity and inhibition of carbonyl reductase. Biochim Biophys Acta 1772(9):1065–1074
Kaito M (2007) Molecular mechanism of iron metabolism and overload in chronic hepatitis C. J Gastroenterol 42(1):96–99
Kaito M, Iwasa M, Kobayashi Y, Fujita N, Tanaka H, Gabazza EC et al (2006) Iron reduction therapy by phlebotomy reduces lipid peroxidation and oxidative stress in patients with chronic hepatitis C. J Gastroenterol 41(9):921–922
Kakhlon O, Breuer W, Munnich A, Cabantchik ZI (2010) Iron redistribution as a therapeutic strategy for treating diseases of localized iron accumulation. Can J Physiol Pharmacol 88(3):187–196
Kala SV, Hasinoff BB, Richardson JS (1996) Brain samples from Alzheimer’s patients have elevated levels of loosely bound iron. Int J Neurosci 86(3–4):263–269
Kallianpur AR (2005) Iron and oxidative injury—a commentary on “Fatty acid-mediated iron translocation: a synergistic mechanism of oxidative injury” by D. Yao et al. Free Radic Biol Med 39(10):1305–1309
Kalpravidh RW, Siritanaratkul N, Insain P, Charoensakdi R, Panichkul N, Hatairaktham S et al (2010) Improvement in oxidative stress and antioxidant parameters in beta-thalassemia/Hb E patients treated with curcuminoids. Clin Biochem 43(4–5):424–429
Kamp DW (2009) Asbestos-induced lung diseases: an update. Transl Res 153(4):143–152
Kamp DW, Graceffa P, Pryor WA, Weitzman SA (1992) The role of free radicals in asbestos-induced diseases. Free Radic Biol Med 12(4):293–315
Kamp DW, Israbian VA, Preusen SE, Zhang CX, Weitzman SA (1995a) Asbestos causes DNA strand breaks in cultured pulmonary epithelial cells: role of iron-catalyzed free radicals. Am J Physiol 268(3 Pt 1): L471–L480
Kamp DW, Israbian VA, Yeldandi AV, Panos RJ, Graceffa P, Weitzman SA (1995b) Phytic acid, an iron chelator, attenuates pulmonary inflammation and fibrosis in rats after intratracheal instillation of asbestos. Toxicol Pathol 23(6):689–695
Kamp DW, Panduri V, Weitzman SA, Chandel N (2002) Asbestos-induced alveolar epithelial cell apoptosis: role of mitochondrial dysfunction caused by iron-derived free radicals. Mol Cell Biochem 234–235(1–2):153–160
Kamp DW, Weitzman SA (1997) Asbestosis: clinical spectrum and pathogenic mechanisms. Proc Soc Exp Biol Med 214(1):12–26
Kamp DW, Weitzman SA (1999) The molecular basis of asbestos induced lung injury. Thorax 54(7):638–652
Kanai Y, Endou H (2003) Functional properties of multispecific amino acid transporters and their implications to transporter-mediated toxicity. J Toxicol Sci 28(1):1–17
Kandaswami C, Middleton E Jr (1994) Free radical scavenging and antioxidant activity of plant flavonoids. Adv Exp Med Biol 366:351–376
Kang JO, Jones C, Brothwell B (1998) Toxicity associated with iron overload found in hemochromatosis: possible mechanism in a rat model. Clin Lab Sci 11(6):350–354
Kaprelyants AS, Gottschal JC, Kell DB (1993) Dormancy in non-sporulating bacteria. FEMS Microbiol Rev 10:271–286
Karbownik M, Reiter RJ, Garcia JJ, Tan D (2000) Melatonin reduces phenylhydrazine-induced oxidative damage to cellular membranes: evidence for the involvement of iron. Int J Biochem Cell Biol 32(10):1045–1054
Karin M (2008) The IkappaB kinase—a bridge between inflammation and cancer. Cell Res 18(3):334–342
Karin M, Greten FR (2005) NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 5(10):749–759
Karlberg T, Schagerlöf U, Gakh O, Park S, Ryde U, Lindahl M et al (2006) The structures of frataxin oligomers reveal the mechanism for the delivery and detoxification of iron. Structure 14(10):1535–1546
Karlsen JR, Borregaard N, Cowland JB (2010) Induction of neutrophil gelatinase-associated lipocalin expression by co-stimulation with IL-17 and TNF-{alpha} is controlled by I{kappa}B-{zeta} but neither by C/EBP-{beta} nor by C/EBP-{delta}. J Biol Chem
Kartikasari AE, Visseren FL, Marx JJ, van Mullekom S, Kats-Renaud JH, Asbeck BS et al (2009) Intracellular labile iron promotes firm adhesion of human monocytes to endothelium under flow and transendothelial migration: iron and monocyte-endothelial cell interactions. Atherosclerosis 205(2):369–375
Kasarskis EJ, Tandon L, Lovell MA, Ehmann WD (1995) Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: a preliminary study. J Neurol Sci 130(2):203–208
Kato J, Kobune M, Nakamura T, Kuroiwa G, Takada K, Takimoto R et al (2001) Normalization of elevated hepatic 8-hydroxy-2′-deoxyguanosine levels in chronic hepatitis C patients by phlebotomy and low iron diet. Cancer Res 61(24):8697–8702
Kato J, Miyanishi K, Kobune M, Nakamura T, Takada K, Takimoto R et al (2007) Long-term phlebotomy with low-iron diet therapy lowers risk of development of hepatocellular carcinoma from chronic hepatitis C. J Gastroenterol 42(10):830–836
Katta S, Kaur I, Chakrabarti S (2009) The molecular genetic basis of age-related macular degeneration: an overview. J Genet 88(4):425–449
Kaur D, Andersen J (2004) Does cellular iron dysregulation play a causative role in Parkinson’s disease? Ageing Res Rev 3(3):327–343
Kaur D, Rajagopalan S, Andersen JK (2009) Chronic expression of H-ferritin in dopaminergic midbrain neurons results in an age-related expansion of the labile iron pool and subsequent neurodegeneration: implications for Parkinson’s disease. Brain Res
Kaur D, Rajagopalan S, Chinta S, Kumar J, Di Monte D, Cherny RA et al (2007) Chronic ferritin expression within murine dopaminergic midbrain neurons results in a progressive age-related neurodegeneration. Brain Res 1140:188–194
Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JO, Boonplueang R et al (2003) Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson’s disease. Neuron 37(6):899–909
Kaur P, Aschner M, Syversen T (2006) Glutathione modulation influences methyl mercury induced neurotoxicity in primary cell cultures of neurons and astrocytes. Neurotoxicology 27(4):492–500
Kaushal V, Schlichter LC (2008) Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci 28(9):2221–2230
Kauwe JSK, Bertelsen S, Mayo K, Cruchaga C, Abraham R, Hollingworth P et al (2010) Suggestive synergy between genetic variants in TF and HFE as risk factors for Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet 153B(4):955–959
Kawabata T, Ma Y, Yamador I, Okada S (1997) Iron-induced apoptosis in mouse renal proximal tubules after an injection of a renal carcinogen, iron-nitrilotriacetate. Carcinogenesis 18(7):1389–1394
Kaysen GA (2009) Biochemistry and biomarkers of inflamed patients: why look, what to assess. Clin J Am Soc Nephrol 4(Suppl 1):S56–S63
Kazi TG, Afridi HI, Kazi N, Jamali MK, Arain MB, Sarfraz RA et al (2008) Distribution of zinc, copper and iron in biological samples of Pakistani myocardial infarction (1st, 2nd and 3rd heart attack) patients and controls. Clin Chim Acta 389(1–2):114–119
Ke Y, Qian ZM (2003) Iron misregulation in the brain: a primary cause of neurodegenerative disorders. Lancet Neurol 2(4):246–253
Keel M, Trentz O (2005) Pathophysiology of polytrauma. Injury 36(6):691–709
Kehrer JP (2000) The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 149(1):43–50
Keith CT, Borisy AA, Stockwell BR (2005) Multicomponent therapeutics for networked systems. Nat Rev Drug Discov 4(1):71–78
Kell DB (2004) Metabolomics and systems biology: making sense of the soup. Curr Op Microbiol 7:296–307
Kell DB (2006a) Metabolomics, modelling, machine learning in systems biology: towards an understanding of the languages of cells. The 2005 Theodor Bücher lecture. FEBS J 273:873–894
Kell DB (2006b) Systems biology, metabolic modelling and metabolomics in drug discovery and development. Drug Disc Today 11(23/24):1085–1092
Kell DB (2007) The virtual human: towards a global systems biology of multiscale, distributed biochemical network models. IUBMB Life 59(11):689–695
Kell DB (2008) Progress being made on standards for use in data sharing. Nature 456(7218):29
Kell DB (2009a) Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med Genomics 2:2. http://www.biomedcentral.com/1755-8794/2/2/
Kell DB (2009b) A systems biologist ponders how disparate ideas can sometimes come together beautifully. Nature 460:669
Kell DB, Darby RM, Draper J (2001) Genomic computing: explanatory analysis of plant expression profiling data using machine learning. Plant Physiol. 126:943–951
Kell DB, Dobson PD (2009) The cellular uptake of pharmaceutical drugs is mainly carrier-mediated and is thus an issue not so much of biophysics but of systems biology. Proceedings of the international Beilstein symposium on systems chemistry, 149–168. http://www.beilstein-institut.de/Bozen2008/Proceedings/Kell/Kell.pdf). Logos Verlag, Berlin
Kell DB, Kaprelyants AS, Grafen A (1995) On pheromones, social behaviour and the functions of secondary metabolism in bacteria. Trends Ecol Evol 10:126–129
Kell DB, Kaprelyants AS, Weichart DH, Harwood CL, Barer MR (1998) Viability and activity in readily culturable bacteria: a review and discussion of the practical issues. Antonie van Leeuwenhoek 73:169–187
Kell DB, King RD (2000) On the optimization of classes for the assignment of unidentified reading frames in functional genomics programmes: the need for machine learning. Trends Biotechnol 18(3):93–98
Kell DB, Knowles JD (2006) The role of modeling in systems biology. In: Szallasi Z, Stelling J, Periwal V (eds) System modeling in cellular biology: from concepts to nuts and bolts. MIT Press, Cambridge, pp 3–18
Kell DB, Mendes P (2008) The markup is the model: reasoning about systems biology models in the Semantic Web era. J Theoret Biol 252:538–543. doi: http://www.sciencedirect.com/science?_ob=MImg&_imagekey=B6WMD-4R0KTJW-4-1&_cdi=6932&_user=121749&_orig=browse&_coverDate=10%2F27%2F2007&_sk=999999999&view=c&wchp=dGLbVtb-zSkWz&md5=464b923e674b26776e8ad407dd17f4ce&ie=/sdarticle.pdf
Kell DB, Oliver SG (2004) Here is the evidence, now what is the hypothesis? The complementary roles of inductive and hypothesis-driven science in the post-genomic era. Bioessays 26:99–105
Kell DB, Westerhoff HV (1986a) Metabolic control theory: its role in microbiology and biotechnology. FEMS Microbiol Rev 39:305–320
Kell DB, Westerhoff HV (1986b) Towards a rational approach to the optimization of flux in microbial biotransformations. Trends Biotechnol 4:137–142
Kelleher P, Pacheco K, Newman LS (2000) Inorganic dust pneumonias: the metal-related parenchymal disorders. Environ Health Perspect 108(Suppl 4):685–696
Kenyon CJ (2010) The genetics of ageing. Nature 464(7288):504–512
Kermer P, Liman J, Weishaupt JH, Bahr M (2004) Neuronal apoptosis in neurodegenerative diseases: from basic research to clinical application. Neurodegener Dis 1(1):9–19
Ketonen L, Kieburtz K, Kazee AM, Tuite M (1996) Putaminal iron deposition in HIV infection. J NeuroAIDS 1(2):33–40
Kew MC, Asare GA (2007) Dietary iron overload in the African and hepatocellular carcinoma. Liver Int 27(6):735–741
Khalil M, Enzinger C, Langkammer C, Tscherner M, Wallner-Blazek M, Jehna M et al (2009) Quantitative assessment of brain iron by R2* relaxometry in patients with clinically isolated syndrome and relapsing-remitting multiple sclerosis. Mult Scler 15(9):1048–1054
Khan MF, Kaphalia BS, Ansari GA (1995) Erythrocyte-aniline interaction leads to their accumulation and iron deposition in rat spleen. J Toxicol Environ Health 44(4):415–421
Khan MF, Wu X, Alcock NW, Boor PJ, Ansari GA (1999) Iron exacerbates aniline-associated splenic toxicity. J Toxicol Environ Health A 57(3):173–184
Kibel A, Belovari T, Drenjančević-Perić I (2008) The role of transferrin in atherosclerosis. Med Hypotheses 70(4):793–797
Kidd PM (2000) Parkinson’s disease as multifactorial oxidative neurodegeneration: implications for integrative management. Altern Med Rev 5(6):502–529
Kiechl S, Willeit J, Egger G, Poewe W, Oberhollenzer F (1997) Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation 96(10):3300–3307
Kim BH, Jun YC, Jin JK, Kim JI, Kim NH, Leibold EA et al (2007) Alteration of iron regulatory proteins (IRP1 and IRP2) and ferritin in the brains of scrapie-infected mice. Neurosci Lett 422(3):158–163
Kim D, Chun BG, Kim YK, Lee YH, Park CS, Jeon I et al (2008a) In vivo tracking of human mesenchymal stem cells in experimental stroke. Cell Transpl 16(10):1007–1012
Kim EY, Ham SK, Shigenaga MK, Han O (2008b) Bioactive dietary polyphenolic compounds reduce nonheme iron transport across human intestinal cell monolayers. J Nutr 138(9):1647–1651
Kim JI, Choi SI, Kim NH, Jin JK, Choi EK, Carp RI et al (2001) Oxidative stress and neurodegeneration in prion diseases. Ann NY Acad Sci 928:182–186
Kim NH, Park SJ, Jin JK, Kwon MS, Choi EK, Carp RI et al (2000) Increased ferric iron content and iron-induced oxidative stress in the brains of scrapie-infected mice. Brain Res 884(1–2):98–103
Kim YW, Lee SM, Shin SM, Hwang SJ, Brooks JS, Kang HE et al (2009) Efficacy of sauchinone as a novel AMPK-activating lignan for preventing iron-induced oxidative stress and liver injury. Free Radic Biol Med 47:1082–1092
King RD, Rowland J, Oliver SG, Young M, Aubrey W, Byrne E et al (2009) The automation of science. Science 324(5923):85–89
King RD, Whelan KE, Jones FM, Reiser PGK, Bryant CH, Muggleton SH et al (2004) Functional genomic hypothesis generation and experimentation by a robot scientist. Nature 427:247–252
Kiokias S, Varzakas T, Oreopoulou V (2008) In vitro activity of vitamins, flavonoids, and natural phenolic antioxidants against the oxidative deterioration of oil-based systems. Crit Rev Food Sci Nutr 48(1):78–93
Kirkwood TB (2008) A systematic look at an old problem. Nature 451(7179):644–647
Kirsch R, Sijtsema HP, Tlali M, Marais AD, Hall P, de la M (2006) Effects of iron overload in a rat nutritional model of non-alcoholic fatty liver disease. Liver Int 26(10):1258–1267
Kitagawa S, Tanaka Y, Tanaka M, Endo K, Yoshii A (2009) Enhanced skin delivery of quercetin by microemulsion. J Pharm Pharmacol 61(7):855–860
Kitano H (2007) A robustness-based approach to systems-oriented drug design. Nat Rev Drug Discov 6(3):202–210
Klipp E, Herwig R, Kowald A, Wierling C, Lehrach H (2005) Systems biology in practice: concepts, implementation and clinical application. Wiley/VCH, Berlin
Klipp E, Liebermeister W, Helbig A, Kowald A, Schaber J (2007) Systems biology standards-the community speaks. Nat Biotechnol 25(4):390–391
Knaapen AM, Borm PJ, Albrecht C, Schins RP (2004) Inhaled particles and lung cancer. Part A: mechanisms. Int J Cancer 109(6):799–809
Knight CG, Platt M, Rowe W, Wedge DC, Khan F, Day P et al (2009) Array-based evolution of DNA aptamers allows modelling of an explicit sequence-fitness landscape. Nucleic Acids Res 37(1):e6
Knight SA, Kim R, Pain D, Dancis A (1999) The yeast connection to Friedreich ataxia. Am J Hum Genet 64(2):365–371
Knovich MA, Storey JA, Coffman LG, Torti SV, Torti FM (2009) Ferritin for the clinician. Blood Rev 23(3):95–104
Ko C, Siddaiah N, Berger J, Gish R, Brandhagen D, Sterling RK et al (2007) Prevalence of hepatic iron overload and association with hepatocellular cancer in end-stage liver disease: results from the national hemochromatosis transplant registry. Liver Int 27(10):1394–1401
Kobayashi K, Yoshino F, Takahashi SS, Todoki K, Maehata Y, Komatsu T et al (2008) Direct assessments of the antioxidant effects of propofol medium chain triglyceride/long chain triglyceride on the brain of stroke-prone spontaneously hypertensive rats using electron spin resonance spectroscopy. Anesthesiology 109(3):426–435
Koc M, Nad’ová Z, Kovář J (2006) Sensitivity of cells to apoptosis induced by iron deprivation can be reversibly changed by iron availability. Cell Prolif 39(6):551–561
Kocyigit A, Armutcu F, Gurel A, Ermis B (2004) Alterations in plasma essential trace elements selenium, manganese, zinc, copper, and iron concentrations and the possible role of these elements on oxidative status in patients with childhood asthma. Biol Trace Elem Res 97(1):31–41
Kohanski MA, DePristo MA, Collins JJ (2010) Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell 37(3):311–320
Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130(5):797–810
Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ (2008) Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135(4):679–690
Kola I, Landis J (2004) Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3(8):711–715
Kolberg JA, Jorgensen T, Gerwien RW, Hamren S, McKenna MP, Moler E et al (2009) Development of a type 2 diabetes risk model from a panel of serum biomarkers from the Inter99 cohort. Diabetes Care 32(7):1207–1212
Kom GD, Schwedhelm E, Nielsen P, Böger RH (2006) Increased urinary excretion of 8-iso-prostaglandin F2alpha in patients with HFE-related hemochromatosis: a case-control study. Free Radic Biol Med 40(7):1194–1200
Kompala SD, Babbs CF, Blaho KE (1986) Effect of deferoxamine on late deaths following CPR in rats. Ann Emerg Med 15(4):405–407
Kon K, Kim JS, Jaeschke H, Lemasters JJ (2004) Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology 40(5):1170–1179
Končić MZ, Kremer D, Gruz J, Strnad M, Biševac G, Kosalec I et al (2010) Antioxidant and antimicrobial properties of Moltkia petraea (Tratt.) Griseb. flower, leaf and stem infusions. Food Chem Toxicol 48(6):1537–1542
Kondapalli KC, Kok NM, Dancis A, Stemmler TL (2008) Drosophila frataxin: an iron chaperone during cellular Fe-S cluster bioassembly. Biochemistry 47(26):6917–6927
Kontoghiorghes GJ, Kolnagou A, Peng CT, Shah SV, Aessopos A (2010) Safety issues of iron chelation therapy in patients with normal range iron stores including thalassaemia, neurodegenerative, renal and infectious diseases. Expert Opin Drug Saf 9(2):201–206
Kooncumchoo P, Sharma S, Porter J, Govitrapong P, Ebadi M (2006) Coenzyme Q(10) provides neuroprotection in iron-induced apoptosis in dopaminergic neurons. J Mol Neurosci 28(2):125–141
Koppenol WH, Moreno JJ, Pryor WA, Ischiropoulos H, Beckman JS (1992) Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem Res Toxicol 5(6):834–842
Kopterides P, Falagas ME (2009) Statins for sepsis: a critical and updated review. Clin Microbiol Infect 15(4):325–334
Korkina LG, Afanas’ev IB (1997) Antioxidant and chelating properties of flavonoids. Adv Pharmacol 38:151–163
Korkmaz A, Kunak ZI, Paredes SD, Yaren H, Tan DX, Reiter RJ (2008) The use of melatonin to combat mustard toxicity. REVIEW. Neuro Endocrinol Lett 29(5):614–619
Korkmaz A, Reiter RJ, Topal T, Manchester LC, Oter S, Tan DX (2009) Melatonin: an established antioxidant worthy of use in clinical trials. Mol Med 15(1–2):43–50
Korkmaz A, Yaren H, Topal T, Oter S (2006) Molecular targets against mustard toxicity: implication of cell surface receptors, peroxynitrite production, and PARP activation. Arch Toxicol 80(10):662–670
Kostoff RN (2002) Overcoming specialization. Bioscience 52(10):937–941
Kostoff RN, Delafuente JC (2006) The unknown impacts of combinations of large numbers of drugs. Drug Saf 29(3):183–185
Kotamraju S, Chitambar CR, Kalivendi SV, Joseph J, Kalyanaraman B (2002) Transferrin receptor-dependent iron uptake is responsible for doxorubicin-mediated apoptosis in endothelial cells—role of oxidant-induced iron signaling in apoptosis. J Biol Chem 277(19):17179–17187
Kotamraju S, Kalivendi SV, Konorev E, Chitambar CR, Joseph J, Kalyanaraman B (2004a) Oxidant-induced iron signaling in doxorubicin-mediated apoptosis. Methods Enzymol 378:362–382
Kotamraju S, Tampo Y, Kalivendi SV, Joseph J, Chitambar CR, Kalyanaraman B (2004b) Nitric oxide mitigates peroxide-induced iron-signaling, oxidative damage, and apoptosis in endothelial cells: role of proteasomal function? Arch Biochem Biophys 423(1):74–80
Koufaki M, Detsi A, Kiziridi C (2009) Multifunctional lipoic acid conjugates. Curr Med Chem 16(35):4728–4742
Kourie JI (2002) Prion channel proteins and their role in vacuolation and neurodegenerative diseases. Eur Biophys J 31(5):409–416
Kovář J, Stunz LL, Stewart BC, Kriegerbecková K, Ashman RF, Kemp JD (1997) Direct evidence that iron deprivation induces apoptosis in murine lymphoma 38C13. Pathobiology 65(2):61–68
Kovář J, Valenta T, Štýbrová H (2001) Differing sensitivity of tumor cells to apoptosis induced by iron deprivation in vitro. In Vitro Cell Dev Biol Anim 37(7):450–458
Kowdley KV (2010) The role of iron in nonalcoholic fatty liver disease: the story continues. Gastroenterology 138(3):817–819
Krukoski DW, Comar SR, Claro LM, Leonart MS, do Nascimento AJ (2009) Effect of vitamin C, deferoxamine, quercetin and rutin against tert-butyl hydroperoxide oxidative damage in human erythrocytes. Hematology 14(3):168–172
Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP (1997) Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci 17(13):5089–5100
Kruszewski M (2003) Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat Res 531(1–2):81–92
Kuhn M, Campillos M, Letunic I, Jensen LJ, Bork P (2010) A side effect resource to capture phenotypic effects of drugs. Mol Syst Biol 6:343
Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE et al (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309(5733):481–484
Kumar O, Sugendran K, Vijayaraghavan R (2003) Oxidative stress associated hepatic and renal toxicity induced by ricin in mice. Toxicon 41(3):333–338
Kumar V, Sinha AK, Makkar HPS, Becker K (2010) Dietary roles of phytate and phytase in human nutrition: a review. Food Chem 120(4):945–959
Kummer S, Hayes GR, Gilbert RO, Beach DH, Lucas JJ, Singh BN (2008) Induction of human host cell apoptosis by Trichomonas vaginalis cysteine proteases is modulated by parasite exposure to iron. Microb Pathog 44(3):197–203
Kunsch C, Medford RM (1999) Oxidative stress as a regulator of gene expression in the vasculature. Circ Res 85(8):753–766
Kuo KL, Hung SC, Wei YH, Tarng DC (2008) Intravenous iron exacerbates oxidative DNA damage in peripheral blood lymphocytes in chronic hemodialysis patients. J Am Soc Nephrol 19(9):1817–1826
Kurz T, Leake A, von Zglinicki T, Brunk UT (2004) Relocalized redox-active lysosomal iron is an important mediator of oxidative-stress-induced DNA damage. Biochem J 378:1039–1045
Kurz T, Terman A, Brunk UT (2007) Autophagy, ageing and apoptosis: the role of oxidative stress and lysosomal iron. Arch Biochem Biophys 462(2):220–230
Kurz T, Terman A, Gustafsson B, Brunk UT (2008a) Lysosomes and oxidative stress in aging and apoptosis. Biochim Biophys Acta 1780(11):1291–1303
Kurz T, Terman A, Gustafsson B, Brunk UT (2008b) Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol 129(4):389–406
Kwiatkowski JL (2010) Oral iron chelators. Hematol Oncol Clin North Am 24(1):229–248
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323(5918):1205–1208
Lachili B, Hininger I, Faure H, Arnaud J, Richard MJ, Favier A et al (2001) Increased lipid peroxidation in pregnant women after iron and vitamin C supplementation. Biol Trace Elem Res 83(2):103–110
Lagan AL, Melley DD, Evans TW, Quinlan GJ (2008) Pathogenesis of the systemic inflammatory syndrome and acute lung injury: role of iron mobilization and decompartmentalization. Am J Physiol Lung Cell Mol Physiol 294(2):L161–174
Lakshmi Devi S, Anuradha CV (2009) Mitochondrial damage, cytotoxicity and apoptosis in iron-potentiated alcoholic liver fibrosis: amelioration by taurine. Amino Acids
Lam WW, Au WY, Chu WC, Tam S, Ha SY, Pennell DJ (2008) One-stop measurement of iron deposition in the anterior pituitary, liver, and heart in thalassemia patients. J Magn Reson Imaging 28(1):29–33
Lamey PJ, Biagioni PA (1995) Relationship between iron status and recrudescent herpes labialis. Eur J Clin Microbiol Infect Dis 14(7):604–605
Lancaster JR Jr (2008) Protein cysteine thiol nitrosation: maker or marker of reactive nitrogen species-induced nonerythroid cellular signaling? Nitric Oxide 19(2):68–72
Landino LM (2008) Protein thiol modification by peroxynitrite anion and nitric oxide donors. Methods Enzymol 440:95–109
Lapenna D, Pierdomenico SD, Ciofani G, Ucchino S, Neri M, Giamberardino MA et al (2007) Association of body iron stores with low molecular weight iron and oxidant damage of human atherosclerotic plaques. Free Rad Biol Med 42(4):492–498
Latunde-Dada GO, Vulpe CD, Anderson GJ, Simpson RJ, McKie AT (2004) Tissue-specific changes in iron metabolism genes in mice following phenylhydrazine-induced haemolysis. Biochim Biophys Acta 1690(2):169–176
Lavados M, Guillón M, Mujica MC, Rojo LE, Fuentes P, Maccioni RB (2008) Mild cognitive impairment and Alzheimer patients display different levels of redox-active CSF iron. J Alzheimers Dis 13(2):225–232
Le Novère N, Finney A, Hucka M, Bhalla US, Campagne F, Collado-Vides J et al (2005) Minimum information requested in the annotation of biochemical models (MIRIAM). Nat Biotechnol 23(12):1509–1515
Le Novère N, Hucka M, Mi H, Moodie S, Schreiber F, Sorokin A et al (2009) The systems biology graphical notation. Nat Biotechnol 27(8):735–741
Leardi A, Caraglia M, Selleri C, Pepe S, Pizzi C, Notaro R et al (1998) Desferioxamine increases iron depletion and apoptosis induced by ara-C of human myeloid leukaemic cells. Br J Haematol 102(3):746–752
LeBel CP, Ali SF, Bondy SC (1992) Deferoxamine inhibits methyl mercury-induced increases in reactive oxygen species formation in rat brain. Toxicol Appl Pharmacol 112(1):161–165
Lecube A, Hernandez C, Pelegri D, Simo R (2008) Factors accounting for high ferritin levels in obesity. Int J Obes (Lond), doi:10.1038/ijo.2008.1154
Lecube A, Hernández C, Simó R (2009) Glucose abnormalities in non-alcoholic fatty liver disease and chronic hepatitis C virus infection: the role of iron overload. Diabetes Metab Res Rev 25(5):403–410
Lee RE, Balcerzak SP, Westerman MP (1967) Gaucher’s disease. A morphologic study and measurements of iron metabolism. Am J Med 42(6):891–898
Lee RE, Peters SP, Glew RH (1977) Gaucher’s disease: clinical, morphologic, and pathogenetic considerations. Pathol Annu (12 Pt 2):309–339
Lee FY, Lee TS, Pan CC, Huang AL, Chau LY (1998) Colocalization of iron and ceroid in human atherosclerotic lesions. Atherosclerosis 138(2):281–288
Lee TS, Shiao MS, Pan CC, Chau LY (1999) Iron-deficient diet reduces atherosclerotic lesions in ApoE-deficient mice. Circulation 99(9):1222–1229
Lee DW, Andersen JK (2010) Iron elevations in the aging Parkinsonian brain: a consequence of impaired iron homeostasis? J Neurochem 112(2):332–339
Lee DH, Zacharski LR, Jacobs DR (2006a) Comparison of the serum ferritin and percentage of transferrin saturation as exposure markers of iron-driven oxidative stress-related disease outcomes. Am Heart J 151(6). doi:10.1016/j.ahj.2006.1003.1009
Lee DW, Andersen JK, Kaur D (2006b) Iron dysregulation and neurodegeneration—the molecular connection. Mol Interv 6(2):89–97
Lee JC, Son YO, Choi KC, Jang YS (2006c) Hydrogen peroxide induces apoptosis of BJAB cells due to formation of hydroxyl radicals via intracellular iron-mediated Fenton chemistry in glucose oxidase-mediated oxidative stress. Mol Cells 22(1):21–29
Lee MT, Piomelli S, Granger S, Miller ST, Harkness S, Brambilla DJ et al (2006d) Stroke prevention trial in sickle cell anemia (STOP): extended follow-up and final results. Blood 108(3):847–852
Lee SK, Jang HJ, Lee HJ, Lee J, Jeon BH, Jun CD et al (2006e) p38 and ERK MAP kinase mediates iron chelator-induced apoptosis and -suppressed differentiation of immortalized and malignant human oral keratinocytes. Life Sci 79(15):1419–1427
Lee SK, Lee JJ, Lee HJ, Lee J, Jeon BH, Jun CD et al (2006f) Iron chelator-induced growth arrest and cytochrome c-dependent apoptosis in immortalized and malignant oral keratinocytes. J Oral Pathol Med 35(4):218–226
Lee KS, Raymond LD, Schoen B, Raymond GJ, Kett L, Moore RA et al (2007a) Hemin interactions and alterations of the subcellular localization of prion protein. J Biol Chem 282(50):36525–36533
Lee MS, Johansen L, Zhang Y, Wilson A, Keegan M, Avery W et al (2007b) The novel combination of chlorpromazine and pentamidine exerts synergistic antiproliferative effects through dual mitotic action. Cancer Res 67(23):11359–11367
Leeson PD, Springthorpe B (2007) The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov 6(11):881–890
Lehár J, Krueger A, Zimmermann G, Borisy A (2008a) High-order combination effects and biological robustness. Mol Syst Biol 4:215
Lehár J, Stockwell BR, Giaever G, Nislow C (2008b) Combination chemical genetics. Nat Chem Biol 4(11):674–681
Lehár J, Zimmermann GR, Krueger AS, Molnar RA, Ledell JT, Heilbut AM et al (2007) Chemical combination effects predict connectivity in biological systems. Mol Syst Biol 3:80
Lehmann DJ, Worwood M, Ellis R, Wimhurst VL, Merryweather-Clarke AT, Warden DR et al (2006) Iron genes, iron load and risk of Alzheimer’s disease. J Med Genet 43(10):e52
Lehmann S (2002) Metal ions and prion diseases. Curr Opin Chem Biol 6(2):187–192
Leitch HA (2007) Improving clinical outcome in patients with myelodysplastic syndrome and iron overload using iron chelation therapy. Leuk Res 31(Suppl 3):S7–S9
Leithead JA, Ferguson JW, Bates CM, Davidson JS, Lee A, Bathgate AJ et al (2009) The systemic inflammatory response syndrome is predictive of renal dysfunction in patients with non-paracetamol-induced acute liver failure. Gut 58(3):443–449
Lemineur T, Deby-Dupont G, Preiser JC (2006) Biomarkers of oxidative stress in critically ill patients: what should be measured, when and how? Curr Op Clin Nutr Metab Care 9(6):704–710
Leuner B, Kozorovitskiy Y, Gross CG, Gould E (2007) Diminished adult neurogenesis in the marmoset brain precedes old age. Proc Natl Acad Sci USA 104(43):17169–17173
Levenson CW (2003) Iron and Parkinson’s disease: chelators to the rescue? Nutr Rev 61(9):311–313
Levenson CW (2005) Trace metal regulation of neuronal apoptosis: from genes to behavior. Physiol Behav 86(3):399–406
Leveugle B, Spik G, Perl DP, Bouras C, Fillit HM, Hof PR (1997) Distribution of the iron-binding protein lactoferrin in the pathological lesions of neurodegenerative diseases. Lactoferrin Interact Biol Funct 28:119–124, 408
LeVine SM (1997) Iron deposits in multiple sclerosis and Alzheimer’s disease brains. Brain Res 760(1–2):298–303
LeVine SM, Chakrabarty A (2004) The role of iron in the pathogenesis of experimental allergic encephalomyelitis and multiple sclerosis. Ann NY Acad Sci 1012:252–266
Levresse V, Renier A, Levy F, Broaddus VC, Jaurand M (2000) DNA breakage in asbestos-treated normal and transformed (TSV40) rat pleural mesothelial cells. Mutagenesis 15(3):239–244
Li D, Shimamura T, Ji H, Chen L, Haringsma HJ, McNamara K et al (2007) Bronchial and peripheral murine lung carcinomas induced by T790M–L858R mutant EGFR respond to HKI-272 and rapamycin combination therapy. Cancer Cell 12(1):81–93
Li H, Rybicki AC, Suzuka SM, von Bonsdorff L, Breuer W, Hall CB et al (2010) Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med 16(2):177–182
Li H, Zhou Q, Wu Y, Fu J, Wang T, Jiang G (2009) Effects of waterborne nano-iron on medaka (Oryzias latipes): antioxidant enzymatic activity, lipid peroxidation and histopathology. Ecotoxicol Environ Saf 72(3):684–692
Li L, Hölscher C (2007) Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev 56(2):384–402
Li Q, Withoff S, Verma IM (2005) Inflammation-associated cancer: NF-kappaB is the lynchpin. Trends Immunol 26(6):318–325
Li T-S, Marbán E (2010) Physiological levels of reactive oxygen species are required to maintain genomic stability in stem cells. Stem Cells
Li XJ, Brazhnik O, Kamal A, Guo D, Lee C, Hoops S (2003) Databases and visualization for metabolomics. In: Harrigan GG, Goodacre R et al (eds) Metabolic profiling: its role in biomarker discovery and gene function analysis. Kluwer Academic Publishers, Boston, pp 293–309
Libby P (2002) Inflammation in atherosclerosis. Nature 420(6917):868–874
Libby P, Ridker PM, Maseri A (2002) Inflammation and atherosclerosis. Circulation 105(9):1135–1143
Liby KT, Yore MM, Sporn MB (2007) Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer 7(5):357–369
Liebermeister W, Klipp E (2006) Bringing metabolic networks to life: integration of kinetic, metabolic, and proteomic data. Theor Biol Med Model 3:42
Lill R (2009) Function and biogenesis of iron-sulphur proteins. Nature 460(7257):831–838
Lill R, Muhlenhoff U (2008) Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu Rev Biochem 77:669–700
Lim CK, Kalinowski DS, Richardson DR (2008) Protection against hydrogen peroxide-mediated cytotoxicity in Friedreich’s ataxia fibroblasts using novel iron chelators of the 2-pyridylcarboxaldehyde isonicotinoyl hydrazone class. Mol Pharmacol 74(1):225–235
Lim MK, Lee CK, Ju YS, Cho YS, Lee MS, Yoo B et al (2001) Serum ferritin as a serologic marker of activity in systemic lupus erythematosus. Rheumatol Int 20(3):89–93
Lim ZY, Fiaccadori V, Gandhi S, Hayden J, Kenyon M, Ireland R et al (2010) Impact of pre-transplant serum ferritin on outcomes of patients with myelodysplastic syndromes or secondary acute myeloid leukaemia receiving reduced intensity conditioning allogeneic haematopoietic stem cell transplantation. Leuk Res 34(6):723–727
Limdi JK, Crampton JR (2004) Hereditary haemochromatosis. QJM 97(6):315–324
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443(7113):787–795
Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR (2008) Physiology of the prion protein. Physiol Rev 88(2):673–728
Linseisen J, Rohrmann S (2008) Biomarkers of dietary intake of flavonoids and phenolic acids for studying diet-cancer relationship in humans. Eur J Nutr 47(Suppl 2):60–68
Liu IF, Annamalai T, Sutherland JH, Tse-Dinh YC (2009a) Hydroxyl radicals are involved in cell killing by the bacterial topoisomerase I cleavage complex. J Bacteriol 191(16):5315–5319
Liu G, Men P, Kudo W, Perry G, Smith MA (2009b) Nanoparticle-chelator conjugates as inhibitors of amyloid-beta aggregation and neurotoxicity: a novel therapeutic approach for Alzheimer disease. Neurosci Lett 455(3):187–190
Liu G, Men P, Perry G, Smith MA (2009c) Metal chelators coupled with nanoparticles as potential therapeutic agents for Alzheimer’s disease. J Nanoneurosci 1(1):42–55
Liu G, Beri R, Mueller A, Kamp DW (2010a) Molecular mechanisms of asbestos-induced lung epithelial cell apoptosis. Chem Biol Interact (in press)
Liu G, Men P, Perry G, Smith MA (2010b) Nanoparticle and iron chelators as a potential novel Alzheimer therapy. Methods Mol Biol 610:123–144
Liu PK, Wang FS (2008) Inverse problems of biological systems using multi-objective optimization. J Chin Inst Chem Eng 39(5):399–406
Ljung L (1987) System identification: theory for the user. Prentice Hall, Englewood Cliffs, NJ
Lledo PM, Alonso M, Grubb MS (2006) Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci 7(3):179–193
Lleó A, Blesa R, Angelopoulos C, Pastor-Rubio P, Villa M, Oliva R et al (2002) Transferrin C2 allele, haemochromatosis gene mutations, and risk for Alzheimer’s disease. J Neurol Neurosurg Psychiatry 72(6):820–821
Lloyd DR, Carmichael PL, Phillips DH (1998) Comparison of the formation of 8-hydroxy-2′-deoxyguanosine and single- and double-strand breaks in DNA mediated by Fenton reactions. Chem Res Toxicol 11(5):420–427
Loft S, Poulsen HE (1996) Cancer risk and oxidative DNA damage in man. J Mol Med 74(6):297–312
Loh A, Hadziahmetovic M, Dunaief JL (2009) Iron homeostasis and eye disease. Biochim Biophys Acta 1790(7):637–649
Loh KP, Huang SH, De Silva R, Tan BK, Zhu YZ (2006) Oxidative stress: apoptosis in neuronal injury. Curr Alzheimer Res 3(4):327–337
Loke WM, Jenner AM, Proudfoot JM, McKinley AJ, Hodgson JM, Halliwell B et al (2009) A metabolite profiling approach to identify biomarkers of flavonoid intake in humans. J Nutr 139(12):2309–2314
Lolis E, Bucala R (2003) Therapeutic approaches to innate immunity: severe sepsis and septic shock. Nat Rev Drug Discov 2(8):635–645
Long LH, Clement MV, Halliwell B (2000) Artifacts in cell culture: Rapid generation of hydrogen peroxide on addition of (−)-epigallocatechin, (−)-epigallocatechin gallate, (+)-catechin, and quercetin to commonly used cell culture media. Biochem Biophys Res Comm 273(1):50–53
Loomis LD, Raymond KN (1991) Solution equilibria of enterobactin and metal enterobactin complexes. Inorg Chem 30(5):906–911
Lopes GKB, Schulman HM, Hermes-Lima M (1999) Polyphenol tannic acid inhibits hydroxyl radical formation from Fenton reaction by complexing ferrous ions. Biochimica Et Biophysica Acta-General Subjects 1472(1–2):142–152
Lorber M (1960) The occurrence of intracellular iron in Gaucher’s disease. Ann Intern Med 53:293–305
Lorber M (1970) Adult-type Gaucher’s disease: a secondary disorder of iron metabolism. Mt Sinai J Med 37(4):404–417
Lorenz DR, Cantor CR, Collins JJ (2009) A network biology approach to aging in yeast. Proc Natl Acad Sci USA 106(4):1145–1150
Lorrain B, Dangles O, Genot C, Dufour C (2010) Chemical modeling of heme-induced lipid oxidation in gastric conditions and inhibition by dietary polyphenols. J Agric Food Chem 58(1):676–683
Lotery A, Trump D (2007) Progress in defining the molecular biology of age related macular degeneration. Hum Genet 122(3–4):219–236
Lou M, Lieb K, Selim M (2009) The relationship between hematoma iron content and perihematoma edema: an MRI study. Cerebrovasc Dis 27(3):266–271
Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR (1998) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158(1):47–52
Lu C, Cortopassi G (2007) Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts. Arch Biochem Biophys 457(1):111–122
Lu JJ, Meng LH, Cai YJ, Chen Q, Tong LJ, Lin LP et al (2008) Dihydroartemisinin induces apoptosis in HL-60 leukemia cells dependent of iron and p38 mitogen-activated protein kinase activation but independent of reactive oxygen species. Cancer Biol Ther 7(7):1017–1023
Lüdtke N, Panzeri S, Brown M, Broomhead DS, Knowles J, Montemurro MA et al (2008) Information-theoretic Sensitivity Analysis: a general method for credit assignment in complex networks. J Roy Soc Interface 5(19):223–235. doi:10.1098/rsif.2007.1079
Luft FC (2006) Doxorubicin toxicity in the Iron Age. J Mol Med 84(7):529–531
Lukosz M, Jakob S, Buchner N, Zschauer TC, Altschmied J, Haendeler J (2010) Nuclear redox signaling. Antioxid Redox Signal 12(6):713–742
Lumsden AL, Henshall TL, Dayan S, Lardelli MT, Richards RI (2007) Huntingtin-deficient zebrafish exhibit defects in iron utilization and development. Hum Mol Genet 16(16):1905–1920
Lund LG, Aust AE (1991) Iron-catalyzed reactions may be responsible for the biochemical and biological effects of asbestos. Biofactors 3(2):83–89
Lund LG, Aust AE (1992) Iron mobilization from crocidolite asbestos greatly enhances crocidolite-dependent formation of DNA single-strand breaks in phi X174 RFI DNA. Carcinogenesis 13(4):637–642
Lusis AJ (2000) Atherosclerosis. Nature 407(6801):233–241
Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E (2004) Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab 81(1):70–73
Ma’ayan A (2009) Insights into the organization of biochemical regulatory networks using graph theory analyses. J Biol Chem 284(9):5451–5455
Ma H, Sorokin A, Mazein A, Selkov A, Selkov E, Demin O et al (2007) The Edinburgh human metabolic network reconstruction and its functional analysis. Mol Syst Biol 3:135
Ma H, Wang J, Abdel-Rahman SZ, Boor PJ, Khan MF (2008) Oxidative DNA damage and its repair in rat spleen following subchronic exposure to aniline. Toxicol Appl Pharmacol 233(2):247–253
Ma HW, Zeng AP (2003) The connectivity structure, giant strong component and centrality of metabolic networks. Bioinformatics 19(11):1423–1430
Ma HW, Zhao XM, Yuan YJ, Zeng AP (2004) Decomposition of metabolic network into functional modules based on the global connectivity structure of reaction graph. Bioinformatics 20(12):1870–1876
Ma YS, Wu SB, Lee WY, Cheng JS, Wei YH (2009) Response to the increase of oxidative stress and mutation of mitochondrial DNA in aging. Biochim Biophys Acta 1790(10):1021–1029
MacKenzie EL, Iwasaki K, Tsuji Y (2008) Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Signal 10(6):997–1030
Madamanchi NR, Hakim ZS, Runge MS (2005a) Oxidative stress in atherogenesis and arterial thrombosis: the disconnect between cellular studies and clinical outcomes. J Thromb Haemost 3(2):254–267
Madamanchi NR, Vendrov A, Runge MS (2005b) Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol 25(1):29–38
Maeda S, Omata M (2008) Inflammation and cancer: role of nuclear factor-kappaB activation. Cancer Sci 99(5):836–842
Maguire JL, deVeber G, Parkin PC (2007) Association between iron-deficiency anemia and stroke in young children. Pediatrics 120(5):1053–1057
Maher B (2008) The case of the missing heritability. Nature 456(7218):18–21
Mahesh S, Ginzburg Y, Verma A (2008) Iron overload in myelodysplastic syndromes. Leuk Lymphoma 49(3):427–438
Mahmoud A, Musallam KM, Taher AT (2008) Iron and hepatitis C: what can we learn from thalassemia major? Liver Int 28(6):892–893; author reply 893
Maioli MA, Alves LC, Campanini AL, Lima MC, Dorta DJ, Groppo M et al (2010) Iron chelating-mediated antioxidant activity of Plectranthus barbatus extract on mitochondria. Food Chem
Mair SM, Weiss G (2009) New pharmacological concepts for the treatment of iron overload disorders. Curr Med Chem 16(5):576–590
Majumdar AP, Banerjee S, Nautiyal J, Patel BB, Patel V, Du J et al (2009) Curcumin synergizes with resveratrol to inhibit colon cancer. Nutr Cancer 61(4):544–553
Malcovati L (2009) Red blood cell transfusion therapy and iron chelation in patients with myelodysplastic syndromes. Clin Lymphoma Myeloma 9(Suppl 3):S305–S311
Malecki EA, Connor JR (2002) The case for iron chelation and/or antioxidant therapy in Alzheimer’s disease. Drug Dev Res 56(3):526–530
Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J (2003) Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302(5646):871–874
Mallucci GR, White MD, Farmer M, Dickinson A, Khatun H, Powell AD et al (2007) Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 53(3):325–335
Manach C, Scalbert A, Morand C, Remesy C, Jimenez L (2004) Polyphenols: food sources and bioavailability. Am J Clin Nutr 79(5):727–747
Manach C, Mazur A, Scalbert A (2005a) Polyphenols and prevention of cardiovascular diseases. Curr Opin Lipidol 16(1):77–84
Manach C, Williamson G, Morand C, Scalbert A, Remesy C (2005b) Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr 81(1 Suppl):230S–242S
Managbanag JR, Witten TM, Bonchev D, Fox LA, Tsuchiya M, Kennedy BK et al (2008) Shortest-path network analysis is a useful approach toward identifying genetic determinants of longevity. PLoS One 3(11):e3802
Mancino AM, Hindo SS, Kochi A, Lim MH (2009) Effects of clioquinol on metal-triggered amyloid-beta aggregation revisited. Inorg Chem 48(20):9596–9598
Mandel S, Grunblatt E, Riederer P, Youdim MB (2003) Genes and oxidative stress in Parkinsonism: cDNA microarray studies. Adv Neurol 91:123–132
Mandel S, Amit T, Bar-Am O, Youdim MB (2007) Iron dysregulation in Alzheimer’s disease: multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog Neurobiol 82(6):348–360
Mandel S, Weinreb O, Amit T, Youdim MB (2005) Mechanism of neuroprotective action of the anti-Parkinson drug rasagiline and its derivatives. Brain Res Brain Res Rev 48(2):379–387
Mandel S, Weinreb O, Amit T, Youdim MBH (2004) Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (-)-epigallocatechin-3-gallate: implications for neurodegenerative diseases. J Neurochem 88(6):1555–1569
Mandel S, Youdim MBH (2004) Catechin polyphenols: neurodegeneration and neuroprotection in neurodegenerative diseases. Free Radic Biol Med 37(3):304–317
Mandel SA, Amit T, Weinreb O, Reznichenko L, Youdim MB (2008) Simultaneous manipulation of multiple brain targets by green tea catechins: a potential neuroprotective strategy for Alzheimer and Parkinson diseases. CNS Neurosci Ther 14(4):352–365
Mandemakers W, Morais VA, De Strooper B (2007) A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J Cell Sci 120(Pt 10):1707–1716
Mansour MA (2000) Protective effects of thymoquinone and desferrioxamine against hepatotoxicity of carbon tetrachloride in mice. Life Sci 66(26):2583–2591
Markesbery WR (1997) Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med 23(1):134–147
Markesbery WR, Lovell MA (1998) 4-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging 19(1):33–36
Marmolino D, Acquaviva F (2009) Friedreich’s Ataxia: from the (GAA)n repeat mediated silencing to new promising molecules for therapy. Cerebellum 8(3):245–259
Marniemi J, Alanen E, Impivaara O, Seppanen R, Hakala P, Rajala T et al (2005) Dietary and serum vitamins and minerals as predictors of myocardial infarction and stroke in elderly subjects. Nutr Metab Cardiovasc Dis 15(3):188–197
Martin-Murphy BV, Holt MP, Ju C (2010) The role of damage associated molecular pattern molecules in acetaminophen-induced liver injury in mice. Toxicol Lett 192(3):387–394
Martin GS, Mannino DM, Eaton S, Moss M (2003) The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 348(16):1546–1554
Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y et al (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465(7295):223–226
Maruyama Y, Nakayama M, Yoshimura K, Nakano H, Yamamoto H, Yokoyama K et al (2007) Effect of repeated intravenous iron administration in haemodialysis patients on serum 8-hydroxy-2′-deoxyguanosine levels. Nephrol Dial Transpl 22(5):1407–1412
Marx JJM (2002) Prevention of organ failure in hereditary haemochromatosis. Neth J Med 60(11):419–422
Marx JJM, Kartikasari AER, Georgiou NA (2008) Can iron chelators influence the progression of atherosclerosis? Hemoglobin 32(1–2):123–134
Mascitelli L, Pezzetta F (2006) High iron stores and risk of ischemic stroke in persons with metabolic syndrome. Intern Med 45(5):347
Mascitelli L, Pezzetta F, Goldstein MR (2009) Iron, type 2 diabetes mellitus, and Alzheimer’s disease. Cell Mol Life Sci 66:2943
Massie HR, Aiello VR, Banziger V (1983) Iron accumulation and lipid peroxidation in aging C57bl/6j mice. Exp Gerontol 18(4):277–285
Masubuchi Y, Suda C, Horie T (2005) Involvement of mitochondrial permeability transition in acetaminophen-induced liver injury in mice. J Hepatol 42(1):110–116
Mateo-Gallego R, Calmarza P, Jarauta E, Burillo E, Cenarro A, Civeira F (2010) Serum ferritin is a major determinant of lipid phenotype in familial combined hyperlipidemia and familial hypertriglyceridemia. Metabolism 59(2):154–158
Matthews AJ, Vercellotti GM, Menchaca HJ, Bloch PH, Michalek VN, Marker PH et al (1997) Iron and atherosclerosis: inhibition by the iron chelator deferiprone (L1). J Surg Res 73(1):35–40
Mattson MP (2004) Metal-catalyzed disruption of membrane protein and lipid signaling in the pathogenesis of neurodegenerative disorders. Ann N Y Acad Sci 1012:37–50
Mattson MP (2006) Neuronal life-and-death signaling, apoptosis, and neurodegenerative disorders. Antioxid Redox Signal 8(11–12):1997–2006
Mattson MP (2008a) Awareness of hormesis will enhance future research in basic and applied neuroscience. Crit Rev Toxicol 38(7):633–639
Mattson MP (2008b) Hormesis defined. Ageing Res Rev 7(1):1–7
Matusch A, Depboylu C, Palm C, Wu B, Hoglinger GU, Schafer MK et al (2010) Cerebral bioimaging of Cu, Fe, Zn, and Mn in the MPTP mouse model of Parkinson’s disease using laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS). J Am Soc Mass Spectrom 21(1):161–171
Mazumdar M, Heeney MM, Sox CM, Lieu TA (2007) Preventing stroke among children with sickle cell anemia: an analysis of strategies that involve transcranial Doppler testing and chronic transfusion. Pediatrics 120(4):e1107–1116
McDermid JM, Jaye A, Schim van der Loeff MF, Todd J, Bates C, Austin S et al (2007) Elevated iron status strongly predicts mortality in West African adults with HIV infection. J Acquir Immune Defic Syndr 46(4):498–507
McDermid JM, Prentice AM (2006) Iron and infection: effects of host iron status and the iron-regulatory genes haptoglobin and NRAMP1 (SLC11A1) on host-pathogen interactions in tuberculosis and HIV. Clin Sci (Lond) 110(5):503–524
McDermid JM, van der Loeff MF, Jaye A, Hennig BJ, Bates C, Todd J et al (2009) Mortality in HIV infection is independently predicted by host iron status and SLC11A1 and HP genotypes, with new evidence of a gene-nutrient interaction. Am J Clin Nutr 90(1):225–233
McLaren GD, Gordeuk VR (2009) Hereditary hemochromatosis: insights from the hemochromatosis and iron overload screening (HEIRS) study. Hematol Am Soc Hematol Educ Program 195–206
McLean CA (2008) Review. The neuropathology of kuru and variant Creutzfeldt-Jakob disease. Philos Trans R Soc Lond B Biol Sci 363(1510):3685–3687
McNeill A, Birchall D, Hayflick SJ, Gregory A, Schenk JF, Zimmerman EA et al (2008) T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation. Neurology 70(18):1614–1619
McRae R, Bagchi P, Sumalekshmy S, Fahrni CJ (2009) In situ imaging of metals in cells and tissues. Chem Rev 109(10):4780–4827
Mehdiratta M, Kumar S, Hackney D, Schlaug G, Selim M (2008) Association between serum ferritin level and perihematoma edema volume in patients with spontaneous intracerebral hemorrhage. Stroke 39(4):1165–1170
Mehrotra B, Li XJ, Kamal A, Hoops S, Swainston N, Mendes P (2010) B-Net: a relational data model for biochemical networks. BMC Bioinform
Mehta SH, Webb RC, Ergul A, Tawak A, Dorrance AM (2004) Neuroprotection by tempol in a model of iron-induced oxidative stress in acute ischemic stroke. Am J Physiol 286(2):R283–R288
Mellert W, Deckardt K, Gembardt C, Zwirner-Baier I, Jackh R, van Ravenzwaay B (2004) Aniline: early indicators of toxicity in male rats and their relevance to spleen carcinogenicity. Hum Exp Toxicol 23(8):379–389
Mendes P, Kell DB (1996) On the analysis of the inverse problem of metabolic pathways using artificial neural networks. Biosystems 38:15–28
Mendes P, Kell DB (1998) Non-linear optimization of biochemical pathways: applications to metabolic engineering and parameter estimation. Bioinformatics 14:869–883
Menke A, Fernández-Real JM, Muntner P, Guallar E (2009) The association of biomarkers of iron status with peripheral arterial disease in US adults. BMC Cardiovasc Disord 9(1):34
Messer JG, Kilbarger AK, Erikson KM, Kipp DE (2009) Iron overload alters iron-regulatory genes and proteins, down-regulates osteoblastic phenotype, and is associated with apoptosis in fetal rat calvaria cultures. Bone
Messner DJ, Sivam G, Kowdley KV (2009) Curcumin reduces the toxic effects of iron loading in rat liver epithelial cells. Liver Int 29(1):63–72
Méthy D, Bertrand N, Prigent-Tessier A, Mossiat C, Stanimirovic D, Beley A et al (2008) Beneficial effect of dipyridyl, a liposoluble iron chelator against focal cerebral ischemia: in vivo and in vitro evidence of protection of cerebral endothelial cells. Brain Res 1193:136–142
Meyer D (2006) Iron chelation as therapy for HIV and Mycobacterium tuberculosis co-infection under conditions of iron overload. Curr Pharm Des 12(16):1943–1947
Michalik A, Van Broeckhoven C (2003) Pathogenesis of polyglutamine disorders: aggregation revisited. Hum Mol Genet 12(2):R173–R186
Miele G, Manson J, Clinton M (2001) A novel erythroid-specific marker of transmissible spongiform encephalopathies. Nature Med 7(3):361–364
Miethke M, Marahiel MA (2007) Siderophore-based iron acquisition and pathogen control. Microbiol Mol Biol Rev 71(3):413–451
Migliore L, Fontana I, Colognato R, Coppede F, Siciliano G, Murri L (2005) Searching for the role and the most suitable biomarkers of oxidative stress in Alzheimer’s disease and in other neurodegenerative diseases. Neurobiol Aging 26(5):587–595
Milhavet O, Lehmann S (2002) Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res Rev 38(3):328–339
Millán M, Sobrino T, Arenillas JF, Rodriguez-Yáñez M, Garcia M, Nombela F et al (2008) Biological signatures of brain damage associated with high serum ferritin levels in patients with acute ischemic stroke and thrombolytic treatment. Dis Markers 25(3):181–188
Millan M, Sobrino T, Castellanos M, Nombela F, Arenillas JF, Riva E et al (2007) Increased body iron stores are associated with poor outcome after thrombolytic treatment in acute stroke. Stroke 38(1):90–95
Millan MJ (2006) Multi-target strategies for the improved treatment of depressive states: Conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol Ther 110(2):135–370
Miller DM, Buettner GR, Aust SD (1990) Transition metals as catalysts of “autoxidation” reactions. Free Radic Biol Med 8(1):95–108
Miller ER III, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E (2005) Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 142(1):37–46
Miller RL, James-Kracke M, Sun GY, Sun AY (2009) Oxidative and inflammatory pathways in Parkinson’s disease. Neurochem Res 34(1):55–65
Millerot-Serrurot E, Bertrand N, Mossiat C, Faure P, Prigent-Tessier A, Garnier P et al (2008) Temporal changes in free iron levels after brain ischemia Relevance to the timing of iron chelation therapy in stroke. Neurochem Int 52(8):1442–1448
Milo R, Jorgensen P, Moran U, Weber G, Springer M (2010) BioNumbers—the database of key numbers in molecular and cell biology. Nucleic Acids Res 38(Database issue):D750–D753
Milton NGN (2004) Role of hydrogen peroxide in the aetiology of Alzheimer’s disease—implications for treatment. Drugs Aging 21(2):81–100
Minihane AM, Rimbach G (2002) Iron absorption and the iron binding and anti-oxidant properties of phytic acid. Int J Food Sci Technol 37(7):741–748
Minqin R, Watt F, Huat BTK, Halliwell B (2003) Correlation of iron and zinc levels with lesion depth in newly formed atherosclerotic lesions. Free Radic Biol Med 34(6):746–752
Miranda S, Opazo C, Larrondo LF, Munoz FJ, Ruiz F, Leighton F et al (2000) The role of oxidative stress in the toxicity induced by amyloid beta-peptide in Alzheimer’s disease. Progr Neurobiol 62(6):633–648
Mirre E, Brousse V, Berteloot L, Lambot-Juhan K, Verlhac S, Boulat C et al (2010) Feasibility and efficacy of chronic transfusion for stroke prevention in children with sickle cell disease. Eur J Haematol 84(3):259–265
Mirshafiey A, Mohsenzadegan M (2009) Antioxidant therapy in multiple sclerosis. Immunopharmacol Immunotoxicol 31(1):13–29
Mishra V (2007) Oxidative stress and role of antioxidant supplementation in critical illness. Clin Lab 53(3–4):199–209
Mitchell KM, Dotson AL, Cool KM, Chakrabarty A, Benedict SH, LeVine SM (2007) Deferiprone, an orally deliverable iron chelator, ameliorates experimental autoimmune encephalomyelitis. Mult Scler 13(9):1118–1126
Miyamoto S, Kuwata G, Imai M, Nagao A, Terao J (2000) Protective effect of phytic acid hydrolysis products on iron-induced lipid peroxidation of liposomal membranes. Lipids 35(12):1411–1413
Mladĕnka P, Kalinowski DS, Hašková P, Bobrovová Z, Hrdina R, Šimůnek T et al (2009) The novel iron chelator, 2-pyridylcarboxaldehyde 2-thiophenecarboxyl hydrazone, reduces catecholamine-mediated myocardial toxicity. Chem Res Toxicol 22(1):208–217
Moalem S, Percy ME, Andrews DF, Kruck TP, Wong S, Dalton AJ et al (2000) Are hereditary hemochromatosis mutations involved in Alzheimer disease? Am J Med Genet 93(1):58–66
Mogilner A, Wollman R, Marshall WF (2006) Quantitative modeling in cell biology: what is it good for? Dev Cell 11(3):279–287
Mohkam M, Shamsian BS, Gharib A, Nariman S, Arzanian MT (2008) Early markers of renal dysfunction in patients with beta-thalassemia major. Pediatr Nephrol 23(6):971–976
Moles CG, Mendes P, Banga JR (2003) Parameter estimation in biochemical pathways: a comparison of global optimization methods. Genome Res 13(11):2467–2474
Molfino A, Kushta I, Tommasi V, Fanelli FR, Muscaritoli M (2009) Amyotrophic lateral sclerosis, enteral nutrition and the risk of iron overload. J Neurol 256(6):1015–1016
Moore JH, Asselbergs FW, Williams SM (2010) Bioinformatics challenges for genome-wide association studies. Bioinformatics 26(4):445–455
Moreira PI, Duarte AI, Santos MS, Rego AC, Oliveira CR (2009) An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. J Alzheimers Dis 16(4):741–761
Morel I, Lescoat G, Cillard P, Cillard J (1994) Role of flavonoids and iron chelation in antioxidant action. Methods Enzymol 234:437–443
Morgan C, Colombres M, Nunez MT, Inestrosa NC (2004) Structure and function of amyloid in Alzheimer’s disease. Prog Neurobiol 74(6):323–349
Morgan MA, Hoffbrand AV, Laulicht M, Luck W, Knowles S (1983) Serum ferritin concentration in Gaucher’s disease. Br Med J (Clin Res Ed) 286(6381):1864
Morohashi M, Winn AE, Borisuk MT, Bolouri H, Doyle J, Kitano H (2002) Robustness as a measure of plausibility in models of biochemical networks. J Theor Biol 216(1):19–30
Morphy R, Rankovic Z (2007) Fragments, network biology and designing multiple ligands. Drug Discov Today 12(3–4):156–160
Mossman BT, Churg A (1998) Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med 157(5 Pt 1):1666–1680
Mossman BT, Marsh JP, Shatos MA, Doherty J, Gilbert R, Hill S (1987) Implication of active oxygen species as second messengers of asbestos toxicity. Drug Chem Toxicol 10(1–2):157–180
Mu D, Chang YS, Vexler ZS, Ferriero DM (2005) Hypoxia-inducible factor 1alpha and erythropoietin upregulation with deferoxamine salvage after neonatal stroke. Exp Neurol 195(2):407–415
Mueller S, Afdhal NH, Schuppan D (2006) Iron, HCV, and liver cancer: hard metal setting the pace? Gastroenterology 130(7):2229–2234
Muldoon DF, Hassoun EA, Stohs SJ (1996) Role of iron in ricin-induced lipid peroxidation and superoxide production. Res Commun Mol Pathol Pharmacol 92(1):107–118
Mullenix PS, Andersen CA, Starnes BW (2005) Atherosclerosis as inflammation. Ann Vasc Surg 19(1):130–138
Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H (2007) Trends in oxidative aging theories. Free Radic Biol Med 43(4):477–503
Muñoz X, Cruz MJ, Freixa A, Guardino X, Morell F (2009) Occupational asthma caused by metal arc welding of iron. Respiration 78(4):455–459
Murakami K, Ishida K, Watakabe K, Tsubouchi R, Naruse M, Yoshino M (2006) Maltol/iron-mediated apoptosis in HL60 cells: participation of reactive oxygen species. Toxicol Lett 161(2):102–107
Murphy MP, Packer MA, Scarlett JL, Martin SW (1998) Peroxynitrite: a biologically significant oxidant. Gen Pharmacol 31(2):179–186
Murray AR, Kisin E, Leonard SS, Young SH, Kommineni C, Kagan VE et al (2009) Oxidative stress and inflammatory response in dermal toxicity of single-walled carbon nanotubes. Toxicology 257(3):161–171
Nagababu E, Rifkind JM, Boindala S, Nakka L (2010) Assessment of antioxidant activity of eugenol in vitro and in vivo. Methods Mol Biol 610:165–180
Nagai Y, Fujikake N, Ohno K, Higashiyama H, Popiel HA, Rahadian J et al (2003) Prevention of polyglutamine oligomerization and neurodegeneration by the peptide inhibitor QBP1 in Drosophila. Hum Mol Genet 12(11):1253–1259
Naghii MR (2002) Sulfur mustard intoxication, oxidative stress, and antioxidants. Mil Med 167(7):573–575
Nahon P, Sutton A, Rufat P, Ziol M, Thabut G, Schischmanoff PO et al (2008) Liver iron, HFE gene mutations, and hepatocellular carcinoma occurrence in patients with cirrhosis. Gastroenterology 134(1):102–110
Nakamura T, Keep RF, Hua Y, Nagao S, Hoff JT, Xi G (2006) Iron-induced oxidative brain injury after experimental intracerebral hemorrhage. Acta Neurochir Suppl 96:194–198
Nakano M, Kawanishi Y, Kamohara S, Uchida Y, Shiota M, Inatomi Y et al (2003) Oxidative DNA damage (8-hydroxydeoxyguanosine) and body iron status: a study on 2507 healthy people. Free Radic Biol Med 35(7):826–832
Napier I, Ponka P, Richardson DR (2005) Iron trafficking in the mitochondrion: novel pathways revealed by disease. Blood 105(5):1867–1874
Napoli E, Taroni F, Cortopassi GA (2006) Frataxin, iron-sulfur clusters, heme, ROS, and aging. Antioxid Redox Signal 8(3–4):506–516
Narula MK, Ahuja GK, Whig J, Narang AP, Soni RK (2007) Status of lipid peroxidation and plasma iron level in bronchial asthmatic patients. Indian J Physiol Pharmacol 51(3):289–292
Nathens AB, Neff MJ, Jurkovich GJ, Klotz P, Farver K, Ruzinski JT et al (2002) Randomized, prospective trial of antioxidant supplementation in critically ill surgical patients. Ann Surg 236(6):814–822
Natoli M, Felsani A, Ferruzza S, Sambuy Y, Canali R, Scarino ML (2009) Mechanisms of defence from Fe(II) toxicity in human intestinal Caco-2 cells. Toxicol In Vitro 23(8):1510–1515
Neale BM, Fagerness J, Reynolds R, Sobrin L, Parker M, Raychaudhuri S et al (2010) Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc Natl Acad Sci USA
Neema M, Arora A, Healy BC, Guss ZD, Brass SD, Duan Y et al (2009a) Deep gray matter involvement on brain MRI scans is associated with clinical progression in multiple sclerosis. J Neuroimaging 19(1):3–8
Neema M, Goldberg-Zimring D, Guss ZD, Healy BC, Guttmann CR, Houtchens MK et al (2009b) 3 T MRI relaxometry detects T2 prolongation in the cerebral normal-appearing white matter in multiple sclerosis. Neuroimage 46(3):633–641
Neema M, Stankiewicz J, Arora A, Dandamudi VS, Batt CE, Guss ZD et al (2007a) T1- and T2-based MRI measures of diffuse gray matter and white matter damage in patients with multiple sclerosis. J Neuroimaging 17(Suppl 1):16S–21S
Neema M, Stankiewicz J, Arora A, Guss ZD, Bakshi R (2007b) MRI in multiple sclerosis: what’s inside the toolbox? Neurotherapeutics 4(4):602–617
Nelson DE, Ihekwaba AEC, Elliott M, Gibney CA, Foreman BE, Nelson G et al (2004) Oscillations in NF-κB signalling control the dynamics of target gene expression. Science 306:704–708
Nemery B (1990) Metal toxicity and the respiratory tract. Eur Respir J 3(2):202–219
Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW (2010) Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 464(7287):441–444
Neumann KF, Rojo L, Navarrete LP, Farias G, Reyes P, Maccioni RB (2008) Insulin resistance and Alzheimer’s disease: molecular links & clinical implications. Curr Alzheimer Res 5(5):438–447
Neupane GP, Kim DM (2010) In vitro time-kill activities of ciprofloxacin alone and in combination with the iron chelator deferasirox against Vibrio vulnificus. Eur J Clin Microbiol Infect Dis 29(4):407–410
Nguyen HB, Rivers EP, Abrahamian FM, Moran GJ, Abraham E, Trzeciak S et al (2006) Severe sepsis and septic shock: Review of the literature and emergency department management guidelines. Ann Emerg Med 48(1):28–54
Nguyen T, Hamby A, Massa SM (2005) Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington’s disease mouse model. Proc Natl Acad Sci USA 102(33):11840–11845
Nidhi G, Glick M, Davies JW, Jenkins JL (2006) Prediction of biological targets for compounds using multiple-category Bayesian models trained on chemogenomics databases. J Chem Inf Model 46(3):1124–1133
Nighoghossian N, Wiart M, Berthezene Y (2008) Novel applications of magnetic resonance imaging to image tissue inflammation after stroke. J Neuroimaging 18(4):349–352
Nigro J, Osman N, Dart AM, Little PJ (2006) Insulin resistance and atherosclerosis. Endocr Rev 27(3):242–259
Nijveldt RJ, van Nood E, van Hoorn DEC, Boelens PG, van Norren K, van Leeuwen PAM (2001) Flavonoids: a review of probable mechanisms of action and potential applications. Am J Clin Nutr 74(4):418–425
Nishi T, Shimizu N, Hiramoto M, Sato I, Yamaguchi Y, Hasegawa M et al (2002) Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem 277(46):44548–44556
Nishina S, Hino K, Korenaga M, Vecchi C, Pietrangelo A, Mizukami Y et al (2008) Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology 134(1):226–238
Noble D (2002) The rise of computational biology. Nat Rev Mol Cell Biol 3(6):460–463
Noble D (2006) The music of life: biology beyond genes. Oxford University Press, Oxford
Noble D (2010) Biophysics and systems biology. Philos Transact A Math Phys Eng Sci 368(1914):1125–1139
Northrop-Clewes CA (2008) Interpreting indicators of iron status during an acute phase response—lessons from malaria and human immunodeficiency virus. Ann Clin Biochem 45(Pt 1):18–32
Nowicki M, Kosacka J, Spanel-Borowski K, Borlak J (2009) Deferoxamine-induced neurite outgrowth and synapse formation in postnatal rat dorsal root ganglion (DRG) cell cultures. Eur J Cell Biol 88(10):551–562
Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA (2006) Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol 65(7):631–641
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK et al (2001) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60(8):759–767
Nymark P, Wikman H, Hienonen-Kempas T, Anttila S (2008) Molecular and genetic changes in asbestos-related lung cancer. Cancer Lett 265(1):1–15
O’Donnell VB, Freeman BA (2001) Interactions between nitric oxide and lipid oxidation pathways—implications for vascular disease. Circ Res 88(1):12–21
O’Hagan A (2006) Bayesian analysis of computer code outputs: a tutorial. Reliab Eng Syst Saf 91(10–11):1290–1300
O’Hagan S, Dunn WB, Broadhurst D, Williams R, Ashworth JA, Cameron M et al (2007) Closed-loop, multi-objective optimisation of two-dimensional gas chromatography (GCxGC-tof-MS) for serum metabolomics. Anal Chem 79(2):464–476
O’Hagan S, Dunn WB, Brown M, Knowles JD, Kell DB (2005) Closed-loop, multiobjective optimisation of analytical instrumentation: gas-chromatography-time-of-flight mass spectrometry of the metabolomes of human serum and of yeast fermentations. Anal Chem 77:290–303
O’Neill HA, Gakh O, Park S, Cui J, Mooney SM, Sampson M et al (2005) Assembly of human frataxin is a mechanism for detoxifying redox-active iron. Biochemistry 44(2):537–545
O’Rourke D, McCarthy R, Lynch B (2008) Iron deficiency anaemia and childhood stroke. Ir Med J 101(6):165
Oakley AE, Collingwood JF, Dobson J, Love G, Perrott HR, Edwardson JA et al (2007) Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology 68(21):1820–1825
Oates AC, Gorfinkiel N, Gonzalez-Gaitan M, Heisenberg CP (2009) Quantitative approaches in developmental biology. Nat Rev Genet 10(8):517–530
Oda K, Kitano H (2006) A comprehensive map of the toll-like receptor signaling network. Mol Syst Biol 2:2006 0015
Ogeturk M, Kus I, Kavakli A, Zararsiz I, Ilhan N, Sarsilmaz M (2004) Effects of melatonin on carbon tetrachloride-induced changes in rat serum. J Physiol Biochem 60(3):205–210
Ohnuma S, Harris WA (2003) Neurogenesis and the cell cycle. Neuron 40(2):199–208
Okauchi M, Hua Y, Keep RF, Morgenstern LB, Schallert T, Xi G (2010) Deferoxamine treatment for intracerebral hemorrhage in aged rats: therapeutic time window and optimal duration. Stroke 41(2):375–382
Okauchi M, Hua Y, Keep RF, Morgenstern LB, Xi G (2009) Effects of deferoxamine on intracerebral hemorrhage-induced brain injury in aged rats. Stroke 40(5):1858–1863
Okouchi M, Ekshyyan O, Maracine M, Aw TY (2007) Neuronal apoptosis in neurodegeneration. Antioxid Redox Signal 9(8):1059–1096
Oksenberg JR, Baranzini SE, Sawcer S, Hauser SL (2008) The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nat Rev Genet 9(7):516–526
Olaleye MT, Rocha BT (2008) Acetaminophen-induced liver damage in mice: effects of some medicinal plants on the oxidative defense system. Exp Toxicol Pathol 59(5):319–327
Olanow CW, Arendash GW (1994) Metals and free radicals in neurodegeneration. Curr Opin Neurol 7(6):548–558
Oldroyd D (1986) The arch of knowledge: an introduction to the history of the philosophy and methodology of science. Methuen, New York
Olivares D, Huang X, Branden L, Greig NH, Rogers JT (2009) Physiological and pathological role of alpha-synuclein in Parkinson’s disease through iron mediated oxidative stress; the role of a putative iron-responsive element. Int J Mol Sci 10(3):1226–1260
Omar RF, Hasinoff BB, Mejilla F, Rahimtula AD (1990) Mechanism of ochratoxin A stimulated lipid peroxidation. Biochem Pharmacol 40(6):1183–1191
Ong WY, Farooqui AA (2005) Iron, neuroinflammation, and Alzheimer’s disease. J Alzheimers Dis 8(2):183–200
Orhan H, van Holland B, Krab B, Moeken J, Vermeulen NP, Hollander P et al (2004) Evaluation of a multi-parameter biomarker set for oxidative damage in man: increased urinary excretion of lipid, protein and DNA oxidation products after one hour of exercise. Free Radic Res 38(12):1269–1279
Orrenius S, Gogvadze A, Zhivotovsky B (2007) Mitochondrial oxidative stress: implications for cell death. Ann Rev Pharmacol Toxicol 47:143–183
Ossowska K, Śmiałowska M, Kuter K, Wierońska J, Zięba B, Wardas J et al (2006) Degeneration of dopaminergic mesocortical neurons and activation of compensatory processes induced by a long-term paraquat administration in rats: implications for Parkinson’s disease. Neuroscience 141(4):2155–2165
Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B (2000) The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci 20(16):6048–6054
Othman AI, El-Missiry MA, Amer MA, Arafa M (2008) Melatonin controls oxidative stress and modulates iron, ferritin, and transferrin levels in adriamycin treated rats. Life Sci 83(15–16):563–568
Otogawa K, Ogawa T, Shiga R, Nakatani K, Ikeda K, Nakajima Y et al (2008) Attenuation of acute and chronic liver injury in rats by iron-deficient diet. Am J Physiol Regul Integr Comp Physiol 294(2):R311–320
Pachauri V, Saxena G, Mehta A, Mishra D, Flora SJ (2009) Combinational chelation therapy abrogates lead-induced neurodegeneration in rats. Toxicol Appl Pharmacol 240(2):255–264
Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87(1):315–424
Packard RR, Libby P (2008) Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem 54(1):24–38
Pae HO, Lee YC, Chung HT (2008) Heme oxygenase-1 and carbon monoxide: emerging therapeutic targets in inflammation and allergy. Recent Pat Inflamm Allergy Drug Discov 2(3):159–165
Palau F (2001) Friedreich’s ataxia and frataxin: molecular genetics, evolution and pathogenesis (Review). Int J Mol Med 7(6):581–589
Palsson BØ (2006) Systems biology: properties of reconstructed networks. Cambridge University Press, Cambridge
Pamplona R, Naudi A, Gavin R, Pastrana MA, Sajnani G, Ilieva EV et al (2008) Increased oxidation, glycoxidation, and lipoxidation of brain proteins in prion disease. Free Radic Biol Med 45(8):1159–1166
Pan YJ, Hopkins RG, Loo G (2004) Increased GADD153 gene expression during iron chelation-induced apoptosis in Jurkat T-lymphocytes. Biochim Biophys Acta 1691(1):41–50
Pandey M, Gupta S (2009) Green tea and prostate cancer: from bench to clinic. Front Biosci (Elite Ed) 1:13–25
Pandolfo M (2009) Friedreich ataxia: the clinical picture. J Neurol 256(Suppl 1):3–8.
Pandolfo M, Pastore A (2009) The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J Neurol 256(Suppl 1):9–17
Panduri V, Surapureddi S, Soberanes S, Weitzman SA, Chandel N, Kamp DW (2006) P53 mediates amosite asbestos-induced alveolar epithelial cell mitochondria-regulated apoptosis. Am J Respir Cell Mol Biol 34(4):443–452
Pantopoulos K (2008) Function of the hemochromatosis protein HFE: Lessons from animal models. World J Gastroenterol 14(45):6893–6901
Paoletti R, Gotto AM Jr, Hajjar DP (2004) Inflammation in atherosclerosis and implications for therapy. Circulation 109(23 Suppl 1):III20–III26
Papanikolaou G, Pantopoulos K (2005) Iron metabolism and toxicity. Toxicol Appl Pharmacol 202(2):199–211
Paris I, Martinez-Alvarado P, Cárdenas S, Perez-Pastene C, Graumann R, Fuentes P et al (2005) Dopamine-dependent iron toxicity in cells derived from rat hypothalamus. Chem Res Toxicol 18(3):415–419
Park SH, Aust AE (1998) Participation of iron and nitric oxide in the mutagenicity of asbestos in hgprt-, gpt+ Chinese hamster V79 cells. Cancer Res 58(6):1144–1148
Park S, Gakh O, Mooney SM, Isaya G (2002) The ferroxidase activity of yeast frataxin. J Biol Chem 277(41):38589–38595
Park JH, Lee KH, Kim TY, Lee SY (2007) Metabolic engineering of Escherichia coli for the production of L-valine based on transcriptome analysis and in silico gene knockout simulation. Proc Natl Acad Sci USA 104(19):7797–7802
Park JH, Lee SY, Kim TY, Kim HU (2008a) Application of systems biology for bioprocess development. Trends Biotechnol 26(8):404–412
Park SJ, Kim NH, Jeong BH, Jin JK, Choi JK, Park YJ et al (2008b) The effect of Fenton reaction on protease-resistant prion protein (PrPSc) degradation and scrapie infectivity. Brain Res 1238:172–180
Park SJ, Chung HY, Lee JH (2010) Rapid in vivo screening system for anti-oxidant activity using bacterial redox sensor strains. J Appl Microbiol 108:1217–1225
Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7(9):710–723
Pastore C, Franzese M, Sica F, Temussi P, Pastore A (2007) Understanding the binding properties of an unusual metal-binding protein—a study of bacterial frataxin. FEBS J 274(16):4199–4210
Patel N, Adewoyin T, Chong NV (2008) Age-related macular degeneration: a perspective on genetic studies. Eye (Lond) 22(6):768–776
Patel PI, Isaya G (2001) Friedreich ataxia: from GAA triplet-repeat expansion to frataxin deficiency. Am J Hum Genet 69(1):15–24
Paterniti I, Genovese T, Crisafulli C, Mazzon E, Di Paola R, Galuppo M et al (2009) Treatment with green tea extract attenuates secondary inflammatory response in an experimental model of spinal cord trauma. Naunyn Schmiedebergs Arch Pharmacol 380(2):179–192
Patil BS, Jayaprakasha GK, Chidambara Murthy KN, Vikram A (2009) Bioactive compounds: historical perspectives, opportunities, and challenges. J Agric Food Chem 57(18):8142–8160
Patt A, Horesh IR, Berger EM, Harken AH, Repine JE (1990) Iron depletion or chelation reduces ischemia/reperfusion-induced edema in gerbil brains. J Pediatr Surg 25(2):224–227 (discussion 227–228)
Pavlica S, Gebhardt R (2010) Protective effects of flavonoids and two metabolites against oxidative stress in neuronal PC12 cells. Life Sci 86(3–4):79–86
Pavlovic R, Santaniello E (2007) Peroxynitrite and nitrosoperoxycarbonate, a tightly connected oxidizing-nitrating couple in the reactive nitrogen-oxygen species family: new perspectives for protection from radical-promoted injury by flavonoids. J Pharm Pharmacol 59(12):1687–1695
Pedchenko TV, LeVine SM (1998) Desferrioxamine suppresses experimental allergic encephalomyelitis induced by MBP in SJL mice. J Neuroimmunol 84(2):188–197
Peden AH, Ironside JW (2004) Review: pathology of variant Creutzfeldt-Jakob disease. Folia Neuropathol 42(Suppl A):85–91
Peng CT, Chang JS, Wu KH, Tsai CH, Lin HS (2008) Mechanisms of and obstacles to iron cardiomyopathy in thalassemia. Front Biosci 13:5975–5987
Peng J, Peng L, Stevenson FF, Doctrow SR, Andersen JK (2007) Iron and paraquat as synergistic environmental risk factors in sporadic Parkinson’s disease accelerate age-related neurodegeneration. J Neurosci 27(26):6914–6922
Peng J, Stevenson FF, Oo ML, Andersen JK (2009) Iron-enhanced paraquat-mediated dopaminergic cell death due to increased oxidative stress as a consequence of microglial activation. Free Radic Biol Med 46(2):312–320
Peng Y, Wang C, Xu HH, Liu YN, Zhou F (2010) Binding of alpha-synuclein with Fe(III) and with Fe(II) and biological implications of the resultant complexes. J Inorg Biochem 104(4):365–370
Pennell DJ, Porter JB, Cappellini MD, El-Beshlawy A, Chan LL, Aydinok Y et al (2010) Efficacy of deferasirox in reducing and preventing cardiac iron overload in {beta}-thalassemia. Blood 115:2364–2371
Pérez-Jiménez J, Neveu V, Vos F, Scalbert A (2010) Systematic analysis of the content of 502 polyphenols in 452 foods and beverages: an application of the phenol-explorer database. J Agric Food Chem 58:4959–4969
Perez CA, Tong Y, Guo M (2008) Iron chelators as potential therapeutic agents for Parkinson’s disease. Curr Bioactive Comp 4(3):150–158
Perez LR, Franz KJ (2010) Minding metals: tailoring multifunctional chelating agents for neurodegenerative disease. Dalton Trans 39(9):2177–2187
Pérez VI, Bokov A, Van Remmen H, Mele J, Ran Q, Ikeno Y et al (2009a) Is the oxidative stress theory of aging dead? Biochim Biophys Acta 1790(10):1005–1014
Pérez VI, Van Remmen H, Bokov A, Epstein CJ, Vijg J, Richardson A (2009b) The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 8(1):73–75
Perez VP, de Lima MN, da Silva RS, Dornelles AS, Vedana G, Bogo MR et al (2010) Iron leads to memory impairment that is associated with a decrease in acetylcholinesterase pathways. Curr Neurovasc Res 7(1):15–22
Perlstein EO, Ruderfer DM, Ramachandran G, Haggarty SJ, Kruglyak L, Schreiber SL (2006) Revealing complex traits with small molecules and naturally recombinant yeast strains. Chem Biol 13(3):319–327
Perron NR, Brumaghim JL (2009) A review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem Biophys 53(2):75–100
Perron NR, Hodges JN, Jenkins M, Brumaghim JL (2008) Predicting how polyphenol antioxidants prevent DNA damage by binding to iron. Inorg Chem 47(14):6153–6161
Persson HL (2005) Iron-dependent lysosomal destabilization initiates silica-induced apoptosis in murine macrophages. Toxicol Lett 159(2):124–133
Persson HL, Yu Z, Tirosh O, Eaton JW, Brunk UT (2003) Prevention of oxidant-induced cell death by lysosomotropic iron chelators. Free Radic Biol Med 34(10):1295–1305
Perutz MF, Windle AH (2001) Cause of neural death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature 412(6843):143–144
Petersen RB, Siedlak SL, Lee HG, Kim YS, Nunomura A, Tagliavini F et al (2005) Redox metals and oxidative abnormalities in human prion diseases. Acta Neuropathologica 110(3):232–238
Petri S, Calingasan NY, Alsaied OA, Wille E, Kiaei M, Friedman JE et al (2007) The lipophilic metal chelators DP-109 and DP-460 are neuroprotective in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem 102(3):991–1000
Petronilho F, Constantino L, de Souza B, Reinke A, Martins MR, Fraga CM et al (2009) Efficacy of the combination of N-acetylcysteine and desferrioxamine in the prevention and treatment of gentamicin-induced acute renal failure in male Wistar rats. Nephrol Dial Transpl 24(7):2077–2082
Petti S, Scully C (2009) Polyphenols, oral health and disease: a review. J Dent 37(6):413–423
Pettifer SR, Thorne D, McDermott P, Marsh J, Villéger A, Kell DB et al (2009) Visualising biological data: a semantic approach to tool and database integration. BMC Bioinform 10(Suppl 6):S19
Pfefferbaum A, Adalsteinsson E, Rohlfing T, Sullivan EV (2009) MRI estimates of brain iron concentration in normal aging: comparison of field-dependent (FDRI) and phase (SWI) methods. Neuroimage 47(2):493–500
Phillips DC, Dias HK, Kitas GD, Griffiths HR (2010) Aberrant reactive oxygen and nitrogen species generation in rheumatoid arthritis (RA): causes and consequences for immune function, cell survival, and therapeutic intervention. Antioxid Redox Signal 12(6):743–785
Phillips R, Milo R (2009) A feeling for the numbers in biology. Proc Natl Acad Sci USA 106(51):21465–21471
Pierron J, Malan C, Creus M, Gradinaru J, Hafner I, Ivanova A et al (2008) Artificial metalloenzymes for asymmetric allylic alkylation on the basis of the biotin-avidin technology. Angew Chem Int Ed Engl 47(4):701–705
Pietrangelo A (2006) Hereditary hemochromatosis. Biochim Biophys Acta 1763(7):700–710
Pietta PG (2000) Flavonoids as antioxidants. J Nat Prod 63(7):1035–1042
Pinho RA, Silveira PC, Silva LA, Luiz Streck E, Dal-Pizzol F, JC FM (2005) N-acetylcysteine and deferoxamine reduce pulmonary oxidative stress and inflammation in rats after coal dust exposure. Environ Res 99(3):355–360
Pirko I, Johnson AJ, Lohrey AK, Chen Y, Ying J (2009) Deep gray matter T2 hypointensity correlates with disability in a murine model of MS. J Neurol Sci 282(1–2):34–38
Platt M, Rowe W, Wedge DC, Kell DB, Knowles J, Day PJR (2009) Aptamer evolution for array-based diagnostics. Anal Biochem 390:203–205
Pócsi I, Jeney V, Kertai P, Pócsi I, Emri T, Gyémánt G et al (2008) Fungal siderophores function as protective agents of LDL oxidation and are promising anti-atherosclerotic metabolites in functional food. Mol Nutr Food Res 52(12):1434–1447
Poland A, Knutson JC (1982) 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu Rev Pharmacol Toxicol 22:517–554
Polla AS, Polla LL, Polla BS (2003) Iron as the malignant spirit in successful ageing. Ageing Res Rev 2(1):25–37
Pollera C, Lucchini B, Formentin E, Bareggi S, Poli G, Ponti W (2005) Evaluation of anti-prionic activity of clioquinol in an in vivo model (Mesocricetus auratus). Vet Res Commun 29(Suppl 2):253–255
Ponraj D, Makjanic J, Thong PSP, Tan BKH, Watt F (1999) The onset of atherosclerotic lesion formation in hypercholesterolemic rabbits is delayed by iron depletion. FEBS Lett 459(2):218–222
Popa C, Netea MG, van Riel PL, van der Meer JW, Stalenhoef AF (2007) The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res 48(4):751–762
Popescu BF, Pickering IJ, George GN, Nichol H (2007) The chemical form of mitochondrial iron in Friedreich’s ataxia. J Inorg Biochem 101(6):957–966
Pordea A, Ward TR (2008) Chemogenetic protein engineering: an efficient tool for the optimization of artificial metalloenzymes. Chem Commun (Camb) (36):4239–4249
Porter J, Galanello R, Saglio G, Neufeld EJ, Vichinsky E, Cappellini MD et al (2008) Relative response of patients with myelodysplastic syndromes and other transfusion-dependent anaemias to deferasirox (ICL670): a 1-yr prospective study. Eur J Haematol 80(2):168–176
Porter MJ, Roberts SM, Skidmore J (1999) Polyamino acids as catalysts in asymmetric synthesis. Bioorg Med Chem 7(10):2145–2156
Poser I, Rahman Q, Lohani M, Yadav S, Becker HH, Weiss DG et al (2004) Modulation of genotoxic effects in asbestos-exposed primary human mesothelial cells by radical scavengers, metal chelators and a glutathione precursor. Mutat Res 559(1–2):19–27
Postgate JR (1967) Viability measurements and the survival of microbes under minimum stress. Adv Micr Physiol 1:1–23
Postgate JR (1976) Death in microbes and macrobes. In: Gray TRG, Postgate JR (eds) In the survival of vegetative microbes. Cambridge University Press, Cambridge, pp 1–19
Prass K, Ruscher K, Karsch M, Isaev N, Megow D, Priller J et al (2002) Desferrioxamine induces delayed tolerance against cerebral ischemia in vivo and in vitro. J Cereb Blood Flow Metab 22(5):520–525
Price L, Kowdley KV (2009) The role of iron in the pathophysiology and treatment of chronic hepatitis C. Can J Gastroenterol 23(12):822–828
Proctor CJ, Gray DA (2010) GSK3 and p53—is there a link in Alzheimer’s disease? Mol Neurodegener 5:7
Proctor CJ, Tsirigotis M, Gray DA (2007) An in silico model of the ubiquitin-proteasome system that incorporates normal homeostasis and age-related decline. BMC Syst Biol 1:17
Protti A, Singer M (2006) Bench-to-bedside review: Potential strategies to protect or reverse mitochondrial dysfunction in sepsis-induced organ failure. Crit Care 10(5):227–233
Prusiner SB (1998) Prions. Proc Natl Acad Sci 95(23):13363–13383
Prusiner SB (2001) Shattuck lecture—neurodegenerative diseases and prions. N Engl J Med 344(20):1516–1526
Pryor WA, Squadrito GL (1995) The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol 268(5 Pt 1): L699–L722
Puccio H (2009) Multicellular models of Friedreich ataxia. J Neurol 256(Suppl 1):18–24
Pulliam JF, Jennings CD, Kryscio RJ, Davis DG, Wilson D, Montine TJ et al (2003) Association of HFE mutations with neurodegeneration and oxidative stress in Alzheimer’s disease and correlation with APOE. Am J Med Genet B Neuropsychiatr Genet 119B(1):48–53
Purdey M (2000) Ecosystems supporting clusters of sporadic TSEs demonstrate excesses of the radical-generating divalent cation manganese and deficiencies of antioxidant co factors Cu, Se, Fe, Zn. Does a foreign cation substitution at prion protein’s Cu domain initiate TSE? Med Hypotheses 54(2):278–306
Que L Jr, Tolman WB (2008) Biologically inspired oxidation catalysis. Nature 455(7211):333–340
Quinlan GJ, Chen Y, Evans TW, Gutteridge JMC (2001) Iron signalling regulated directly and through oxygen: implications for sepsis and the acute respiratory distress syndrome. Clinical Science 100(2):169–182
Quinlan GJ, Evans TW, Gutteridge JM (2002) Iron and the redox status of the lungs. Free Radic Biol Med 33(10):1306–1313
Quinlan TR, Marsh JP, Janssen YM, Borm PA, Mossman BT (1994) Oxygen radicals and asbestos-mediated disease. Environ Health Perspect 102(Suppl 10):107–110
Quintana C (2007) Contribution of analytical microscopies to human neurodegenerative diseases research (PSP and AD). Mini Rev Med Chem 7(9):961–975
Quintana C, Bellefqih S, Laval JY, Guerquin-Kern JL, Wu TD, Avila J et al (2006) Study of the localization of iron, ferritin, and hemosiderin in Alzheimer’s disease hippocampus by analytical microscopy at the subcellular level. J Struct Biol 153(1):42–54
Quintanilla RA, Johnson GVW (2009) Role of mitochondrial dysfunction in the pathogenesis of Huntington’s disease. Brain Res Bull 80(4–5):242–247
Qureshi AI, Mendelow AD, Hanley DF (2009) Intracerebral haemorrhage. Lancet 373(9675):1632–1644
Qureshi M, Brown RH Jr, Rogers JT, Cudkowicz ME (2008) Serum ferritin and metal levels as risk factors for amyotrophic lateral sclerosis. Open Neurol J 2:51–54
Radak Z, Chung HY, Koltai E, Taylor AW, Goto S (2008) Exercise, oxidative stress and hormesis. Ageing Res Rev 7(1):34–42
Rader DJ, Daugherty A (2008) Translating molecular discoveries into new therapies for atherosclerosis. Nature 451(7181):904–913
Radi R, Cassina A, Hodara R, Quijano C, Castro L (2002) Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med 33(11):1451–1464
Radi R, Peluffo G, Alvarez MN, Naviliat M, Cayota A (2001) Unraveling peroxynitrite formation in biological systems. Free Radic Biol Med 30(5):463–488
Rajendran R, Ren M, Ning P, Tan Kwong Huat B, Halliwell B, Watt F (2007) Promotion of atherogenesis by copper or iron—which is more likely? Biochem Biophys Res Commun 353(1):6–10
Rajpathak SN, Crandall JP, Wylie-Rosett J, Kabat GC, Rohan TE, Hu FB (2009) The role of iron in type 2 diabetes in humans. Biochim Biophys Acta 1790:671–681
Ramagopalan SV, Cukjati M, Cernilec M, DeLuca GC, Dyment DA, Degenhardt A et al (2008) Mutations in the hemochromatosis gene and the clinical outcome of multiple sclerosis. J Neuroimmunol 203(1):104–107
Ramakrishna G, Rooke TW, Cooper LT (2003) Iron and peripheral arterial disease: revisiting the iron hypothesis in a different light. Vasc Med 8(3):203–210
Ramesh E, Jayakumar T, Elanchezhian R, Sakthivel M, Geraldine P, Thomas PA (2009) Green tea catechins, alleviate hepatic lipidemic-oxidative injury in Wistar rats fed an atherogenic diet. Chem Biol Interact 180(1):10–19
Rana A, Gnaneswari D, Bansal S, Kundu B (2009) Prion metal interaction: is prion pathogenesis a cause or a consequence of metal imbalance? Chem Biol Interact 181(3):282–291
Rao PS, Liu XK, Das DK, Weinstein GS, Tyras DH (1991) Protection of ischemic heart from reperfusion injury by myo-inositol hexaphosphate, a natural antioxidant. Ann Thorac Surg 52(4):908–912
Ratan RR, Siddiq A, Aminova L, Langley B, McConoughey S, Karpisheva K et al (2008) Small molecule activation of adaptive gene expression: tilorone or its analogs are novel potent activators of hypoxia inducible factor-1 that provide prophylaxis against stroke and spinal cord injury. Ann NY Acad Sci 1147:383–394
Rattan SI (2008) Hormesis in aging. Ageing Res Rev 7(1):63–78
Rattan SI, Demirovic D (2009) Hormesis can and does work in humans. Dose Response 8(1):58–63
Rattan SIS (2004) Aging, anti-aging, and hormesis. Mech Ageing Dev 125(4):285–289
Rattner A, Nathans J (2006) Macular degeneration: recent advances and therapeutic opportunities. Nat Rev Neurosci 7(11):860–872
Ravindran J, Prasad S, Aggarwal BB (2009) Curcumin and cancer cells: how many ways can curry kill tumor cells selectively? AAPS J 11(3):495–510
Rawat R, Humphrey J, Ntozini R, Mutasa K, Iliff P, Stoltzfus R (2008) Elevated iron stores are associated with HIV disease severity and mortality among postpartum women in Zimbabwe. Public Health Nutr 12:1–9
Rayman MP, Barlis J, Evans RW, Redman CW, King LJ (2002) Abnormal iron parameters in the pregnancy syndrome preeclampsia. Am J Obstet Gynecol 187(2):412–418
Raymond KN, Dertz EA, Kim SS (2003) Enterobactin: an archetype for microbial iron transport. Proc Natl Acad Sci USA 100(7):3584–3588
Reddy PH (2008) Mitochondrial medicine for aging and neurodegenerative diseases. Neuromol Med 10(4):291–315
Reddy PH, Beal MF (2008) Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med 14(2):45–53
Reddy VP, Zhu X, Perry G, Smith MA (2009) Oxidative stress in diabetes and Alzheimer’s disease. J Alzheimers Dis 16(4):763–774
Redl H, Gasser H, Schlag G, Marzi I (1993) Involvement of oxygen radicals in shock related cell injury. Br Med Bull 49(3):556–565
Reid AB, Kurten RC, McCullough SS, Brock RW, Hinson JA (2005) Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J Pharmacol Exp Ther 312(2):509–516
Reif DW (1992) Ferritin as a source of iron for oxidative damage. Free Radic Biol Med 12(5):417–427
Reiter RJ, Paredes SD, Manchester LC, Tan DX (2009) Reducing oxidative/nitrosative stress: a newly-discovered genre for melatonin. Crit Rev Biochem Mol Biol 44(4):175–200
Ren MQ, Rajendran R, Pan N, Tan BKH, Ong WY, Watt F et al (2005) The iron chelator desferrioxamine inhibits atherosclerotic lesion development and decreases lesion iron concentrations in the cholesterol-fed rabbit. Free Radic Biol Med 38(9):1206–1211
Ren MQ, Watt F, Huat BTK, Halliwell B (2003) Trace elemental distributions in induced atherosclerotic lesions using nuclear microscopy. Nuclear Instr Meth Phys Res B 210:336–342
Reynolds A, Laurie C, Mosley RL, Gendelman HE (2007) Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol 82:297–325
Reynolds FD, Dauchy R, Blask D, Dietz PA, Lynch D, Zuckerman R (2003) The pineal gland hormone melatonin improves survival in a rat model of sepsis/shock induced by zymosan A. Surgery 134(3):474–479
Reznichenko L, Amit T, Zheng H, Avramovich-Tirosh Y, Youdim MB, Weinreb O et al (2006) Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (-)-epigallocatechin-3-gallate in cell cultures: implications for iron chelation in Alzheimer’s disease. J Neurochem 97(2):527–536
Reznichenko L, Kalfon L, Amit T, Youdim MB, Mandel SA (2010) Low dosage of rasagiline and epigallocatechin gallate synergistically restored the nigrostriatal axis in MPTP-induced Parkinsonism. Neurodegener Dis
Rice-Evans CA, Miller J, Paganga G (1997) Antioxidant properties of phenolic compounds. Trends Plant Sci 2(4):152–159
Rice-Evans CA, Packer L (eds) (2003) Flavonoids in health and disease, 2nd edn. Marcel Dekker, New York
Richardson DR, Huang ML, Whitnall M, Becker EM, Ponka P, Suryo Rahmanto Y (2009a) The ins and outs of mitochondrial iron-loading: the metabolic defect in Friedreich’s ataxia. J Mol Med
Richardson DR, Kalinowski DS, Lau S, Jansson PJ, Lovejoy DB (2009b) Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim Biophys Acta 1790(7):702–717
Richer S, Rudy D, Statkute L, Karofty K, Frankowski J (2002) Serum iron, transferrin saturation, ferritin, and dietary data in age-related macular degeneration. Am J Ther 9(1):25–28
Ridker PM, Brown NJ, Vaughan DE, Harrison DG, Mehta JL (2004) Established and emerging plasma biomarkers in the prediction of first atherothrombotic events. Circulation 109(25 Suppl 1):6–19
Riedl MA, Nel AE (2008) Importance of oxidative stress in the pathogenesis and treatment of asthma. Curr Opin Allergy Clin Immunol 8(1):49–56
Rigamonti C, Andorno S, Maduli E, Capelli F, Boldorini R, Sartori M (2005) Gender and liver fibrosis in chronic hepatitis: the role of iron status. Aliment Pharmacol Ther 21(12):1445–1451
Rimbach G, Pallauf J, Moehring J, Kraemer K, Minihane AM (2008) Effect of dietary phytate and microbial phytase on mineral and trace element bioavailability: a literature review. Curr Topics Nutraceut Res 6:131–144
Ristow M, Zarse K (2010) How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp Gerontol
Ritter C, Andrades ME, Reinke A, Menna-Barreto S, Moreira JC, Dal-Pizzol F (2004) Treatment with N-acetylcysteine plus deferoxamine protects rats against oxidative stress and improves survival in sepsis. Crit Care Med 32(2):342–349
Ritter C, da Cunha AA, Echer IC, Andrades M, Reinke A, Lucchiari N et al (2006) Effects of N-acetylcysteine plus deferoxamine in lipopolysaccharide-induced acute lung injury in the rat. Crit Care Med 34(2):471–477
Rittirsch D, Flierl MA, Ward PA (2008) Harmful molecular mechanisms in sepsis. Nat Rev Immunol 8(10):776–787
Rival T, Page RM, Chandraratna DS, Sendall TJ, Ryder E, Liu B et al (2009) Fenton chemistry and oxidative stress mediate the toxicity of the beta-amyloid peptide in a Drosophila model of Alzheimer’s disease. Eur J Neurosci 29(7):1335–1347
Rivera-Espinoza Y, Muriel P (2009) Pharmacological actions of curcumin in liver diseases or damage. Liver Int 29(10):1457–1466
Robinson SR, Dang TN, Dringen R, Bishop GM (2009) Hemin toxicity: a preventable source of brain damage following hemorrhagic stroke. Redox Rep 14(6):228–235
Robson KJ, Lehmann DJ, Wimhurst VL, Livesey KJ, Combrinck M, Merryweather-Clarke AT et al (2004) Synergy between the C2 allele of transferrin and the C282Y allele of the haemochromatosis gene (HFE) as risk factors for developing Alzheimer’s disease. J Med Genet 41(4):261–265
Rodríguez-Caso C, Corominas-Murtra B, Solé RV (2009) On the basic computational structure of gene regulatory networks. Mol Biosyst 5(12):1617–1629
Rodriguez-Fernandez M, Mendes P, Banga JR (2006) A hybrid approach for efficient and robust parameter estimation in biochemical pathways. Biosystems 83(2–3):248–265
Roijers RB, Dutta RK, Mutsaers PHA, Gijbels MJJ, de Winther MPJ, de Goeij JJM et al (2005) Spatial correlation of trace elements with morphological features of atherosclerotic plaques. Nuclear Instrum Methods Phys Res Sect B-Beam Interact Mater Atoms 231:239–244
Rokushima M, Omi K, Araki A, Kyokawa Y, Furukawa N, Itoh F et al (2007a) A toxicogenomic approach revealed hepatic gene expression changes mechanistically linked to drug-induced hemolytic anemia. Toxicol Sci 95(2):474–484
Rokushima M, Omi K, Imura K, Araki A, Furukawa N, Itoh F et al (2007b) Toxicogenomics of drug-induced hemolytic anemia by analyzing gene expression profiles in the spleen. Toxicol Sci 100(1):290–302
Romeo AM, Christen L, Niles EG, Kosman DJ (2001) Intracellular chelation of iron by bipyridyl inhibits DNA virus replication: ribonucleotide reductase maturation as a probe of intracellular iron pools. J Biol Chem 276(26):24301–24308
Roostaee AR, Roostaee MH, Roucou X (2010) An update on prion biology and proteomics. Curr Proteom 7(1):36–48
Rooyakkers TM, Stroes ES, Kooistra MP, van Faassen EE, Hider RC, Rabelink TJ et al (2002) Ferric saccharate induces oxygen radical stress and endothelial dysfunction in vivo. Eur J Clin Invest 32(Suppl 1):9–16
Roriz-Filho JS, Sa-Roriz TM, Rosset I, Camozzato AL, Santos AC, Chaves ML et al (2009) (Pre)diabetes, brain aging, and cognition. Biochim Biophys Acta 1792(5):432–443
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362(6415):59–62
Ross CA (1995) When more is less: pathogenesis of glutamine repeat neurodegenerative diseases. Neuron 15(3):493–496
Ross OA, Meschia JF (2009) Genetics of ischemic stroke: inheritance of a sporadic disorder. Curr Neurol Neurosci Rep 9(1):19–27
Ross R (1999) Atherosclerosis: an inflammatory disease. N Engl J Med 340(2):115–126
Rötig A, Sidi D, Munnich A, Rustin P (2002) Molecular insights into Friedreich’s ataxia and antioxidant-based therapies. Trends Mol Med 8(5):221–224
Rottkamp CA, Nunomura A, Raina AK, Sayre LM, Perry G, Smith MA (2000) Oxidative stress, antioxidants, and Alzheimer disease. Alzheimer Dis Assoc Disord 14(Suppl 1):S62–S66
Rouault TA (2006) The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol 2(8):406–414
Rowe W, Platt M, Wedge D, Day PJ, Kell DB, Knowles J (2010) Analysis of a complete DNA-protein affinity landscape. J R Soc Interface 7(44):397–408
Rubbo H, O’Donnell V (2005) Nitric oxide, peroxynitrite and lipoxygenase in atherogenesis: mechanistic insights. Toxicology 208(2):305–317
Rubbo H, Trostchansky A, O’Donnell VB (2009) Peroxynitrite-mediated lipid oxidation and nitration: mechanisms and consequences. Arch Biochem Biophys 484(2):167–172
Sadrzadeh SM, Saffari Y (2004) Iron and brain disorders. Am J Clin Pathol 121(Suppl):S64–S70
Sahle S, Mendes P, Hoops S, Kummer U (2008) A new strategy for assessing sensitivities in biochemical models. Phil Trans Royal Soc A 366:3619–3631. doi:10.1098/rsta.2008.0108
Saito K, Ishizaka N, Aizawa T, Sata M, Iso-o N, Noiri E et al (2005) Iron chelation and a free radical scavenger suppress angiotensin II-induced upregulation of TGF-beta1 in the heart. Am J Physiol Heart Circ Physiol 288(4):H1836–1843
Sakaida I, Kayano K, Wasaki S, Nagatomi A, Matsumura Y, Okita K (1995) Protection against acetaminophen-induced liver injury in vivo by an iron chelator, deferoxamine. Scand J Gastroenterol 30(1):61–67
Sakudo A, Ikuta K (2009a) Fundamentals of prion diseases and their involvement in the loss of function of cellular prion protein. Protein Pept Lett 16(3):217–229
Sakudo A, Ikuta K (2009b) Prion protein functions and dysfunction in prion diseases. Curr Med Chem 16(3):380–389
Saleh A, Schroeter M, Ringelstein A, Hartung HP, Siebler M, Modder U et al (2007) Iron oxide particle-enhanced MRI suggests variability of brain inflammation at early stages after ischemic stroke. Stroke 38(10):2733–2737
Salonen JT, Nyyssonen K, Korpela H, Tuomilehto J, Seppanen R, Salonen R (1992) High stored iron levels are associated with excess risk of myocardial infarction in eastern Finnish men. Circulation 86(3):803–811
Saltelli A, Ratto M, Andres T, Campolongo F, Cariboni J, Gatelli D et al (2008) Global sensitivity analysis: the primer. WileyBlackwell, New York
Saltelli A, Tarantola S, Campolongo F, Ratto M (2004) Sensitivity analysis in practice: a guide to assessing scientific models. Wiley, New York
Salvador GA (2010) Iron in neuronal function and dysfunction. Biofactors 36:103–110
Sampietro M, Caputo L, Casatta A, Meregalli M, Pellagatti A, Tagliabue J et al (2001) The hemochromatosis gene affects the age of onset of sporadic Alzheimer’s disease. Neurobiol Aging 22(4):563–568
Sandberg AS, Brune M, Carlsson NG, Hallberg L, Skoglund E, Rossander-Hulthen L (1999) Inositol phosphates with different numbers of phosphate groups influence iron absorption in humans. Am J Clin Nutr 70(2):240–246
Sandy M, Butler A (2009) Microbial iron acquisition: marine and terrestrial siderophores. Chem Rev 109(10):4580–4595
Sanfeliu C, Sebastià J, Cristòfol R, Rodríguez-Farré E (2003) Neurotoxicity of organomercurial compounds. Neurotox Res 5(4):283–305
Santhosh K, Kesavadas C, Thomas B, Gupta AK, Thamburaj K, Kapilamoorthy TR (2009) Susceptibility weighted imaging: a new tool in magnetic resonance imaging of stroke. Clin Radiol 64(1):74–83
Santos R, Lefevre S, Sliwa D, Seguin A, Camadro JM, Lesuisse E (2010) Friedreich’s Ataxia: molecular mechanisms, redox considerations and therapeutic opportunities. Antioxid Redox Signal
Sarafian T, Verity MA (1991) Oxidative mechanisms underlying methyl mercury neurotoxicity. Int J Dev Neurosci 9(2):147–153
Sarafian TA, Vartavarian L, Kane DJ, Bredesen DE, Verity MA (1994) bcl-2 expression decreases methyl mercury-induced free-radical generation and cell killing in a neural cell line. Toxicol Lett 74(2):149–155
Sarkar FH, Li Y (2009) Harnessing the fruits of nature for the development of multi-targeted cancer therapeutics. Cancer Treat Rev 35(7):597–607
Sarkar FH, Li Y, Wang Z, Kong D (2009) Cellular signaling perturbation by natural products. Cell Signal 21(11):1541–1547
Savarino A, Pescarmona GP, Boelaert JR (1999) Iron metabolism and HIV infection: reciprocal interactions with potentially harmful consequences? Cell Biochem Funct 17(4):279–287
Sawyers CL (2007) Cancer: mixing cocktails. Nature 449(7165):993–996
Saxena K, Ranalli M, Khan N, Blanchong C, Kahwash SB (2005) Fatal stroke in a child with severe iron deficiency anemia and multiple hereditary risk factors for thrombosis. Clin Pediatr (Phila) 44(2):175–180
Sayre LM, Moreira PI, Smith MA, Perry G (2005) Metal ions and oxidative protein modification in neurological disease. Ann Ist Super Sanita 41(2):143–164
Sayre LM, Perry G, Atwood CS, Smith MA (2000a) The role of metals in neurodegenerative diseases. Cell Mol Biol (Noisy-le-grand) 46(4):731–741
Sayre LM, Perry G, Harris PLR, Liu YH, Schubert KA, Smith MA (2000b) In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: A central role for bound transition metals. J Neurochem 74(1):270–279
Scalbert A, Manach C, Morand C, Remesy C, Jimenez L (2005) Dietary polyphenols and the prevention of diseases. Crit Rev Food Sci Nutr 45(4):287–306
Scalbert A, Williamson G (2000) Dietary intake and bioavailability of polyphenols. J Nutr 130(8S Suppl):2073S–2085S
Schagerlöf U, Elmlund H, Gakh O, Nordlund G, Hebert H, Lindahl M et al (2008) Structural basis of the iron storage function of frataxin from single-particle reconstruction of the iron-loaded oligomer. Biochemistry 47(17):4948–4954
Schenck JF, Zimmerman EA (2004) High-field magnetic resonance imaging of brain iron: birth of a biomarker? NMR Biomed 17(7):433–445
Schiano TD, Grinberg M, Nawabi I, Grabowski G (1993) Blue nasal secretions: a presentation of Gaucher’s disease and concurrent hemosiderosis. Am J Hematol 44(3):219–220
Schlawe D, Majdalani A, Velcicky J, Hessler E, Wieder T, Prokop A et al (2004) Iron-containing nucleoside analogues with pronounced apoptosis-inducing activity. Angew Chem Int Ed Engl 43(13):1731–1734
Schleicher E, Friess U (2007) Oxidative stress, AGE, and atherosclerosis. Kidney Int Suppl(106):S17–S26
Schlemmer U, Frolich W, Prieto RM, Grases F (2009) Phytate in foods and significance for humans: food sources, intake, processing, bioavailability, protective role and analysis. Mol Nutr Food Res 53(Suppl 2):S330–S375
Schmitt-Ulms G, Ehsani S, Watts JC, Westaway D, Wille H (2009) Evolutionary descent of prion genes from the ZIP family of metal ion transporters. PLoS One 4(9):e7208
Schnaider Beeri M, Goldbourt U, Silverman JM, Noy S, Schmeidler J, Ravona-Springer R et al (2004) Diabetes mellitus in midlife and the risk of dementia three decades later. Neurology 63(10):1902–1907
Schnellmann JG, Pumford NR, Kusewitt DF, Bucci TJ, Hinson JA (1999) Deferoxamine delays the development of the hepatotoxicity of acetaminophen in mice. Toxicol Lett 106(1):79–88
Schrag M, McAuley G, Pomakian J, Jiffry A, Tung S, Mueller C et al (2009) Correlation of hypointensities in susceptibility-weighted images to tissue histology in dementia patients with cerebral amyloid angiopathy: a postmortem MRI study. Acta Neuropathol
Schrattenholz A, Šoškić V (2008) What does systems biology mean for drug development? Curr Med Chem 15(15):1520–1528
Schreck R, Albermann K, Baeuerle PA (1992a) Nuclear factor kappa-B—an oxidative stress-responsive transcription factor of eukaryotic cells (a Review). Free Rad Res Comm 17(4):221–237
Schreck R, Meier B, Mannel DN, Droge W, Baeuerle PA (1992b) Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J Exp Med 175(5):1181–1194
Schreck R, Baeuerle PA (1994) Assessing oxygen radicals as mediators in activation of inducible eukaryotic transcription factor NF-kappa B. Methods Enzymol 234:151–163
Schreck R, Rieber P, Baeuerle PA (1991) Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-Kappa-B transcription Factor and HIV-1. EMBO J 10(8):2247–2258
Schulz JB, Boesch S, Burk K, Durr A, Giunti P, Mariotti C et al (2009) Diagnosis and treatment of Friedreich ataxia: a European perspective. Nat Rev Neurol 5(4):222–234
Schulz M, Uhlendorf J, Klipp E, Liebermeister W (2006) SBMLmerge, a system for combining biochemical network models. Genome Inform 17(1):62–71
Scott LE, Orvig C (2009) Medicinal inorganic chemistry approaches to passivation and removal of aberrant metal ions in disease. Chem Rev 109(10):4885–4910
Sebai H, Ben-Attia M, Sani M, Aouani E, Ghanem-Boughanmi N (2009) Protective effect of resveratrol in endotoxemia-induced acute phase response in rats. Arch Toxicol 83(4):335–340
Sebastiani G, Walker AP (2007) HFE gene in primary and secondary hepatic iron overload. World J Gastroenterol 13(35):4673–4689
Seet RC, Lee CY, Lim EC, Tan JJ, Quek AM, Chong WL et al (2010) Oxidative damage in Parkinson disease: measurement using accurate biomarkers. Free Radic Biol Med 48(4):560–566
Seguin A, Sutak R, Bulteau AL, Garcia-Serres R, Oddou JL, Lefevre S et al (2010) Evidence that yeast frataxin is not an iron storage protein in vivo. Biochim Biophys Acta 1802(6):531–538
Seifried HE, Anderson DE, Fisher EI, Milner JA (2007) A review of the interaction among dietary antioxidants and reactive oxygen species. J Nutr Biochem 18(9):567–579
Selim M (2009) Deferoxamine mesylate: a new hope for intracerebral hemorrhage: from bench to clinical trials. Stroke 40(3 Suppl):S90–91
Selim MH, Ratan RR (2004) The role of iron neurotoxicity in ischemic stroke. Ageing Res Rev 3(3):345–353
Sen CK, Packer L (1996) Antioxidant and redox regulation of gene transcription. FASEB J 10(7):709–720
Sener A, Ozsavci D, Bingol-Ozakpinar O, Cevik O, Yanikkaya-Demirel G, Yardimci T (2009) Oxidized-LDL and Fe3+/Ascorbic Acid-Induced Oxidative Modifications and Phosphatidylserine Exposure in Human Platelets are Reduced by Melatonin. Folia Biol (Praha) 55(2):45–52
Serra JA, Domínguez RO, Marschoff ER, Guareschi EM, Famulari AL, Boveris A (2009) Systemic oxidative stress associated with the neurological diseases of aging. Neurochem Res
Shah SV, Alam MG (2003) Role of iron in atherosclerosis. Am J Kidney Dis 41(3 Suppl 1):S80–S83
Shamoto-Nagai M, Maruyama W, Yi H, Akao Y, Tribl F, Gerlach M et al (2006) Neuromelanin induces oxidative stress in mitochondria through release of iron: mechanism behind the inhibition of 26S proteasome. J Neural Transm 113(5):633–644
Shamsuddin AM (1995) Inositol phosphates have novel anticancer function. J Nutr 125(3 Suppl):725S–732S
Shanker G, Aschner M (2003) Methylmercury-induced reactive oxygen species formation in neonatal cerebral astrocytic cultures is attenuated by antioxidants. Brain Res Mol Brain Res 110(1):85–91
Shannahan JH, Schladweiler MC, Richards JH, Ledbetter AD, Ghio AJ, Kodavanti UP (2010) Pulmonary oxidative stress, inflammation, and dysregulated iron homeostasis in rat models of cardiovascular disease. J Toxicol Environ Health A 73(10):641–656
Sharifi F, Nasab NM, Zadeh HJ (2008) Elevated serum ferritin concentrations in prediabetic subjects. Diab Vasc Dis Res 5(1):15–18
Sharma S, Haldar C (2009) Comparative effect of melatonin and vitamin E on phenylhydrazine-induced toxicity in the spleen of Funambulus pennanti. Environ Toxicol 24(1):1–9
Sharom JR, Bellows DS, Tyers M (2004) From large networks to small molecules. Curr Opin Chem Biol 8(1):81–90
Shatos MA, Doherty JM, Marsh JP, Mossman BT (1987) Prevention of asbestos-induced cell death in rat lung fibroblasts and alveolar macrophages by scavengers of active oxygen species. Environ Res 44(1):103–116
Shcherbatykh I, Carpenter DO (2007) The role of metals in the etiology of Alzheimer’s disease. J Alzheimers Dis 11(2):191–205
She H, Xiong S, Lin M, Zandi E, Giulivi C, Tsukamoto H (2002) Iron activates NF-kappaB in Kupffer cells. Am J Physiol Gastrointest Liver Physiol 283(3):G719–726
Sheftel A, Stehling O, Lill R (2010) Iron-sulfur proteins in health and disease. Trends Endocrinol Metab 21(5):302–314
Shen Z, Parker VD, Aust AE (1995) Mediated, thin-layer cell, coulometric determination of redox-active iron on the surface of asbestos fibers. Anal Chem 67(2):307–311
Sheth S, Brittenham GM (2000) Genetic disorders affecting proteins of iron metabolism: clinical implications. Annu Rev Med 51:443–464
Shi T, Knaapen AM, Begerow J, Birmili W, Borm PJ, Schins RP (2003) Temporal variation of hydroxyl radical generation and 8-hydroxy-2′-deoxyguanosine formation by coarse and fine particulate matter. Occup Environ Med 60(5):315–321
Shi Z, Nie G, Duan XL, Rouault T, Wu WS, Ning B et al (2010) Neuroprotective mechanism of mitochondrial ferritin on 6-hydroxydopamine induced dopaminergic cell damage: implication for neuroprotection in Parkinson’s disease. Antioxid Redox Signal
Shin SM, Kim SG (2009) Inhibition of arachidonic acid and iron-induced mitochondrial dysfunction and apoptosis by oltipraz and novel 1, 2-dithiole-3-thione congeners. Mol Pharmacol 75(1):242–253
Shiomi M, Ito T, Tsukada T, Tsujita Y, Horikoshi H (1999) Combination treatment with troglitazone, an insulin action enhancer, and pravastatin, an inhibitor of HMG-CoA reductase, shows a synergistic effect on atherosclerosis of WHHL rabbits. Atherosclerosis 142(2):345–353
Shoelson SE, Goldfine AB (2009) Getting away from glucose: fanning the flames of obesity-induced inflammation. Nat Med 15(4):373–374
Shoelson SE, Herrero L, Naaz A (2007) Obesity, inflammation, and insulin resistance. Gastroenterology 132(6):2169–2180
Shore SA (2008) Obesity and asthma: possible mechanisms. J Allergy Clin Immunol 121(5):1087–1093 (quiz 1094–1085)
Shukla A, Gulumian M, Hei TK, Kamp D, Rahman Q, Mossman BT (2003a) Multiple roles of oxidants in the pathogenesis of asbestos-induced diseases. Free Radic Biol Med 34(9):1117–1129
Shukla A, Jung M, Stern M, Fukagawa NK, Taatjes DJ, Sawyer D et al (2003b) Asbestos induces mitochondrial DNA damage and dysfunction linked to the development of apoptosis. Am J Physiol Lung Cell Mol Physiol 285(5):L1018–L1025
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361(17):1651–1661
Signorini C, Ciccoli L, Leoncini S, Carloni S, Perrone S, Comporti M et al (2009) Free iron, total F-isoprostanes and total F-neuroprostanes in a model of neonatal hypoxic-ischemic encephalopathy: neuroprotective effect of melatonin. J Pineal Res 46(2):148–154
Sikorska K, Stalke P, Izycka-Swieszewska E, Romanowski T, Bielawski KP (2010) The role of iron overload and HFE gene mutations in the era of pegylated interferon and ribavirin treatment of chronic hepatitis C. Med Sci Monit 16(3):CR137–CR143
Silver H, Chertkow Y, Weinreb O, Danovich L, Youdim M (2009) Multifunctional pharmacotherapy: what can we learn from study of selective serotonin reuptake inhibitor augmentation of antipsychotics in negative-symptom schizophrenia? Neurotherapeutics 6(1):86–93
Silver LL (2007) Multi-targeting by monotherapeutic antibacterials. Nat Rev Drug Discov 6(1):41–55
Silvestri L, Camaschella C (2008) A potential pathogenetic role of iron in Alzheimer’s Disease. J Cell Mol Med 12(5a):1548–1550
Simka M, Rybak Z (2008) Hypothetical molecular mechanisms by which local iron overload facilitates the development of venous leg ulcers and multiple sclerosis lesions. Med Hypotheses 71(2):293–297
Simmonds RE, Foxwell BM (2008) NF-{kappa}B and its relevance to arthritis and inflammation. Rheumatology (Oxford)
Simmons-Willis TA, Koh AS, Clarkson TW, Ballatori N (2002) Transport of a neurotoxicant by molecular mimicry: the methylmercury-L-cysteine complex is a substrate for human L-type large neutral amino acid transporter (LAT) 1 and LAT2. Biochem J 367(Pt 1):239–246
Simonart T, Degraef C, Andrei G, Mosselmans R, Hermans P, Van Vooren JP et al (2000) Iron chelators inhibit the growth and induce the apoptosis of Kaposi’s sarcoma cells and of their putative endothelial precursors. J Invest Dermatol 115(5):893–900
Šimůnek T, Štĕrba M, Popelova O, Adamcova M, Hrdina R, Geršl V (2009) Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol Rep 61(1):154–171
Singh AV, Zamboni P (2009) Anomalous venous blood flow and iron deposition in multiple sclerosis. J Cereb Blood Flow Metab 29(12):1867–1878
Singh N, Pillay V, Choonara YE (2007) Advances in the treatment of Parkinson’s disease. Prog Neurobiol 81(1):29–44
Singh A, Isaac AO, Luo X, Mohan ML, Cohen ML, Chen F et al (2009a) Abnormal brain iron homeostasis in human and animal prion disorders. PLoS Pathog 5(3):e1000336
Singh A, Kong Q, Luo X, Petersen RB, Meyerson H, Singh N (2009b) Prion protein (PrP) knock-out mice show altered iron metabolism: a functional role for PrP in iron uptake and transport. PLoS One 4(7):e6115
Singh A, Mohan ML, Isaac AO, Luo X, Petrak J, Vyoral D et al (2009c) Prion protein modulates cellular iron uptake: a novel function with implications for prion disease pathogenesis. PLoS ONE 4(2):e4468
Singh N, Das D, Singh A, Mohan ML (2010a) Prion protein and metal interaction: physiological and pathological implications. Curr Issues Mol Biol 12(2):99–108
Singh N, Singh A, Das D, Mohan ML (2010b) Redox control of prion and disease pathogenesis. Antioxid Redox Signal 12:1271–1294
Skinner AC, Steiner MJ, Henderson FW, Perrin EM (2010) Multiple markers of inflammation and weight status: cross-sectional analyses throughout childhood. Pediatrics 125(4):e801–809
Slemmer JE, Shacka JJ, Sweeney MI, Weber JT (2008) Antioxidants and free radical scavengers for the treatment of stroke, traumatic brain injury and aging. Curr Med Chem 15(4):404–414
Smallbone K, Simeonidis E, Broomhead DS, Kell DB (2007) Something from nothing: bridging the gap between constraint-based and kinetic modelling. FEBS J 274:5576–5585
Smirnova GV, Samoylova ZY, Muzyka NG, Oktyabrsky ON (2009) Influence of polyphenols on Escherichia coli resistance to oxidative stress. Free Radic Biol Med 46(6):759–768
Smith AG, Chernova T (2009) Disruption of heme synthesis by polyhalogenated aromatics. Adv Mol Toxicaol 3:169–218
Smith AG, Clothier B, Robinson S, Scullion MJ, Carthew P, Edwards R et al (1998) Interaction between iron metabolism and 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin in mice with variants of the Ahr gene: a hepatic oxidative mechanism. Mol Pharmacol 53(1):52–61
Smith AG, Elder GH (2010) Complex gene-chemical interactions: hepatic uroporphyria as a paradigm. Chem Res Toxicol 23(4):712–723
Smith C, Mitchinson MJ, Aruoma OI, Halliwell B (1992) Stimulation of lipid peroxidation and hydroxyl radical generation by the contents of human atherosclerotic lesions. Biochem J 286:901–905
Smith DG, Cappai R, Barnham KJ (2007) The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim Biophys Acta 1768(8):1976–1990
Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF et al (1996) Oxidative damage in Alzheimer’s. Nature 382(6587):120–121
Smith MA, Harris PLR, Sayre LM, Perry G (1997a) Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci 94(18):9866–9868
Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G (1997b) Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J Neurosci 17(8):2653–2657
Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G (2000) Oxidative stress in Alzheimer’s disease. Biochim Biophys Acta 1502(1):139–144
Smith MA, Zhu X, Tabaton M, Liu G, McKeel DW Jr, Cohen ML et al (2010) Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis 19(1):363–372
Sobrin L, Maller JB, Neale BM, Reynolds RC, Fagerness JA, Daly MJ et al (2010) Genetic profile for five common variants associated with age-related macular degeneration in densely affected families: a novel analytic approach. Eur J Hum Genet 18(4):496–501
Sofic E, Paulus W, Jellinger K, Riederer P, Youdim MB (1991) Selective increase of iron in substantia nigra zona compacta of parkinsonian brains. J Neurochem 56(3):978–982
Sohn YS, Breuer W, Munnich A, Cabantchik ZI (2008) Redistribution of accumulated cell iron: a modality of chelation with therapeutic implications. Blood 111(3):1690–1699
Soloniuk DS, Perkins E, Wilson JR (1992) Use of allopurinol and deferoxamine in cellular protection during ischemia. Surg Neurol 38(2):110–113
Song MK, Chung JS, Seol YM, Shin HJ, Choi YJ, Cho GJ (2009) Elevation of serum ferritin is associated with the outcome of patients with newly diagnosed multiple myeloma. Korean J Intern Med 24(4):368–373
Sørensen AR, Thulstrup AM, Hansen J, Ramlau-Hansen CH, Meersohn A, Skytthe A et al (2007) Risk of lung cancer according to mild steel and stainless steel welding. Scand J Work Environ Health 33(5):379–386
Sørensen JG, Holmstrup M, Sarup P, Loeschcke V (2009) Evolutionary theory and studies of model organisms predict a cautiously positive perspective on the therapeutic use of hormesis for healthy aging in humans. Dose Response 8(1):53–57
Sow FB, Alvarez GR, Gross RP, Satoskar AR, Schlesinger LS, Zwilling BS et al (2009) Role of STAT1, NF-kappaB, and C/EBPbeta in the macrophage transcriptional regulation of hepcidin by mycobacterial infection and IFN-gamma. J Leukoc Biol 86(5):1247–1258
Sparaco M, Gaeta LM, Santorelli FM, Passarelli C, Tozzi G, Bertini E et al (2009) Friedreich’s ataxia: oxidative stress and cytoskeletal abnormalities. J Neurol Sci 287(1–2):111–118
Spasojević I, Stević Z, Nikolić-Kokić A, Jones DR, Blagojević D, Spasić MB (2010) Different roles of radical scavengers–ascorbate and urate in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Redox Rep 15(2):81–86
Spencer JP, Abd El Mohsen MM, Minihane AM, Mathers JC (2008) Biomarkers of the intake of dietary polyphenols: strengths, limitations and application in nutrition research. Br J Nutr 99(1):12–22
Sporn MB, Liby K, Yore MM, Suh N, Albini A, Honda T et al (2007) Platforms and networks in triterpenoid pharmacology. Drug Dev Res 68(4):174–182
Squadrito GL, Pryor WA (1998) Oxidative chemistry of nitric oxide: the roles of superoxide, peroxynitrite, and carbon dioxide. Free Radic Biol Med 25(4–5):392–403
Sriram K, Lin GX, Jefferson AM, Roberts JR, Chapman RS, Chen BT et al (2010) Dopaminergic neurotoxicity following pulmonary exposure to manganese-containing welding fumes. Arch Toxicol
Sriskandan S, Altmann DM (2008) The immunology of sepsis. J Pathol 214(2):211–223
Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J et al (2009) Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol 83(8):3719–3733
Stadler N, Lindner RA, Davies MJ (2004) Direct detection and quantification of transition metal ions in human atherosclerotic plaques: evidence for the presence of elevated levels of iron and copper. Arterioscler Thromb Vasc Biol 24(5):949–954
Stadler N, Stanley N, Heeneman S, Vacata V, Daemen MJ, Bannon PG et al (2008) Accumulation of zinc in human atherosclerotic lesions correlates with calcium levels but does not protect against protein oxidation. Arterioscler Thromb Vasc Biol 28(5):1024–1030
Stankiewicz J, Panter SS, Neema M, Arora A, Batt CE, Bakshi R (2007) Iron in chronic brain disorders: imaging and neurotherapeutic implications. Neurotherapeutics 4(3):371–386
Stanley N, Stadler N, Woods AA, Bannon PG, Davies MJ (2006) Concentrations of iron correlate with the extent of protein, but not lipid, oxidation in advanced human atherosclerotic lesions. Free Radic Biol Med 40(9):1636–1643
Starley BQ, Calcagno CJ, Harrison SA (2010) Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 51(5):1820–1832
Stegmann J, Grohmann G (2003) Hypothesis generation guided by co-word clustering. Scientometrics 56(1):111–135
Stein SE, Heller SR, Tchekhovski D (2003) An open standard for chemical structure representation—the IUPAC chemical identifier. In: Proceedings of 2003 Nimes international chemical information conference, pp 131–143
Stelling J, Sauer U, Szallasi Z, Doyle F J 3rd, Doyle J (2004) Robustness of cellular functions. Cell 118(6):675–685
Stevens KB, Del Rio Vilas VJ, Guitian J (2009) Classical sheep scrapie in Great Britain: spatial analysis and identification of environmental and farm-related risk factors. BMC Vet Res 5:33
Stintzi A, Barnes C, Xu J, Raymond KN (2000) Microbial iron transport via a siderophore shuttle: a membrane ion transport paradigm. Proc Natl Acad Sci USA 97(20):10691–10696
Stocker R, Keaney JF (2004) Role of oxidative modifications in atherosclerosis. Physiol Rev 84(4):1381–1478
Stocker R, Keaney JF (2005) New insights on oxidative stress in the artery wall. J Thromb Haemostasis 3(8):1825–1834
Stohs SJ, Bagchi D (1995) Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med 18(2):321–336
Stolovitzky G, Califano A (2006) Reverse engineering biological networks: opportunities and challenges in computational methods for pathway inference, vol 1158. New York Academy of Sciences, New York
Stolovitzky G, Kahlem P, Califano A (2009) The challenges of systems biology: community efforts to harness biological complexity, vol 1158. New York Academy of Sciences, New York
Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R et al (2007) Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 318(5848):287–290
Storey JA, Connor RF, Lewis ZT, Hurd D, Pomper G, Keung YK et al (2009) The transplant iron score as a predictor of stem cell transplant survival. J Hematol Oncol 2:44
Størmer FC, Hoiby EA (1996) Citrinin, ochratoxin A and iron. Possible implications for their biological function and induction of nephropathy. Mycopathologia 134(2):103–107
Streck EL, Comim CM, Barichello T, Quevedo J (2008) The septic brain. Neurochem Res 33(11):2171–2177
Sturm B, Bistrich U, Schranzhofer M, Sarsero JP, Rauen U, Scheiber-Mojdehkar B et al (2005) Friedreich’s ataxia, no changes in mitochondrial labile iron in human lymphoblasts and fibroblasts: a decrease in antioxidative capacity? J Biol Chem 280(8):6701–6708
Subramanian V, Ferrante AW Jr (2009) Obesity, inflammation, and macrophages. Nestle Nutr Workshop Ser Pediatr Program 63:151–159 (discussion 159–162, 259–168)
Suchy J, Chan A, Shea TB (2009) Dietary supplementation with a combination of alpha-lipoic acid, acetyl-L-carnitine, glycerophosphocoline, docosahexaenoic acid, and phosphatidylserine reduces oxidative damage to murine brain and improves cognitive performance. Nutr Res 29(1):70–74
Sugihara N, Arakawa T, Ohnishi M, Furuno K (1999) Anti- and pro-oxidative effects of flavonoids on metal-induced lipid hydroperoxide-dependent lipid peroxidation in cultured hepatocytes loaded with alpha-linolenic acid. Free Radic Biol Med 27(11–12):1313–1323
Sugimoto R, Fujita N, Tomosugi N, Hara N, Miyachi H, Tanaka H et al (2009) Impaired regulation of serum hepcidin during phlebotomy in patients with chronic hepatitis C. Hepatol Res 39(6):619–624
Suh KS, Chon S, Oh S, Kim SW, Kim JW, Kim YS et al (2009) Prooxidative effects of green tea polyphenol (−)-epigallocatethin-3-gallate on the HIT-T15 pancreatic beta cell line. Cell Biol Toxicol
Sullivan JL (1981) Iron and the sex difference in heart-disease risk. Lancet 1(8233):1293–1294
Sullivan JL (2001) Misconceptions in the debate on the iron hypothesis. J Nutr Biochem 12(1):33–37
Sullivan JL (2003) Are menstruating women protected from heart disease because of, or in spite of, estrogen? Relevance to the iron hypothesis. Amer Heart J 145(2):190–194
Sullivan JL (2004) Is stored iron safe? J Lab Clin Med 144(6):280–284
Sullivan JL (2007) Macrophage iron, hepcidin, and atherosclerotic plaque stability. Exp Biol Med (Maywood) 232(8):1014–1020
Sullivan JL (2009) Iron in arterial plaque: a modifiable risk factor for atherosclerosis. Biochim Biophys Acta 1790(7):718–723
Sumida Y, Yoshikawa T, Okanoue T (2009) Role of hepatic iron in non-alcoholic steatohepatitis. Hepatol Res 39(3):213–222
Sun L, Franco OH, Hu FB, Cai L, Yu Z, Li H et al (2008) Ferritin concentrations, metabolic syndrome, and type 2 diabetes in middle-aged and elderly chinese. J Clin Endocrinol Metab 93(12):4690–4696
Sun X, Ge R, Cai Z, Sun H, He QY (2009) Iron depletion decreases proliferation and induces apoptosis in a human colonic adenocarcinoma cell line, CaCO2. J Inorg Biochem 103(7):1074–1081
Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD et al (2006) Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 355(10):1018–1028
Surh YJ, Kundu JK, Na HK, Lee JS (2005) Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J Nutr 135(12 Suppl):2993S–3001S
Surh YJ, Na HK (2008) NF-kappaB and Nrf2 as prime molecular targets for chemoprevention and cytoprotection with anti-inflammatory and antioxidant phytochemicals. Genes Nutr 2(4):313–317
Sutedja NA, Sinke RJ, Van Vught PW, Van der Linden MW, Wokke JH, Van Duijn CM et al (2007) The association between H63D mutations in HFE and amyotrophic lateral sclerosis in a Dutch population. Arch Neurol 64(1):63–67
Sweeny GD, Jones KG, Cole FM, Basford D, Krestynski F (1979) Iron deficiency prevents liver toxicity of 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. Science 204(4390):332–335
Switzer JA, Hess DC, Nichols FT, Adams RJ (2006) Pathophysiology and treatment of stroke in sickle-cell disease: present and future. Lancet Neurol 5(6):501–512
Szabo C (1996) The pathophysiological role of peroxynitrite in shock, inflammation, and ischemia-reperfusion injury. Shock 6(2):79–88
Szabó C, Ischiropoulos H, Radi R (2007) Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6(8):662–680
Szajerka T, Jablecki J (2007) Kaposi’s sarcoma revisited. AIDS Rev 9(4):230–236
Tabner BJ, El-Agnaf OM, German MJ, Fullwood NJ, Allsop D (2005) Protein aggregation, metals and oxidative stress in neurodegenerative diseases. Biochem Soc Trans 33(Pt 5):1082–1086
Tacchini L, Gammella E, De Ponti C, Recalcati S, Cairo G (2008) Role of HIF-1 and NF-kappaB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J Biol Chem 283(30):20674–20686
Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S et al (2010) Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood 115(10):1886–1892
Tamgüney G, Giles K, Glidden DV, Lessard P, Wille H, Tremblay P et al (2008) Genes contributing to prion pathogenesis. J Gen Virol 89(Pt 7):1777–1788
Tan JS, Wang JJ, Flood V, Rochtchina E, Smith W, Mitchell P (2008) Dietary antioxidants and the long-term incidence of age-related macular degeneration: the Blue Mountains eye study. Ophthalmology 115(2):334–341
Tan KT, Lip GY (2008) Imaging of the unstable plaque. Int J Cardiol 127(2):157–165
Tanaka N, Horiuchi A, Yamaura T, Komatsu M, Tanaka E, Kiyosawa K (2007) Efficacy and safety of 6-month iron reduction therapy in patients with hepatitis C virus-related cirrhosis: a pilot study. J Gastroenterol 42(1):49–55
Tanaka N, Horiuchi A, Yamaura T, Komatsu M, Yokoyama T, Okaniwa S et al (2009) Efficacy and safety of addition of minor bloodletting (petit phlebotomy) in hepatitis C virus-infected patients receiving regular glycyrrhizin injections. J Gastroenterol 44(6):577–582
Tanaka N, Kiyosawa K (2004) Phlebotomy: a promising treatment for chronic hepatitis C. J Gastroenterol 39(6):601–603
Tang TY, Muller KH, Graves MJ, Li ZY, Walsh SR, Young V et al (2009) Iron oxide particles for atheroma imaging. Arterioscler Thromb Vasc Biol 29(7):1001–1008
Taqueti VR, Mitchell RN, Lichtman AH (2006) Protecting the pump: controlling myocardial inflammatory responses. Annu Rev Physiol 68:67–95
Taupin P (2007) Protocols for studying adult neurogenesis: insights and recent developments. Regen Med 2(1):51–62
Tedgui A, Mallat Z (2006) Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev 86(2):515–581
Teixeira KC, Soares FS, Rocha LG, Silveira PC, Silva LA, Valenca SS et al (2008) Attenuation of bleomycin-induced lung injury and oxidative stress by N-acetylcysteine plus deferoxamine. Pulm Pharmacol Ther 21(2):309–316
Tenopoulou M, Doulias PT, Barbouti A, Brunk U, Galaris D (2005) Role of compartmentalized redox-active iron in hydrogen peroxide-induced DNA damage and apoptosis. Biochem J 387(Pt 3):703–710
Tenopoulou M, Kurz T, Doulias PT, Galaris D, Brunk UT (2007) Does the calcein-AM method assay the total cellular ‘labile iron pool’ or only a fraction of it? Biochem J 403(2):261–266
Terao J (2009) Dietary flavonoids as antioxidants. Forum Nutr 61:87–94
Terman A, Brunk UT (2004) Lipofuscin. Int J Biochem Cell Biol 36(8):1400–1404
Terman A, Brunk UT (2006) Oxidative stress, accumulation of biological ‘garbage’, and aging. Antioxid Redox Signal 8(1–2):197–204
Terman A, Gustafsson B, Brunk UT (2006a) The lysosomal-mitochondrial axis theory of postmitotic aging and cell death. Chem Biol Interact 163(1–2):29–37
Terman A, Kurz T, Gustafsson B, Brunk UT (2006b) Lysosomal labilization. IUBMB Life 58(9):531–539
Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT (2010) Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal 12(4):503–535
Thannickal VJ, Fanburg BL (2000) Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 279(6):L1005–1028
Theil EC (2007) Coordinating responses to iron and oxygen stress with DNA and mRNA promoters: the ferritin story. Biometals 20(3–4):513–521
Thephinlap C, Phisalaphong C, Fucharoen S, Porter JB, Srichairatanakool S (2009) Efficacy of curcuminoids in alleviation of iron overload and lipid peroxidation in thalassemic mice. Med Chem 5:475–482
Thiele I, Palsson BØ (2010) A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc 5(1):93–121
Thomas M, Jankovic J (2004) Neurodegenerative disease and iron storage in the brain. Curr Opin Neurol 17(4):437–442
Thomas C, Mackey MM, Diaz AA, Cox DP (2009a) Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: implications for diseases associated with iron accumulation. Redox Rep 14(3):102–108
Thomas P, Wang YJ, Zhong JH, Kosaraju S, O’Callaghan NJ, Zhou XF et al (2009b) Grape seed polyphenols and curcumin reduce genomic instability events in a transgenic mouse model for Alzheimer’s disease. Mutat Res 661(1–2):25–34
Thomas SR, Schulz E, Keaney JF (2006) Hydrogen peroxide restrains endothelium-derived nitric oxide bioactivity—role for iron-dependent oxidative stress. Free Radic Biol Med 41(4):681–688
Thompson KJ, Shoham S, Connor JR (2001) Iron and neurodegenerative disorders. Brain Res Bull 55(2):155–164
Thougaard AV, Langer SW, Hainau B, Grauslund M, Juhl BR, Jensen PB et al (2010) A murine experimental anthracycline extravasation model: pathology and study of the involvement of topoisomerase II alpha and iron in the mechanism of tissue damage. Toxicology 269(1):67–72
Tian H, Maddox IS, Ferguson LR, Shu Q (2010) Evaluation of the cytoprotective effects of bovine lactoferrin against intestinal toxins using cellular model systems. Biometals 23:589–592
Tielsch JM, Javitt JC, Coleman A, Katz J, Sommer A (1995) The prevalence of blindness and visual impairment among nursing home residents in Baltimore. N Engl J Med 332(18):1205–1209
Tjoa CW, Benedict RH, Weinstock-Guttman B, Fabiano AJ, Bakshi R (2005) MRI T2 hypointensity of the dentate nucleus is related to ambulatory impairment in multiple sclerosis. J Neurol Sci 234(1–2):17–24
Tomlin CJ, Axelrod JD (2007) Biology by numbers: mathematical modelling in developmental biology. Nat Rev Genet 8(5):331–340
Torreilles F, Salman-Tabcheh S, Guérin M, Torreilles J (1999) Neurodegenerative disorders: the role of peroxynitrite. Brain Res Brain Res Rev 30(2):153–163
Toyokuni S (2002) Iron and carcinogenesis: from Fenton reaction to target genes. Redox Rep 7(4):189–197
Toyokuni S (2009a) Mechanisms of asbestos-induced carcinogenesis. Nagoya J Med Sci 71(1–2):1–10
Toyokuni S (2009b) Role of iron in carcinogenesis: cancer as a ferrotoxic disease. Cancer Sci 100(1):9–16
Toyokuni S, Sagripanti JL (1996) Association between 8-hydroxy-2’-deoxyguanosine formation and DNA strand breaks mediated by copper and iron. Free Radic Biol Med 20(6):859–864
Trachootham D, Alexandre J, Huang P (2009) Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov 8(7):579–591
Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P (2008) Redox regulation of cell survival. Antioxid Redox Signal 10(8):1343–1374
Traoré, H. N., & Meyer, D. (2004). The effect of iron overload on in vitro HIV-1 infection. J Clin Virol 31(Suppl 1):S92-S98.
Traoré HN, Meyer D (2007) Necrosis of host cells and survival of pathogens following iron overload in an in vitro model of co-infection with human immunodeficiency virus (HIV) and Mycobacterium tuberculosis. Int J Antimicrob Agents 29(4):465–470
Trinder D, Ayonrinde OT, Olynyk JK (2008) HCV, iron, and oxidative stress: the new choreography of hepcidin. Gastroenterology 134(1):348–351
Truksa J, Kovář J, Valenta T, Ehrlichová M, Polák J, Naumann PW (2003) Iron deprivation induces apoptosis independently of p53 in human and murine tumour cells. Cell Prolif 36(4):199–213
Tsimikas S, Willeit J, Knoflach M, Mayr M, Egger G, Notdurfter M et al (2009) Lipoprotein-associated phospholipase A2 activity, ferritin levels, metabolic syndrome, and 10-year cardiovascular and non-cardiovascular mortality: results from the Bruneck study. Eur Heart J 30(1):107–115
Tung BY, Emond MJ, Bronner MP, Raaka SD, Cotler SJ, Kowdley KV (2003) Hepatitis C, iron status, and disease severity: relationship with HFE mutations. Gastroenterology 124(2):318–326
Tuntipopipat S, Zeder C, Siriprapa P, Charoenkiatkul S (2009) Inhibitory effects of spices and herbs on iron availability. Int J Food Sci Nutr 60(Suppl 1):43–55
Tuomainen TP, Loft S, Nyyssonen K, Punnonen K, Salonen JT, Poulsen HE (2007) Body iron is a contributor to oxidative damage of DNA. Free Radic Res 41(3):324–328
Tuomainen TP, Punnonen K, Nyyssonen K, Salonen JT (1998) Association between body iron stores and the risk of acute myocardial infarction in men. Circulation 97(15):1461–1466
Turner NJ (2009) Directed evolution drives the next generation of biocatalysts. Nat Chem Biol 5(8):567–573
Ulloa L, Brunner M, Ramos L, Deitch EA (2009) Scientific and clinical challenges in sepsis. Curr Pharm Des 15(16):1918–1935
Ulrich-Merzenich G, Zeitler H, Vetter H, Kraft K (2009) Synergy research: vitamins and secondary plant components in the maintenance of the redox-homeostasis and in cell signaling. Phytomedicine 16(1):2–16
Upadhyay D, Kamp DW (2003) Asbestos-induced pulmonary toxicity: role of DNA damage and apoptosis. Exp Biol Med (Maywood) 228(6):650–659
Usenko CY, Harper SL, Tanguay RL (2008) Fullerene C60 exposure elicits an oxidative stress response in embryonic zebrafish. Toxicol Appl Pharmacol 229(1):44–55
Usuki F, Yasutake A, Umehara F, Tokunaga H, Matsumoto M, Eto K et al (2001) In vivo protection of a water-soluble derivative of vitamin E, Trolox, against methylmercury-intoxication in the rat. Neurosci Lett 304(3):199–203
Utzschneider KM, Kowdley KV (2010) Hereditary hemochromatosis and diabetes mellitus: implications for clinical practice. Nat Rev Endocrinol 6(1):26–33
Valavanidis A, Vlachogianni T, Fiotakis C (2009) 8-hydroxy-2′-deoxyguanosine (8-OHdG): a critical biomarker of oxidative stress and carcinogenesis. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 27(2):120–139
Valavanidis A, Vlahoyianni T, Fiotakis K (2005) Comparative study of the formation of oxidative damage marker 8-hydroxy-2′-deoxyguanosine (8-OHdG) adduct from the nucleoside 2′-deoxyguanosine by transition metals and suspensions of particulate matter in relation to metal content and redox reactivity. Free Radic Res 39(10):1071–1081
Valdmanis PN, Daoud H, Dion PA, Rouleau GA (2009) Recent advances in the genetics of amyotrophic lateral sclerosis. Curr Neurol Neurosci Rep 9(3):198–205
Valenti L, Dongiovanni P, Fargion S, Fracanzani AL (2008) Iron genes, dysmetabolism and fibrosis in chronic hepatitis C. J Hepatol 48(3):513–514 (author reply 514–515)
Valenti L, Swinkels DW, Burdick L, Dongiovanni P, Tjalsma H, Motta BM et al (2010) Serum ferritin levels are associated with vascular damage in patients with nonalcoholic fatty liver disease. Nutr Metab Cardiovasc Dis (in press)
Valenti L, Valenti G, Como G, Burdick L, Santorelli G, Dongiovanni P et al (2007) HFE gene mutations and oxidative stress influence serum ferritin, associated with vascular damage, in hemodialysis patients. Am J Nephrol 27(1):101–107
Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39(1):44–84
Valko M, Morris H, Cronin MTD (2005) Metals, toxicity and oxidative stress. Curr Med Chem 12(10):1161–1208
Valvassori SS, Petronilho FC, Reus GZ, Steckert AV, Oliveira VB, Boeck CR et al (2008) Effect of N-acetylcysteine and/or deferoxamine on oxidative stress and hyperactivity in an animal model of mania. Prog Neuropsychopharmacol Biol Psychiatry 32(4):1064–1068
van Asbeck BS, Georgiou NA, van der Bruggen T, Oudshoorn M, Nottet HS, Marx JJ (2001) Anti-HIV effect of iron chelators: different mechanisms involved. J Clin Virol 20(3):141–147
van Laar VS, Berman SB (2009) Mitochondrial dynamics in Parkinson’s disease. Exp Neurol 218(2):247–256
van Leuven SI, Franssen R, Kastelein JJ, Levi M, Stroes ES, Tak PP (2008) Systemic inflammation as a risk factor for atherothrombosis. Rheumatology (Oxford) 47(1):3–7
van Oostrom AJ, van Wijk J, Cabezas MC (2004) Lipaemia, inflammation and atherosclerosis: novel opportunities in the understanding and treatment of atherosclerosis. Drugs 64(Suppl 2):19–41
Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323(5918):1208–1211
Vanden Berghe T, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N et al (2009) Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ 17:922–930
Vanlangenakker N, Vanden Berghe T, Krysko DV, Festjens N, Vandenabeele P (2008) Molecular mechanisms and pathophysiology of necrotic cell death. Curr Mol Med 8(3):207–220
Vaupel JW (2010) Biodemography of human ageing. Nature 464(7288):536–542
Vaz SM, Augusto O (2008) Inhibition of myeloperoxidase-mediated protein nitration by tempol: kinetics, mechanism, and implications. Proc Natl Acad Sci USA 105(24):8191–8196
Vázquez-Manrique RP, González-Cabo P, Ros S, Aziz H, Baylis HA, Palau F (2006) Reduction of Caenorhabditis elegans frataxin increases sensitivity to oxidative stress, reduces lifespan, and causes lethality in a mitochondrial complex II mutant. Faseb J 20(1):172–174
Vázquez Roncero A, Janer del Valle C, Maestro Durán R, Graciani Constante E (1983) New aniline derivatives in cooking oils associated with toxic oil syndrome. Lancet 2(8357):1024–1025
Vélez-Pardo C, Del Río MJ, Verschueren H, Ebinger G, Vauquelin G (1997) Dopamine and iron induce apoptosis in PC12 cells. Pharmacol Toxicol 80(2):76–84
Venturini D, Simao AN, Barbosa DS, Lavado EL, Narciso VE, Dichi I et al (2010) Increased oxidative stress, decreased total antioxidant capacity, and iron overload in untreated patients with chronic hepatitis C. Dig Dis Sci 55:1120–1127
Verduzco LA, Nathan DG (2009) Sickle cell disease and stroke. Blood 114(25):5117–5125
Vichinsky E, Butensky E, Fung E, Hudes M, Theil E, Ferrell L et al (2005) Comparison of organ dysfunction in transfused patients with SCD or beta thalassemia. Am J Hematol 80(1):70–74
Victor VM, Rocha M, De la Fuente M (2004) Immune cells: free radicals and antioxidants in sepsis. Int Immunopharmacol 4(3):327–347
Victor VM, Rocha M, Esplugues JV, De la Fuente M (2005) Role of free radicals in sepsis: antioxidant therapy. Curr Pharm Des 11(24):3141–3158
Villéger AC, Pettifer SR, Kell DB (2010) Arcadia: a visualization tool for metabolic pathways. Bioinformatics 20(11):1470–1471
Visseren FLJ, Verkerk MSA, van der Bruggen T, Marx JJM, van Asbeck BS, Diepersloot RJA (2002) Iron chelation and hydroxyl radical scavenging reduce the inflammatory response of endothelial cells after infection with Chlamydia pneumoniae or influenza A. Eur J Clin Invest 32(Suppl 1):84–90
Vlachodimitropoulou E, Naftalin RJ, Sharp PA (2010) Quercetin is a substrate for the transmembrane oxidoreductase, Dcytb. Free Radic Biol Med 48:1366–1369
Vlašić Ž, Dodig S, Čepelak I, Zrinski Topić RZ, Živčić J, Nogalo B et al (2009) Iron and ferritin concentrations in exhaled breath condensate of children with asthma. J Asthma 46(1):81–85
Vlessis AA, Goldman RK, Trunkey DD (1995) New concepts in the pathophysiology of oxygen metabolism during sepsis. Br J Surg 82(7):870–876
Voloboueva LA, Killilea DW, Atamna H, Ames BN (2007) N-tert-butyl hydroxylamine, a mitochondrial antioxidant, protects human retinal pigment epithelial cells from iron overload: relevance to macular degeneration. FASEB J 21(14):4077–4086
Vucenik I, Shamsuddin AM (2006) Protection against cancer by dietary IP6 and inositol. Nutr Cancer 55(2):109–125
Vucic S, Kiernan MC (2009) Pathophysiology of neurodegeneration in familial amyotrophic lateral sclerosis. Curr Mol Med 9(3):255–272
Vyshemirsky V, Girolami MA (2008) Bayesian ranking of biochemical system models. Bioinformatics 24(6):833–839
Wagner A (2005) Robustness, evolvability, and neutrality. FEBS Lett 579(8):1772–1778
Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF (2003) Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab 23(6):629–652
Wahba ZZ, Murray WJ, Stohs SJ (1990) Desferrioxamine-induced alterations in hepatic iron distribution, DNA damage and lipid peroxidation in control and 2,3,7,8-tetrachlorodibenzo-p-dioxin-treated rats. J Appl Toxicol 10(2):119–124
Waldron KJ, Rutherford JC, Ford D, Robinson NJ (2009) Metalloproteins and metal sensing. Nature 460(7257):823–830
Walker NM, Stuart KA, Ryan RJ, Desai S, Saab S, Nicol JA et al (2010) Serum ferritin concentration predicts mortality in patients awaiting liver transplantation. Hepatology 51(5):1683–1691
Walter U, Wagner S, Horowski S, Benecke R, Zettl UK (2009) Transcranial brain sonography findings predict disease progression in multiple sclerosis. Neurology 73(13):1010–1017
Walters AS, Rye DB (2009) Review of the relationship of restless legs syndrome and periodic limb movements in sleep to hypertension, heart disease, and stroke. Sleep 32(5):589–597
Walters RH, Murphy RM (2009) Examining polyglutamine peptide length: a connection between collapsed conformations and increased aggregation. J Mol Biol 393(4):978–992
Wandersman C, Delepelaire P (2004) Bacterial iron sources: from siderophores to hemophores. Annu Rev Microbiol 58:611–647
Wang H, Ma S (2008) The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am J Emerg Med 26(6):711–715
Wang XS, Lee S, Simmons Z, Boyer P, Scott K, Liu W et al (2004) Increased incidence of the Hfe mutation in amyotrophic lateral sclerosis and related cellular consequences. J Neurol Sci 227(1):27–33
Wang J, Gines S, MacDonald ME, Gusella JF (2005) Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci 6:1
Wang J, Wang G, Ansari GA, Khan MF (2008) Activation of oxidative stress-responsive signaling pathways in early splenotoxic response of aniline. Toxicol Appl Pharmacol 230(2):227–234
Wang T, Craig EA (2008) Binding of yeast frataxin to the scaffold for Fe-S cluster biogenesis, Isu. J Biol Chem 283(18):12674–12679
Wang X, Zhao X (2009) Contribution of oxidative damage to antimicrobial lethality. Antimicrob Agents Chemother 53(4):1395–1402
Wang HQ, Xu YX, Zhao XY, Zhao H, Yan J, Sun XB et al (2009a) Overexpression of FoF1-ATP synthase alpha suppresses mutant huntingtin aggregation and toxicity in vitro. Biochem Biophys Res Commun 390(4):1294–1298
Wang J, Du XX, Jiang H, Xie JX (2009b) Curcumin attenuates 6-hydroxydopamine-induced cytotoxicity by anti-oxidation and nuclear factor-kappaB modulation in MES23.5 cells. Biochem Pharmacol 78(2):178–183
Wang ZY, Zhao KK, Song ZM, Shen LJ, Qu J (2009c) Erythropoietin as a novel therapeutic agent for atrophic age-related macular degeneration. Med Hypotheses 72(4):448–450
Wang YC, Chen BS (2010) Integrated cellular network of transcription regulations and protein-protein interactions. BMC Syst Biol 4(1):20
Wang J, Ma H, Boor PJ, Ramanujam VM, Ansari GA, Khan MF (2010a) Up-regulation of heme oxygenase-1 in rat spleen after aniline exposure. Free Radic Biol Med 48(4):513–518
Wang W, Knovich MA, Coffman LG, Torti FM, Torti SV (2010b) Serum ferritin: past, present and future. Biochim Biophys Acta
Wang X, Moualla D, Wright JA, Brown DR (2010c) Copper binding regulates intracellular alpha-synuclein localisation, aggregation and toxicity. J Neurochem 113(3):704–714
Wardman P, Candeias LP (1996) Fenton chemistry: an introduction. Rad Res 145(5):523–531
Warkentin LM, Auriat AM, Wowk S, Colbourne F (2010) Failure of deferoxamine, an iron chelator, to improve outcome after collagenase-induced intracerebral hemorrhage in rats. Brain Res 1309:95–103
Watanabe J, Nakamachi T, Ogawa T, Naganuma A, Nakamura M, Shioda S et al (2009) Characterization of antioxidant protection of cultured neural progenitor cells (NPC) against methylmercury (MeHg) toxicity. J Toxicol Sci 34(3):315–325
Watt F, Rajendran R, Ren MQ, Tan BKH, Halliwell B (2006) A nuclear microscopy study of trace elements Ca, Fe, Zn and Cu in atherosclerosis. Nucl Instr Meth Phys Res B 249:646–652
Watterson S, Marshall S, Ghazal P (2008) Logic models of pathway biology. Drug Discov Today 13(9–10):447–456
Watts JC, Westaway D (2007) The prion protein family: diversity, rivalry, and dysfunction. Biochim Biophys Acta 1772(6):654–672
Wayne Martin WR (2009) Quantitative estimation of regional brain iron with magnetic resonance imaging. Parkinsonism Rel Disord 15S3:S215–S218
Wedge D, Kell DB (2008) Rapid prediction of optimum population size in genetic programming using a novel genotype-fitness correlation. In: Keizer M et al (eds) GECCO 2008, pp 1315–1322
Wedge D, Rowe W, Kell DB, Knowles J (2009) In silico modelling of directed evolution: implications for experimental design and stepwise evolution. J Theor Biol 257:131–141
Weinberg ED (1989) Iron, asbestos, and carcinogenicity. Lancet 1(8651):1399–1400
Weinberg ED (2004) Exposing the hidden dangers of iron: what every medical professional should know about the impact of iron on the disease process. Cumberland House
Weinberg F, Chandel NS (2009) Reactive oxygen species-dependent signaling regulates cancer. Cell Mol Life Sci 66(23):3663–3673
Weinberg GA (2006) Iron status and mortality in HIV infection. Haematologica 91(6):721
Weininger D (1988) SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci 28(1):31–36
Weinreb O, Amit T, Mandel S, Youdim MB (2009) Neuroprotective molecular mechanisms of (−)-epigallocatechin-3-gallate: a reflective outcome of its antioxidant, iron chelating and neuritogenic properties. Genes Nutr
Weinreb O, Amit T, Youdim MB (2007) A novel approach of proteomics and transcriptomics to study the mechanism of action of the antioxidant-iron chelator green tea polyphenol (−)-epigallocatechin-3-gallate. Free Radic Biol Med 43(4):546–556
Weinreb O, Mandel S, Bar-Am O, Yogev-Falach M, Avramovich-Tirosh Y, Amit T et al (2009b) Multifunctional neuroprotective derivatives of rasagiline as anti-Alzheimer’s disease drugs. Neurotherapeutics 6(1):163–174
Weisberger J, Emmons F, Gorczyca W (2004) Cytochemical diagnosis of Gaucher’s disease by iron stain. Br J Haematol 124(6):696
Weiss G (2010) Genetic mechanisms and modifying factors in hereditary hemochromatosis. Nat Rev Gastroenterol Hepatol 7(1):50–58
Weitzman SA, Graceffa P (1984) Asbestos catalyzes hydroxyl and superoxide radical generation from hydrogen peroxide. Arch Biochem Biophys 228(1):373–376
Weng LC, Yeh WT, Bai CH, Chen HJ, Chuang SY, Chang HY et al (2008) Is ischemic stroke risk related to folate status or other nutrients correlated with folate intake? Stroke 39(12):3152–3158
Wermuth CG (2004) Selective optimization of side activities: another way for drug discovery. J Med Chem 47(6):1303–1314
West AK, Hidalgo J, Eddins D, Levin ED, Aschner M (2008) Metallothionein in the central nervous system: roles in protection, regeneration and cognition. Neurotoxicology 29(3):489–503
Westaway D, DeArmond SJ, Cayetano-Canlas J, Groth D, Foster D, Yang SL et al (1994) Degeneration of skeletal muscle, peripheral nerves, and the central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell 76(1):117–129
Westergard L, Christensen HM, Harris DA (2007) The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta 1772(6):629–644
Westerhoff HV, Kell DB (2007) The methodologies of systems biology. In: Boogerd FC, Bruggeman FJ, Hofmeyr J-HS, Westerhoff HV (eds) Systems biology: philosophical foundations. Elsevier, Amsterdam, pp 23–70
White BC, Nayini NR, Krause GS, Aust SD, March GG, Bicknell JS et al (1988) Effect on biochemical markers of brain injury of therapy with deferoxamine or superoxide dismutase following cardiac arrest. Am J Emerg Med 6(6):569–576
White CR, Brock TA, Chang LY, Crapo J, Briscoe P, Ku D et al (1994) Superoxide and peroxynitrite in atherosclerosis. Proc Natl Acad Sci USA 91(3):1044–1048
Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M et al (2007) Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 446(7137):815–819
Wijesekera LC, Leigh PN (2009) Amyotrophic lateral sclerosis. Orphanet J Rare Dis 4:3
Wilkinson DJ (2007) Bayesian methods in bioinformatics and computational systems biology. Brief Bioinform 8(2):109–116
Wilkinson SJ, Benson N, Kell DB (2008) Proximate parameter tuning for biochemical networks with uncertain kinetic parameters. Mol Biosyst 4:74–97. doi:10.1039/b707506e
Willerson JT, Ridker PM (2004) Inflammation as a cardiovascular risk factor. Circulation 109(21 Suppl 1):II2–II10
Williams A, Lucassen PJ, Ritchie D, Bruce M (1997) PrP deposition, microglial activation, and neuronal apoptosis in murine scrapie. Exp Neurol 144(2):433–438
Wilson JG, Lindquist JH, Grambow SC, Crook ED, Maher JF (2003) Potential role of increased iron stores in diabetes. Am J Med Sci 325(6):332–339
Wilson RB (2006) Iron dysregulation in Friedreich ataxia. Semin Pediatr Neurol 13(3):166–175
Wimazal F, Nosslinger T, Baumgartner C, Sperr WR, Pfeilstocker M, Valent P (2009) Deferasirox induces regression of iron overload in patients with myelodysplastic syndromes. Eur J Clin Invest 39(5):406–411
Winkelmann G (2002) Microbial siderophore-mediated transport. Biochem Soc Trans 30(4):691–696
Winterbourn CC (1995) Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett 82–83:969–974
Wisniewski T, Sigurdsson EM (2007) Therapeutic approaches for prion and Alzheimer’s diseases. FEBS J 274(15):3784–3798
Wolff B, Volzke H, Ludemann J, Robinson D, Vogelgesang D, Staudt A et al (2004) Association between high serum ferritin levels and carotid atherosclerosis in the study of health in Pomerania (SHIP). Stroke 35(2):453–457
Wolozin B, Golts N (2002) Iron and Parkinson’s disease. Neuroscientist 8(1):22–32
Won JE, Jeong SH, Chung JI, Lee JH, Hwang SH, Kim JW et al (2009) Hepatic iron, serum ferritin, HFE mutation, and hepatic fibrosis in chronic hepatitis C. Intervirology 52(5):239–246
Wondrak GT (2009) Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid Redox Signal 11(12):3013–3069
Wong BS, Brown DR, Pan T, Whiteman M, Liu T, Bu X et al (2001a) Oxidative impairment in scrapie-infected mice is associated with brain metals perturbations and altered antioxidant activities. J Neurochem 79(3):689–698
Wong BS, Brown DR, Sy MS (2001b) A Yin-Yang role for metals in prion disease. Panminerva Medica 43(4):283–287
Wong PK, Yu F, Shahangian A, Cheng G, Sun R, Ho CM (2008) Closed-loop control of cellular functions using combinatory drugs guided by a stochastic search algorithm. Proc Natl Acad Sci USA 105(13):5105–5110
Wong RW, Richa DC, Hahn P, Green WR, Dunaief JL (2007) Iron toxicity as a potential factor in AMD. Retina 27(8):997–1003
Woo PC, Tung ET, Chan KH, Lau CC, Lau SK, Yuen KY (2010) Cytokine profiles induced by the novel swine-origin influenza A/H1N1 virus: implications for treatment strategies. J Infect Dis 201(3):346–353
Wu J, Hua Y, Keep RF, Nakamura T, Hoff JT, Xi G (2003) Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke 34(12):2964–2969
Wu X, Kannan S, Ramanujam VM, Khan MF (2005) Iron release and oxidative DNA damage in splenic toxicity of aniline. J Toxicol Environ Health A 68(8):657–666
Wu X, Patel D, Hasinoff BB (2004) The iron chelating cardioprotective prodrug dexrazoxane does not affect the cell growth inhibitory effects of bleomycin. J Inorg Biochem 98(11):1818–1823
Wu XD, Kumar V, Quinlan JR, Ghosh J, Yang Q, Motoda H et al (2008) Top 10 algorithms in data mining. Knowl Inf Syst 14(1):1–37
Wypijewska A, Galazka-Friedman J, Bauminger ER, Wszolek ZK, Schweitzer KJ, Dickson DW et al (2010) Iron and reactive oxygen species activity in parkinsonian substantia nigra. Parkinsonism Relat Disord
Xiong SG, She HY, Takeuchi H, Han B, Engelhardt JF, Barton CH et al (2003) Signaling role of intracellular iron in NF-kappa B activation. J Biol Chem 278(20):17646–17654
Xu A, Wu LJ, Santella RM, Hei TK (1999) Role of oxyradicals in mutagenicity and DNA damage induced by crocidolite asbestos in mammalian cells. Cancer Res 59(23):5922–5926
Xu Q, Kanthasamy AG, Reddy MB (2008a) Neuroprotective effect of the natural iron chelator, phytic acid in a cell culture model of Parkinson’s disease. Toxicology 245(1–2):101–108
Xu X, Sutak R, Richardson DR (2008b) Iron chelation by clinically relevant anthracyclines: alteration in expression of iron-regulated genes and atypical changes in intracellular iron distribution and trafficking. Mol Pharmacol 73(3):833–844
Xu WL, von Strauss E, Qiu CX, Winblad B, Fratiglioni L (2009) Uncontrolled diabetes increases the risk of Alzheimer’s disease: a population-based cohort study. Diabetologia 52(6):1031–1039
Xu J, Marzetti E, Seo AY, Kim JS, Prolla TA, Leeuwenburgh C (2010) The emerging role of iron dyshomeostasis in the mitochondrial decay of aging. Mech Ageing Dev
Yadav AS, Bhatnagar D (2007a) Free radical scavenging activity, metal chelation and antioxidant power of some of the Indian spices. Biofactors 31(3–4):219–227
Yadav AS, Bhatnagar D (2007b) Modulatory effect of spice extracts on iron-induced lipid peroxidation in rat liver. Biofactors 29(2–3):147–157
Yadav AS, Bhatnagar D (2010) Inhibition of iron induced lipid peroxidation and antioxidant activity of Indian spices and Acacia in vitro. Plant Foods Hum Nutr 65:18–24
Yamamoto K, Kawanishi S (1992) Site-specific DNA damage by phenylhydrazine and phenelzine in the presence of Cu(II) ion or Fe(III) complexes: roles of active oxygen species and carbon radicals. Chem Res Toxicol 5(3):440–446
Yamashita T, Ando Y, Nakamura M, Obayashi K, Terazaki H, Haraoka K et al (2004) Inhibitory effect of α-tocopherol on methylmercury-induced oxidative stress. Environ Health Prev Med 9(3):111–117
Yang F, Stonehuerner JG, Richards JH, Nguyen NB, Callaghan KD, Haile DJ et al (2010) Deficiency in the divalent metal transporter 1 increases bleomycin-induced lung injury. Biometals
Yasuda T, Mochizuki H (2010) The regulatory role of alpha-synuclein and parkin in neuronal cell apoptosis; possible implications for the pathogenesis of Parkinson’s disease. Apoptosis
Yeager MP, Coleman RA (2010) In silico evidence for glutathione- and iron-related pathogeneses in Parkinson’s disease. J Neurosci Methods
Yee S, Choi BH (1994) Methylmercury poisoning induces oxidative stress in the mouse brain. Exp Mol Pathol 60(3):188–196
Yee S, Choi BH (1996) Oxidative stress in neurotoxic effects of methylmercury poisoning. Neurotoxicology 17(1):17–26
Yeh P, Tschumi AI, Kishony R (2006) Functional classification of drugs by properties of their pairwise interactions. Nat Genet 38(4):489–494
Yeh PJ, Hegreness MJ, Aiden AP, Kishony R (2009) Drug interactions and the evolution of antibiotic resistance. Nat Rev Microbiol 7(6):460–466
Yin H, Cheng L, Holt M, Hail N Jr, Maclaren R, Ju C (2010) Lactoferrin protects against acetaminophen-induced liver injury in mice. Hepatology 51(3):1007–1016
Yin Z, Milatovic D, Aschner JL, Syversen T, Rocha JB, Souza DO et al (2007) Methylmercury induces oxidative injury, alterations in permeability and glutamine transport in cultured astrocytes. Brain Res 1131(1):1–10
Yin ZB, Jiang HY, Syversen T, Rocha JBT, Farina M, Aschner M (2008) The methylmercury-L-cysteine conjugate is a substrate for the L-type large neutral amino acid transporter. J Neurochem 107(4):1083–1090
Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J et al (2008) The ground state of embryonic stem cell self-renewal. Nature 453(7194):519–523
Yokoyama H, Kuroiwa H, Yano R, Araki T (2008) Targeting reactive oxygen species, reactive nitrogen species and inflammation in MPTP neurotoxicity and Parkinson’s disease. Neurol Sci 29(5):293–301
Yoneda M, Nozaki Y, Endo H, Mawatari H, Iida H, Fujita K et al (2010) Serum ferritin is a clinical biomarker in Japanese patients with nonalcoholic steatohepatitis (NASH) independent of HFE gene mutation. Dig Dis Sci 55:808–814
Yoshino M, Murakami K (1998) Interaction of iron with polyphenolic compounds: application to antioxidant characterization. Anal Biochem 257(1):40–44
You SA, Archacki SR, Angheloiu G, Moravec CS, Rao S, Kinter M et al (2003) Proteomic approach to coronary atherosclerosis shows ferritin light chain as a significant marker: evidence consistent with iron hypothesis in atherosclerosis. Physiol Genomics 13(1):25–30
You SA, Wang Q (2005) Ferritin in atherosclerosis. Clin Chim Acta 357(1):1–16
Youdim MB (2003) What have we learnt from CDNA microarray gene expression studies about the role of iron in MPTP induced neurodegeneration and Parkinson’s disease? J Neural Transm Suppl (65):73–88
Youdim MBH, Fridkin M, Zheng H (2004a) Novel bifunctional drugs targeting monoamine oxidase inhibition and iron chelation as an approach to neuroprotection in Parkinson’s disease and other neurodegenerative diseases. J Neural Transm 111(10–11):1455–1471
Youdim MBH, Stephenson G, Ben Shachar D (2004b) Ironing iron out in Parkinson’s disease and other neurodegenerative diseases with iron chelators—a lesson from 6-hydroxydopamine and iron chelators, desferal and VK-28. Redox-Active Metals Neurol Disord 1012:306–325
Young JL, Libby P, Schonbeck U (2002) Cytokines in the pathogenesis of atherosclerosis. Thromb Haemost 88(4):554–567
Yu Z, Persson HL, Eaton JW, Brunk UT (2003) Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radic Biol Med 34(10):1243–1252
Yuan XM, Li W (2008) Iron involvement in multiple signaling pathways of atherosclerosis: a revisited hypothesis. Curr Med Chem 15(21):2157–2172
Yue H, Brown M, Knowles J, Wang H, Broomhead DS, Kell DB (2006) Insights into the behaviour of systems biology models from dynamic sensitivity and identifiability analysis: a case study of an NF-kappaB signalling pathway. Mol Biosyst 2:640–649
Yue H, Wang Y, Broomhead DS, Brown M, Kell DB (2007) Sensitivity analysis of an oscillatory signal transduction pathway. FOSBE 2007:99–104
Yue H, Brown M, He F, Jia J, Kell DB (2008) Sensitivity analysis and robust experimental design of a signal transduction pathway system. Int J Chem Kinet 40(11):730–741
Yun SW, Gerlach M, Riederer P, Klein MA (2006) Oxidative stress in the brain at early preclinical stages of mouse scrapie. Exp Neurol 201(1):90–98
Zacharski LR, Chow B, Lavori PW, Howes PS, Bell MR, DiTommaso MA et al (2000) The iron (Fe) and atherosclerosis study (FeAST): a pilot study of reduction of body iron stores in atherosclerotic peripheral vascular disease. Am Heart J 139(2):337–345
Zacharski LR, Chow BK, Howes PS, Shamayeva G, Baron JA, Dalman RL et al (2008) Decreased cancer risk after iron reduction in patients with peripheral arterial disease: results from a randomized trial. J Natl Cancer Inst 100(14):996–1002
Zacharski LR, Gerhard GS (2003) Atherosclerosis: a manifestation of chronic iron toxicity? Vasc Med 8(3):153–155
Zamboni P (2006) The big idea: iron-dependent inflammation in venous disease and proposed parallels in multiple sclerosis. J R Soc Med 99(11):589–593
Zamin LL, Filippi-Chiela EC, Dillenburg-Pilla P, Horn F, Salbego C, Lenz G (2009) Resveratrol and quercetin cooperate to induce senescence-like growth arrest in C6 rat glioma cells. Cancer Sci 100(9):1655–1662
Zandman-Goddard G, Shoenfeld Y (2008) Hyperferritinemia in autoimmunity. Isr Med Assoc J 10(1):83–84
Zanella I, Derosas M, Corrado M, Cocco E, Cavadini P, Biasiotto G et al (2008) The effects of frataxin silencing in HeLa cells are rescued by the expression of human mitochondrial ferritin. Biochim Biophys Acta 1782(2):90–98
Zapelini PH, Rezin GT, Cardoso MR, Ritter C, Klamt F, Moreira JC et al (2008) Antioxidant treatment reverses mitochondrial dysfunction in a sepsis animal model. Mitochondrion 8(3):211–218
Zarember KA, Cruz AR, Huang CY, Gallin JI (2009) Antifungal activities of natural and synthetic iron chelators alone and in combination with azole and polyene antibiotics against Aspergillus fumigatus. Antimicrob Agents Chemother 53(6):2654–2656
Zatta P, Drago D, Bolognin S, Sensi SL (2009) Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol Sci 30(7):346–355
Zecca L, Youdim MBH, Riederer P, Connor JR, Crichton RR (2004) Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 5(11):863–873
Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG et al (1999) Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 154(5):1423–1429
Zhang HY (2006) Same causes, same cures. Biochem Biophys Res Commun 351(3):578–581
Zhang SB, Wang Y, Liu CL (2009) Metal-based therapeutic strategies of neurodegenerative diseases. Progr Chem 21(5):903–910
Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE et al (2010a) Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 463(7280):501–506
Zhang J, Zhang Y, Wang J, Cai P, Luo C, Qian Z et al (2010b) Characterizing iron deposition in Parkinson’s disease using susceptibility-weighted imaging: an in vivo MR study. Brain Res
Zhang X, Smith DL, Meriin AB, Engemann S, Russel DE, Roark M et al (2005a) A potent small molecule inhibits polyglutamine aggregation in Huntington’s disease neurons and suppresses neurodegeneration in vivo. Proc Natl Acad Sci USA 102(3):892–897
Zhang X, Xie W, Qu S, Pan T, Wang X, Le W (2005b) Neuroprotection by iron chelator against proteasome inhibitor-induced nigral degeneration. Biochem Biophys Res Commun 333(2):544–549
Zhang Z, Rigas B (2006) NF-kappaB, inflammation and pancreatic carcinogenesis: NF-kappaB as a chemoprevention target (review). Int J Oncol 29(1):185–192
Zhang Z, Wei T, Hou J, Li G, Yu S, Xin W (2003) Iron-induced oxidative damage and apoptosis in cerebellar granule cells: attenuation by tetramethylpyrazine and ferulic acid. Eur J Pharmacol 467(1–3):41–47
Zhao C, Deng W, Gage FH (2008) Mechanisms and functional implications of adult neurogenesis. Cell 132(4):645–660
Zhao J, Geng C, Tao L, Zhang D, Jiang Y, Tang K et al (2010) Reconstruction and analysis of human liver-specific metabolic network based on CNHLPP data. J Proteome Res
Zhao J, Yu H, Luo JH, Cao ZW, Li YX (2006) Hierarchical modularity of nested bow-ties in metabolic networks. BMC Bioinform 7:386
Zhao M, Laissue JA, Zimmermann A (1997) Hepatocyte apoptosis in hepatic iron overload diseases. Histol Histopathol 12(2):367–374
Zhao R, Planalp RP, Ma R, Greene BT, Jones BT, Brechbiel MW et al (2004) Role of zinc and iron chelation in apoptosis mediated by tachpyridine, an anti-cancer iron chelator. Biochem Pharmacol 67(9):1677–1688
Zheng H, Weiner LM, Bar-Am O, Epsztejn S, Cabantchik ZI, Warshawsky A et al (2005) Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer’s, Parkinson’s, and other neurodegenerative diseases. Bioorg Med Chem 13(3):773–783
Zhu X, Su B, Wang X, Smith MA, Perry G (2007) Causes of oxidative stress in Alzheimer disease. Cell Mol Life Sci 64(17):2202–2210
Ziegler JL, Simonart T, Snoeck R (2001) Kaposi’s sarcoma, oncogenic viruses, and iron. J Clin Virol 20(3):127–130
Zimmermann GR, Lehár J, Keith CT (2007) Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discov Today 12(1–2):34–42
Zimmet JM, Hare JM (2006) Nitroso-redox interactions in the cardiovascular system. Circulation 114(14):1531–1544
Zivadinov R, Bakshi R (2004) Role of MRI in multiple sclerosis II: brain and spinal cord atrophy. Front Biosci 9:647–664
Zivadinov R, Minagar A (2009) Evidence for gray matter pathology in multiple sclerosis: a neuroimaging approach. J Neurol Sci 282(1–2):1–4
Zuliani G, Cherubini A, Ranzini M, Ruggiero C, Atti AR, Fellin R (2006) Risk factors for short-term mortality in older subjects with acute ischemic stroke. Gerontology 52(4):231–236
Zurich MG, Monnet-Tschudi F (2009) Contribution of in vitro neurotoxicology studies to the elucidation of neurodegenerative processes. Brain Res Bull 80(4–5):211–216
Acknowledgments
I thank a great many colleagues and correspondents for their contribution to the refinement of my thinking about these issues, and Julie Cowley for assistance with literature, etc. Previous work that has led to this review has been funded by the BBSRC, EPSRC and MRC.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kell, D.B. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch Toxicol 84, 825–889 (2010). https://doi.org/10.1007/s00204-010-0577-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-010-0577-x