
Abstract
A novel acyl-CoA dehydrogenase involved in degradation of the auxin indoleacetate by Aromatoleum aromaticum was identified as a decarboxylating benzylmalonyl-CoA dehydrogenase (IaaF). It is encoded within the iaa operon coding for enzymes of indoleacetate catabolism. Using enzymatically produced benzylmalonyl-CoA, the reaction was characterized as simultaneous oxidation and decarboxylation of benzylmalonyl-CoA to cinnamoyl-CoA and CO2. Oxygen served as electron acceptor and was reduced to H2O2, whereas electron transfer flavoprotein or artificial dyes serving as electron acceptors for other acyl-CoA dehydrogenases were not used. The enzyme is homotetrameric, contains an FAD cofactor and is enantiospecific in benzylmalonyl-CoA turnover. It shows high catalytic efficiency and strong substrate inhibition with benzylmalonyl-CoA, but otherwise accepts only a few medium-chain alkylmalonyl-CoA compounds as alternative substrates with low activities. Its reactivity of oxidizing 2-carboxyacyl-CoA with simultaneous decarboxylation is unprecedented and indicates a modified reaction mechanism for acyl-CoA dehydrogenases, where elimination of the 2-carboxy group replaces proton abstraction from C2.
Similar content being viewed by others


Introduction
The C3-dicarboxylic acid malonate and its thioesters malonyl-CoA or malonyl-acyl carrier protein (ACP) are important intermediates in the anabolic metabolism of all bacteria and eukarya. Malonyl-CoA is usually produced from acetyl-CoA in an ATP-dependent reaction by the biotin-containing acetyl-CoA carboxylase complex (Gago et al. 2006). Malonyl-CoA then serves (directly or after transfer to ACP) as building block for the synthesis of fatty acids and many other compounds such as flavonoids, stilbenes, and further secondary metabolites (Foster 2012). In addition, some substituted derivatives of malonyl-CoA (Tohge et al. 2007) are used as building blocks for the biosynthesis of special polyketide metabolites (Chan et al. 2009). Examples are methylmalonyl-CoA for producing erythromycin (Staunton and Wilkinson 1997), ethylmalonyl-CoA for phoslactomycin (Chen et al. 2012) or benzylmalonyl-CoA for splenocin biosynthesis (Chang et al. 2015). These compounds are either produced via biotin-dependent carboxylases similar to acetyl-CoA carboxylase (Erb 2011) or via coenzyme B12-dependent rearrangement reactions of the carbon skeletons of the respective CoA-activated dicarboxylic acids (Weichler et al. 2015). Substituted malonyl-CoA derivatives also occur in other important metabolic pathways, e.g., propionate metabolism via methylmalonyl-CoA, the ethylmalonyl-CoA pathway of acetate assimilation (Erb 2011), or anaerobic degradation pathways of alkanes and indoleacetate, which produce alkylmalonyl-CoA (Wilkes et al. 2002) or (2-aminobenzyl)malonyl-CoA as intermediates (Ebenau-Jehle et al. 2012; Schühle et al. 2016).
In this study, we investigate benzylmalonyl-CoA dehydrogenase, a member of the acyl-CoA dehydrogenase family, which is encoded by the gene iaaF and participates in bacterial indoleacetate degradation (Fig. 1; Ebenau-Jehle et al. 2012; Schühle et al. 2016). Most known acyl-CoA dehydrogenases participate in β-oxidation processes and catalyze the FAD-dependent oxidation of CoA-activated acids to α,β-unsaturated enoyl-CoA intermediates. They usually use an electron-transfer flavoprotein (ETF) as electron acceptor, which transfers the redox equivalents further to ubi- or menaquinones via ETF:quinone oxidoreductases (Ghisla and Thorpe 2004; Frerman 1987). Other members of the family are involved in β-condensation reactions as enoyl-CoA or enoyl-acyl carrier protein (ACP) reductases, using NAD(P)H as reductant (Ghisla and Thorpe 2004), or catalyze apparently unrelated reactions such as desulfination or γ,δ-double bond generation (Schürmann et al. 2015; Blake-Hedges et al. 2020). The function of IaaF in the proposed indoleacetate catabolic pathway is the oxidative decarboxylation of (2-aminobenzyl)malonyl-CoA to 2-aminocinnamoyl-CoA and CO2 as shown in Fig. 1. We show in this report that IaaF indeed catalyzes the proposed reaction with benzylmalonyl-CoA, a chemical analogue of the physiological substrate, and propose a catalytic mechanism based on its biochemical properties and sequence comparisons to other acyl-CoA dehydrogenases.

A Organisation of the A. aromatoleum iaa gene cluster. The genes iaaQ and iaaR code for transcriptional regulators, iaaD for a non-functional truncated protein. B Reactions of indoleacetate degradation, starting after uptake (involving the binding protein IaaM) and activation to indoleacetyl-CoA by the CoA ligase IaaB. The reactions of phenylsuccinyl-CoA transferase (IaaL), benzylmalonyl-CoA mutase (IaaGH) and benzylmalonyl-CoA dehydrogenase (IaaF) are shown in detail (IaaF-catalysed reaction highlighted by bold print). Arrows indicate reversible or irreversible steps. Note that in the case of IaaL, both intra- and intermolecular CoA-transfer reactions (with succinate) have been reported (Schühle et al. 2016)
Materials and methods
Cloning, heterologous gene expression and preparation of cell-free extracts
The iaaF gene (ebA2055) was amplified via PCR from chromosomal DNA of A. aromaticum EbN1 using forward and reverse primers (AAGCTCTTCAATGGACTTCGATCTCACCGACG and AAGCTCTTCACCCTGAGCTCACTTCCTTCGCAATG, respectively) and cloned into the vector pAsg-IBA5 (IBA Lifesciences, Göttingen, Germany). The resulting gene codes for a fusion protein of IaaF with a N-terminal Strep-tag. It was produced in E. coli DH5α, which was grown in LB medium at room temperature and protein synthesis was induced by anhydrotetracycline addition as reported previously (Schühle et al. 2016). Cells were harvested by centrifugation and resuspended in two volumes of 10 mM Tris/HCl pH 7.5 containing 0.1 mg/ml DNase I. Cell-free extracts were prepared by sonification at 4 °C, followed by ultracentrifugation (100,000 × g, 60 min). IaaF activity was present in the soluble fraction, which was used for further purification.
Protein purification and characterization
Cell-free extracts with overproduced IaaF were applied on a Strep-tag affinity column (IBA Lifesciences, Göttingen, Germany), which was washed with buffer A (25 mM Tris, 150 mM NaCl, pH 7.9) at a flow rate of 1 ml min−1 for 5 column volumes, and the enzyme was eluted with buffer A containing 2.5 mM desthiobiotin. The buffer of the purified protein was exchanged into protein storage buffer (30% glycerol, 150 mM NaCl, 25 mM Tris/Cl pH 7.9). Proteins were stored at – 20 °C until further use. Production protocols for the auxiliary enzymes used in this study were previously described as follows: a recombinant Ccr variant from Caulobacter crescentus (WP_010920921) carrying three mutations (C146P, I169A, F373G) to open the active site for large substrates (Vögeli et al. 2018); ETF from A. aromaticum (CAI09844, CAI09844; Vogt et al. 2019); acyl-CoA oxidase Acx4 from Arabidopsis thaliana (NP_160752; Schwander et al. 2016; Vögeli et al. 2018); Etr1P from Saccharomyces cerevisiae (Q8WZM3; Rosenthal et al. 2015); and methylmalonyl-CoA epimerase from Rhodobacter sphaeroides (ABA79990; Erb et al. 2008). Native molecular masses were determined by applying the proteins to a calibrated gel filtration column (Superdex 200PG, calibration kit HMW, GE Healthcare). Cofactors were extracted from the purified IaaF protein by heat treatment (30 min, 99 °C) and removing the precipitated protein by centrifugation. The supernatant was then analyzed by paper chromatography with Whatman paper (3 mm) using a mobile phase of n-butanol:acetic acid:H2O (4:1:5) as described in Yagi and Oishi (1971). Retention times were compared with those of standard compounds (FAD, FMN, riboflavin). The quantitation of bound FAD from UV–Vis spectra was as described in Aliverti et al. (1999). Further standard protein analytic techniques, such as SDS–PAGE and protein concentration determinations using Coomassie G-250 dye-binding with bovine serum albumin as standard were performed as described in Coligan et al. (1995).
Synthesis of benzylmalonyl-CoA and other CoA-thioesters
Benzylmalonyl-CoA was synthesized in a buffer containing 100 mM Tris–HCl pH 8.0, 100 mM KHCO3, 20 mM NADPH, 20 µM acyl-CoA oxidase, and 40 μg mL−1 carbonic anhydrase, which was supplied with 2.1 μM of the Ccr variant and 1 mM cinnamoyl-CoA (Vögeli et al. 2018). The mixture was incubated for 60 min at 30 °C and then quenched by the addition of 5% (v/v) formic acid. The synthesized benzylmalonyl-CoA was purified by HPLC, using a 1260 Infinity LC system (Agilent) and a Gemini 10 μm NX-C18 AXOA packed column (110 Å, 100 × 21.2 mm, Phenomenex). The protocol used a flow rate of 25 ml*min−1 with a basal buffer of 50 mM NH4HCO3, pH 8.2 containing 5% methanol for the initial 5 min, followed by a gradual increase from 5 to 40 % MeOH over 15 min, a 2 min washing step at 95% MeOH and re-equilibration of 3 min at 5% MeOH. The purified benzylmalonyl-CoA was lyophilized and stored as powder at – 20 °C until further use. Further substituted malonyl-CoA compounds were generated analogously, as described in Vögeli et al. (2018). Phenylpropionyl-CoA and cinnamoyl-CoA were prepared from the respective acids in two steps: first, phenylpropionic or cinnamic acid was converted to the respective anhydride by solving 200 µmol of the acid and 100 µmol acetic anhydride in 0.5 ml of 100% acetic acid and heating the mixture to 120 °C until the acetic acid was evaporated. Subsequently, the formed anhydrides were converted to CoA-thioesters as described by Schachter and Taggart (1976). The thioesters were further purified by HPLC and lyophilized as described for benzylmalonyl-CoA.
IaaF enzymatic assays and product analysis
IaaF activity was assayed in 100 mM Tris/HCl buffer at pH 7.8 with added IaaF (5–50 µg/ml) and racemic benzylmalonyl-CoA (0–60 µM). Activity was routinely measured in a continuous photometric assay by directly following the formation of cinnamoyl-CoA at 308 nm (ε = 15.4 mM−1 cm−1). If indicated, methylmalonyl-CoA epimerase from Rhodobacter sphaeroides (Erb et al. 2008; accession number B8XVS7; 0.5 mg/ml) was added after the reaction came to an end. Some experiments also contained artificial electron acceptors such as ferricenium hexafluorophosphate (200 µM), phenazine-methosulfate (100 µM) and dichlorophenyl-indophenol (500 µM), or purified ETF from A. aromaticum (1 mg/ml; Vogt et al. 2019) as potential electron acceptors under aerobic or anaerobic conditions. Product formation was confirmed by HPLC analysis after stopping the reactions after defined time periods with added NaHSO4 (0.5 M final concentration). The precipitated proteins were removed by centrifugation, and supernatants were applied to a Kinetex 5 µ C18 column (250 × 4.6 mm, Phenomenex). A linear gradient (5–30% acetonitrile in 50 mM NH4-formate buffer pH 5.6 over 15 min, flow 0.75 ml/min) was applied, and products were detected by their absorption at 220 and 260 nm. Observed retention times were 7.7 min for benzylmalonyl-CoA, 15.9 min for phenylpropionyl-CoA and 14.2 min for cinnamoyl-CoA. Formation of H2O2 was quantified using the fluorescence-based Ampliflu Red “easy to use” kit (Sigma–Aldrich; excitation 571 nm, emission 630 nm) according to the user manual.
An alternative coupled photometric assay for IaaF activity was coupled to NADH reduction in the presence of the unspecific enoyl-CoA reductase Etr1P (Rosenthal et al. 2015), which catalyzes the NADH-dependent reduction of a broad variety of enoyl thioesters to acyl thioesters. The assay mixture contained 0.1 mg/ml IaaF, 0.6 mg/ml Etr1P and 0.4 mM NADH and was started by addition of 50–100 µM of various substituted malonyl-CoA-thioesters. Oxidation of NADH was followed at 340 nm (ε = 6.22 mM−1 cm−1).
Reduction of cinnamoyl-CoA by IaaF (reverse reaction) was tested under anaerobic conditions using 100 mM MOPS/KOH buffer pH 6.8 (for non-carboxylating conditions) or 100 mM bicarbonate/HCl buffer pH 6.8 (for carboxylating conditions). The assays contained 5–25 µg IaaF and 1 mM benzyl- or methyl viologen, which had been fully reduced by adding portions of Na-dithionite from a 10 mM stock solution. Reactions were started by addition of cinnamoyl-CoA (0.5 mM) and oxidation of the reduced viologens was followed photometrically at 600 nm. After reaction times of > 10 min, the assays were also tested for product formation by HPLC analysis.
The rate of spontaneous epimerization of benzylmalonyl-CoA was approximated by determining the rate of hydrogen–deuterium exchange in D2O. To this end, 100 µM lyophilized benzylmalonyl-CoA was dissolved in 100 mM Tris-DCl buffer in D2O (pD 8.0) and incubated at 15 °C in an HPLC–MS autosampler. At defined time points, samples were analyzed via HPLC–MS (1260 Infinity LC system from Agilent using a Gemini 10 mm NX-C18 110 A˚, 100 × 21.2 mm, AXOA packed column from Phenomenex) and the relative amount of deuterium incorporation into the α-position of benzylmalonyl-CoA was determined from the isotopomer ratios. Even if the benzylmalonyl-CoA used was already a racemic mixture, the determined deuteration rate is expected to reflect the approximate racemization rate, because both processes depend on deprotonation of Cα of benzylmalonyl-CoA.
Sequence alignment
Amino acid sequences of IaaF orthologues from bacteria and archaea and of further members of the acyl-CoA dehydrogenase family were aligned using Clustal Omega (www.ebi.ac.uk/Tools/msa/clustalo and avermitilis.ls.kitasato-u.ac.jp/clustalo) and bootstrap values were calculated in 1000 replications. A neighbour-joining tree was constructed based on the alignment, using the Program iTOL (itol.embl.de/).
Results
Enzymatic synthesis of benzylmalonyl-CoA
Since no CoA ligases or CoA-transferases are available to activate benzylmalonate to the CoA-thioester, we assayed several variants of the Ecr enzyme family for synthesizing benzylmalonyl-CoA via reductive carboxylation of cinnamoyl-CoA (Peter et al. 2015). The prototype enzyme of this family is crotonyl-CoA carboxylase/reductase (Ccr), which catalyzes the reductive carboxylation of crotonyl-CoA to ethylmalonyl-CoA in the recently discovered ethylmalonyl-CoA cycle of acetyl-CoA assimilation (Erb et al. 2007; Erb 2011), but these enzymes also convert CoA-thioesters of various other α,β-unsaturated acids to the corresponding saturated 2-carboxyacyl thioesters (Peter et al. 2015), including cinnamoyl-CoA to benzylmalonyl-CoA (Peter et al. 2016). We obtained the best yields of benzylmalonyl-CoA with a previously reported Ccr from Caulobacter crescentus containing three amino acid exchanges and, therefore, used this enzyme to produce the substrate for benzylmalonyl-CoA dehydrogenase (Vögeli et al. 2018; Schwander et al. 2016).
Stereochemistry of benzylmalonyl-CoA
Based on the conserved active site geometry of Ccr, we expected that the enzyme generated stereospecifically (S)- benzylmalonyl-CoA, yet the obtained product appeared to be a racemic mixture when applied in our further experiments (see below). Therefore, we determined whether benzylmalonyl-CoA spontaneously racemizes in solution by determining the time-dependent exchange of the C-2 proton in D2O-based Tris–Cl buffer (pD 8.0) as proxy for the analogous racemization reaction. A gradual increase of deuterium content in benzylmalonyl-CoA from 0 to almost 100% was observed within 13 h at room temperature, indicating a spontaneous deuteration rate of 4.2 × 10−5 s−1 (Fig. 2), assuming first-order kinetics. Racemization does not involve kinetic isotope effects and, therefore, should occur even faster than deuteration. Because of the time needed for extraction and preparation of benzylmalonyl-CoA, we expect that our experiments were always performed with racemic mixtures, regardless of the enantiomer specificity of Ccr. Unfortunately, the lack of standards prohibited us from identifying which enantiomer was produced by the enzymes.

Spontaneous deuteration of enzymatically synthesized benzylmalonyl-CoA. Time-dependent incorporation of deuterium into benzylmalonyl-CoA in D2O-based buffer is shown. Curve fitting indicates a half life time of 4.5 h and a rate constant of 4.2 × 10−5 s−1
Identification of IaaF as a benzylmalonyl-CoA oxidase
The iaaF gene from Aromatoleum aromaticum was cloned into vector pAsg5 with a 5’ strep-tag fusion (IBA Lifesciences, Göttingen, Germany), expressed in E. coli, and the produced protein was purified by affinity chromatography. We obtained a yellow protein which consisted of a single subunit of the expected size (41 kDa after SDS–PAGE). The native mass of the enzyme was determined as 159 kDa by gel filtration chromatography, suggesting a homotetrameric quaternary structure. Spectrophotometric characterization of the purified protein confirmed the presence of a flavin cofactor, which was extracted by acid-precipitation of the protein and identified by comigration with an FAD reference via paper chromatography. The FAD content was calculated as 4.2 per homotetramer, assuming a molar extinction coefficient of 11.3 mM−1 cm−1 at the absorption maximum of 450 nm (Leutwein and Heider 2002). The cofactor was fully reduced by stepwise addition of either benzylmalonyl-CoA or dithionite as reductants (Fig. 3). Changes of the spectra occurred only after about 50 µM of benzylmalonyl-CoA or 200 µM of dithionite had been added (Fig. 3), suggesting that dissolved oxygen in the buffer initially acted as electron acceptor, before reduction of the enzyme took effect. After adjusting for this effect, the calculated molar ratios between added reductant and IaaF reduction revealed stoichiometries of 1.2 dithionite and 1.9 benzylmalonyl-CoA needed to reduce one IaaF monomer, respectively (Fig. 3). Although both reductants are two-electron donors, only the dithionite showed the expected stoichiometry for full reduction of the FAD, whereas the value for benzylmalonyl-CoA is about twice as large. This indicates that IaaF reacts stereospecifically with only one enantiomer of the racemic benzylmalonyl-CoA obtained after synthesis and storage (see above). Although the non-reactive enantiomer racemizes spontaneously (see above), the observed rate of deuterium exchange is 1,700-fold lower than the maximum kcat value of IaaF and, therefore, racemization does not take effect within the time-frame of the experiment.

Absorption spectra of IaaF. IaaF concentrations were at 4.5 µM (per holoenzyme), and the tests were done in the absence of methylmalonyl-CoA epimerase. The enzyme was titrated with A benzylmalonyl-CoA or B dithionite, until fully quenched. Thicker lines indicate the initial (topmost) and final spectra (lowermost, broken line), as well as control spectra of benzylmalonyl-CoA without added enzyme (dotted lines). Note that the strong absorption increase at < 350 nm in A represents the increasing production of cinnamoyl-CoA. Inserts indicate the absorption decrease at 450 nm with added concentrations of the reductants
Catalytic properties of benzylmalonyl-CoA dehydrogenase
Activity of IaaF was assayed photometrically essentially as previously described for benzylsuccinyl-CoA dehydrogenase (Leutwein and Heider 2002). The assays were started with enzymatically produced benzylmalonyl-CoA (see above) with O2 serving as electron acceptor, and were evaluated by recording the absorption increase at 308 nm due to the production of cinnamoyl-CoA (using an experimentally determined ε308 = 15.4 mM−1 cm−1). The expected physiological substrate of IaaF during indoleacetate degradation is (2-aminobenzyl)malonyl-CoA, which was not available for testing. However, the missing amino group in benzylmalonyl-CoA does not appear crucial for substrate recognition, since the compound was readily accepted by IaaF and yielded a maximum turnover rate of 2.7 U mg−1. We only observed activity in the presence of oxygen and did not observe benzylmalonyl-CoA oxidation coupled to the reduction of typical artificial electron acceptors for acyl-CoA dehydrogenases, such as the ferricenium cation or phenazine-methosulfate/dichlorophenyl-indophenol under aerobic or anaerobic assay conditions. Moreover, IaaF did not react with purified recombinant electron transfer flavoprotein (ETF) from A. aromaticum, which serves as physiological electron acceptor for other acyl-CoA dehydrogenases, as recently described by Vogt et al. (2019). While oxygen may be used as electron acceptor for aerobic growth on indoleacetate, it is not clear yet what replaces ETF under denitrifying growth conditions. In contrast to other known acyl-oxidases, e.g., from rat peroxisomes or from Arabidopsis (accession numbers 1IS2, 2IX5), IaaF contains a sequence motif very close to the characterized ETF-interaction site of human medium chain acyl-CoA dehydrogenase (Toogood et al. 2004). However, such a motif is also present in sulfinopropionyl-CoA desulfinase (Schürmann et al. 2015), a member of the enzyme family not even catalysing a redox reaction.
The photometric assay was confirmed by following the turnover of benzylmalonyl-CoA by HPLC analysis. After starting the reaction by adding the substrate, we observed the decrease of its concentration and the formation of a new CoA-thioester over time which was identified as cinnamoyl-CoA by its UV–Vis spectrum (Peter et al. 2016; Johns 1974). Because A. aromaticum grows almost equally well on indoleacetate under aerobic or anaerobic conditions, IaaF may act as an oxidase if oxygen is available. However, it is obvious that it also needs another physiological electron acceptor, which remains unknown. Because of its demonstrated activity with benzylmalonyl-CoA, we propose benzylmalonyl-CoA dehydrogenase as enzyme name, referring to its affiliation to the acyl-CoA dehydrogenase family, which also includes the acyl-CoA oxidases (Kim and Miura 2004). After the reaction with benzylmalonyl-CoA ended, it could be restarted by adding methylmalonyl-CoA epimerase to the assay. This confirms that IaaF is strictly stereospecific and initially converts only one of the benzylmalonyl-CoA enantiomers, while the other one is used only after it is enzymatically epimerized. To our knowledge, this is the first time benzylmalonyl-CoA is reported to be converted by a methylmalonyl-CoA epimerase. As noted before, the non-enzymatic racemization rate of benzylmalonyl-CoA is too slow to interfere in this experiment. None of our experiments produced any trace of phenylpropionyl-CoA, indicating a strict coupling of decarboxylation and oxidation of benzylmalonyl-CoA in the IaaF reaction. The use of oxygen as electron acceptor for benzylmalonyl-CoA oxidation by IaaF suggested the production of H2O2 in the enzyme assay. Using a fluorescence-based detection system, we indeed confirmed the release of H2O2 by IaaF in approximately equimolar ratio with the benzylmalonyl-CoA oxidized (Fig. 4A).

H2O2 production by IaaF and steady state enzyme kinetics of benzylmalonyl-CoA (BMalCoA) oxidation. A Time-dependent H2O2 production after starting the reaction with 20 µM benzylmalonyl-CoA and different IaaF concentrations (triangles, squares and circles represent 18, 36 and 72 nM IaaF, respectively). B Steady state kinetics of IaaF. Standard deviations are indicated by error bars
Apparent kinetic parameters of IaaF were determined for conversion of benzylmalonyl-CoA to cinnamoyl-CoA, using freshly prepared substrate to prevent significant hydrolysis to benzylmalonate. The data fitted well to a strongly substrate-inhibited Michaelis–Menten enzyme kinetics with a rather low apparent Km value of 1.6 ± 0.3 µM for benzylmalonyl-CoA (racemic mixture), an apparent Vmax value of 5.1 ± 0.4 U/mg (equivalent to an apparent kcat of 3.5 s−1), and a low apparent substrate inhibition parameter Kis of 9.1 ± 1.3 µM. Because of the low Kis value, the enzyme falls short of its theoretical maximum rate, exhibiting a maximal observed rate of only 2.7 U/mg at 7 µM benzylmalonyl-CoA (Fig. 4B). Regarding the racemic nature of the substrate, we cannot discriminate which of the enantiomers is responsible for the observed substrate inhibition.
IaaF was also tested for its reverse activity, i.e., reduction of cinnamoyl-CoA under anaerobic conditions in the presence or absence of CO2 (supplied from a 100 mM bicarbonate/CO2 buffer at pH 6.8). Dithionite-reduced benzyl or methyl viologen have previously been used as low-potential reductants to provide exergonic redox conditions for driving the reverse reaction of benzylsuccinyl-CoA dehydrogenase (Vogt et al. 2019), but benzylmalonyl-CoA dehydrogenase did not convert cinnamoyl-CoA to either benzylmalonyl-CoA (with carboxylation) or phenylpropionyl-CoA (without carboxylation) under these conditions, as determined by HPLC analysis.
Finally, we performed assays to determine the substrate spectrum of IaaF. To accomplish this, we synthesized various alkyl- or aryl-substituted malonyl-CoA derivatives using several previously described ECR family variants (Vögeli et al. 2018). These compounds were isolated, lyophilized and used as substrates. Experiments were performed as coupled photometric enzyme assays with the enoyl-CoA reductase Etr1p which unspecifically reduces all unsaturated decarboxylation products produced by IaaF with NADH as electron donor (Rosenthal et al. 2015). Activities were measured by recording the absorption decrease at 340 nm due to NADH oxidation, after the assays were started by adding the respective malonyl-CoA derivatives at concentrations of 50 and 100 µM. These concentrations are high enough to cause already strong substrate inhibition in the case of benzylmalonyl-CoA (Fig. 4B) and represent a compromise between providing still measurable IaaF activities, both in controls with benzylmalonyl-CoA and in assays with sterically restrictive alternative substrates with high expected Km values. The results are shown in Table 1, indicating that IaaF still showed by far the highest activity with benzylmalonyl-CoA, even under substrate-inhibited conditions.
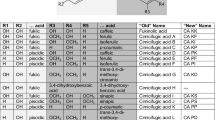
The observed 30% decrease in benzylmalonyl-CoA turnover rates between the assays with 50 and 100 µM substrate is consistent with the recorded substrate inhibition kinetics (Fig. 4B). Using the alternative substrates, we detected low activities of IaaF with hexylmalonyl-CoA, (3-methyl)butylmalonyl-CoA, and butylmalonyl-CoA, whereas no activity was observed with ethylmalonyl-CoA, methylmalonyl-CoA, or phenylpropionyl-CoA. Therefore, IaaF seems to accept several aliphatic alkylmalonyl-CoA analogues with a chain length of four or more C-atoms in straight or branched alkyl chains, but to reject analogues with side chains of only one or two C-atoms. Moreover, it appears crucial that the substrate carries an α-carboxy group, as apparent by the complete inactivity of IaaF with phenylpropionyl-CoA. All alternative substrates show higher turnover rates at 100 µM than at 50 µM concentration, suggesting very high apparent Km values and no substrate inhibition effects at the applied concentrations. Because all measured activities with these substrates were rather low and did not appear to have physiological impact, we did not continue with a full enzyme kinetic characterization.
Discussion
In this manuscript we report on benzylmalonyl-CoA dehydrogenase (IaaF), a new member of the acyl-CoA dehydrogenase family, which catalyses the irreversible oxidative decarboxylation of benzylmalonyl-CoA to cinnamoyl-CoA. The enzyme is involved in the degradation of indoleacetate in A. aromaticum and several other bacteria or archaea and is encoded in a common operon with other enzymes of the proposed degradation pathway (Fig. 1; Ebenau-Jehle et al. 2012, Schühle et al. 2016). The enzyme has been proposed to catalyze a coupled oxidation and decarboxylation reaction of (2-aminobenzyl)malonyl-CoA, producing 2-aminocinnamoyl-CoA for further degradation via β-oxidation (Fig. 1). We show here that the enzyme indeed performs the previously proposed reaction, using benzylmalonyl-CoA as proxy for the unavailable physiological substrate. Although the enzyme participates in an anaerobic degradation pathway, the only accepted electron acceptor we could identify is oxygen, which is reduced to H2O2. Since IaaF accepts neither ETF nor artificial redox dyes such as the ferricenium ion or phenazine-methosulfate as electron acceptors, the re-oxidation mechanism for IaaF still remains unknown. The redox potentials of NAD or NADP are too low to allow direct electron transfer, and a hypothetical electron confurcation mechanism would be dependent of a bifurcation-competent ETF, while the genome of A. aromaticum only codes for a single non-bifurcating ETF (Rabus et al. 2005), which is not reactive with IaaF. Remarkably, the predicted iaa operons of several strictly anaerobic bacteria like Desulfatiglans anilini, other sulfate reducing bacteria or the archaeon Ferroglobus placidus contain genes coding for a non-bifurcating ETF paralogue in addition to an iaaF orthologue, as previously reported (Schühle et al. 2016). It may be speculated that the iaa genes originated in sulfate-reducing bacteria together with a dedicated ETF as electron acceptor for IaaF and were laterally transferred to denitrifyers. These new hosts still use IaaF, but their ETF:quinone oxidoreductases (Frerman 1987) are fundamentally different from those of the strictly anaerobic species (Vogt et al. 2019), and, therefore, a new mechanism for IaaF re-oxidation was needed. We have to assume from our anaerobic growth experiments, that alternative oxidants can be used, but their nature remains unknown for now.
The substrate specificity of IaaF appears to be quite narrow. The observed activities with benzylmalonyl-CoA and several aliphatic alkylmalonyl-CoA analogues with a minimum alkyl chain length of four C-atoms indicate that IaaF is able to convert malonyl-CoA derivatives with substituents of similar sizes as an aromatic ring. Therefore, we expect that it also efficiently turns over substituted variants of benzylmalonyl-CoA, such as the physiologically occurring (2-aminobenzyl)malonyl-CoA, although these compounds were not available for testing. The enzyme showed high activity and a very low apparent Km value with benzylmalonyl-CoA, as well as severe substrate inhibition at low substrate concentrations. It still needs to be seen whether this inhibition is a physiological effect or is exerted by the “wrong” enantiomer present in the racemic mixture. We have previously also reported substrate inhibition for another enzyme of the pathway, indoleacetate-CoA ligase IaaB (Schühle et al. 2016), and propose that the inhibition properties of these two enzymes may prevent the production of too much CoA-activated benzoate analogues, which may overwhelm the further metabolic reactions, e.g., the rather slow reduction of the aromatic ring via benzoyl-CoA reductase (Fuchs et al. 2011). If (2-aminobenzyl)malonyl-CoA accumulates because of a stalled benzoyl-CoA metabolism, it is expected to be converted backwards to (2-aminophenyl)succinyl-CoA via the two preceding reversible reactions of the pathway (phenylsuccinyl-CoA mutase IaaGH and phenylsuccinyl-CoA transferase IaaL; Fig. 1). Because of the labile nature and fast hydrolytic degradation of the phenylsuccinyl-CoA regioisomers (Schühle et al. 2016), accumulation of these intermediates by a blocked degradation pathway would lead to the loss of the CoA-thioester activation. However, the known dual activities of IaaL as intramolecular phenylsuccinyl-CoA transferase and intermolecular CoA transferase between phenylsuccinyl-CoA and succinate would allow the cells to preserve both the activation energy of the thioester groups (via succinyl-CoA to ATP) and the degradation intermediate (in form of phenylsuccinate, resp. (2-aminophenyl)succinate), which can easily be re-activated using succinyl-CoA as CoA-donor when the metabolic block is overcome (Fig. 1). As indicated by data base searches and analyzing for related proteins, IaaF is a member of a special subclass of acyl-CoA dehydrogenases from bacterial and archaeal strains, which are all encoded in gene clusters coding for enzymes of indoleacetate metabolism (Schühle et al. 2016). A multiple alignment of these IaaF-related sequences with representative acyl-CoA dehydrogenases of various other subclasses revealed interesting variations of a highly conserved sequence motif close to their C-termini, which is known to be part of the active site in structurally characterized acyl-CoA dehydrogenases (Ghisla and Thorpe 2004). While most members of subclasses catalyzing oxidation of straight-chain alkyl-CoAs to α,β-unsaturated alkenoyl-CoA derivatives contain a conserved YEG motif, all members of the IaaF subclass deviate from this consensus and instead contain a GGG motif (Fig. 5).

Alignment of the conserved YEG motifs of acyl-CoA dehydrogenases with IaaF orthologues and other deviating members of the enzyme family (deviations highlighted in bold print). IaaFAa and IaaFFp, IaaF orthologues of A. aromaticum and Ferroglobus placidus, respectively; GdhAa and GdhHs, glutaryl-CoA dehydrogenase from A. aromaticum and human, respectively; ACD_srat, ACD_mpig and ACDPx acyl-CoA dehydrogenases from rat (short chain), pig (medium chain), and Paraburkholderia xenovorans, respectively; IbuHs and IvalHs, human isobutyryl- and isovaleryl-CoA dehydrogenases, respectively; BbsGTa, (E)-benzylidenesuccinyl-CoA dehydrogenase from Thauera aromatica. Numbers indicate the positions in the amino acid sequences. Accession numbers: WP_011236983, WP_012966786, CAI07810, 1SIR_A, 1JQI_A, NP_999204, 5JSC_A, NP_055199, 1IVH_A, AAF89842
A similarity tree of IaaF orthologues with selected members from other acyl-CoA dehydrogenase subclasses shows that they occupy an isolated subbranch (labeled by BMal) separate from all other members of the family (Fig. 6). All enzymes of the BMal subbranch are encoded within operons containing further conserved genes of the iaa operon. Acyl-CoA dehydrogenases with other specificities as well as enzymes with unknown function occupy other subbranches of the family and can nicely be grouped based on their sequence conservation. Remarkably, most biochemically characterized enzymes retain either the active site YEG motif (But, MCA, Glut, SCO, MMProp in Fig. 6) or very similar motifs such as GEG (MSucc), GDG (CHCx), LEG (IBu), VEG (BSucc, LCA), IEG (TcdD) or FEG (ACO, TcdD). Substitution of the conserved Tyr, which is involved in binding the CoA-thioester substrates via main chain contacts, may help to accommodate α-substituted or otherwise bulky substrates like isobutyryl-CoA, isovaleryl-CoA, cyclohexanecarboxy-CoA, methyl- or benzylsuccinyl-CoA (Tiffany et al. 1997; Leuthner and Heider 2000; Battailem et al. 2004) in most of the latter enzymes. The conserved Glu of the YEG motif abstracts a proton from C-2 of the substrate and thereby initiates the reaction (Ghisla and Thorpe 2004). In addition to the IaaF orthologues, the Glu is missing in the enzymes of the subbranches IVal (GAG), SProp (AGG), Carn (SGG), and Pim (YGG), which can be explained by altered catalytic mechanisms in the former two cases, where structures are available. In isovaleryl-CoA dehydrogenases, another Glu takes over the function as proton-abstracting group (Tiffany et al. 1997) whereas sulfinopropionyl-CoA desulfinase attacks the sulfino group and eliminates it as sulfite in a completely different overall reaction (Schürmann et al. 2015). Remarkably, many of the still unassigned acyl-CoA dehydrogenases encoded in the A. aromaticum genome (CAI07201, 08576, 08182, 09333, 09396, 10529, 10530, 07221) also contain deviations from the YEG motif, possibly suggesting further unconventional roles of these enzymes.

Similarity tree of acyl-CoA dehydrogenases and oxidases. Bootstrapping results ranging between 700 and 1000 (of 1000 repetitions) are shown as circles as indicated. Benzylmalonyl-CoA oxidizing IaaF orthologues have been labeled with BMal, and several representative source microbes are indicated as follows (with accession numbers): *, A. aromaticum (CAI07262); Rp, Rhodopseudomonas palustris (WP_011664634); Fp, Ferroglobus placidus (WP_012966786); N, strain NaphS2 (WP_006425528); Di1-3, three paralogues from Desulfocarbo indianensis (WP_049676703, WP_049676756, WP_049676580), Tp, Thermoplasmatales (EMR74870). Furthermore, all acyl-CoA dehydrogenases encoded in A. aromatoleum have been included and are indicated by asterisks (and accession numbers for those of unknown function). In analogy to BMal, all acyl-CoA dehydrogenase (DH) subfamilies with biochemically identified members have been labeled as follows (with representative accession numbers), whereas subfamilies consisting of enzymes of unknown function are unlabeled. ACOperox, peroxisomal acyl-CoA oxidase (1IS2); BSucc, benzylsuccinyl-CoA DH (CAI07171); But, butyryl-CoA DH (WP_010965998); Carn, butyrobetainyl-CoA DH (ANK05333); CHCx, cyclohexancarboxyl-CoA DH (CAI07205); Glut, glutaryl-CoA DH (CAI07810); IBu, isobutyl-CoA DH (1RX0); IVal, isovaleryl-CoA DH (CAI08760); LCA, long chain acyl-CoA DH (CAI07411); MCA, medium chain acyl-CoA DH (P11310); MMProp, methylmercaptopropionyl-CoA DH (Q5LLW7); MSucc, methylsuccinyl-CoA DH (CAX24659); PhProp, phenylpropionyl-CoA DH (CAI09150); Pim, pimelyl-CoA DH (CAI10529, CAI09347); SCO, short chain acyl-CoA oxidase (2IX5); SProp, sulfinopropionyl-CoA desulfinase (5AHS); TcdD, γ,δ-acyl-CoA DH (WP_006350839). EutE, propionaldehyde DH as outgroup (WP_011388669)
The only other known decarboxylating acyl-CoA dehydrogenase in addition to IaaF is glutaryl-CoA dehydrogenase (Fu et al. 2004; Rao et al. 2006), which oxidizes glutaryl-CoA to crotonyl-CoA and CO2. This enzyme is well studied and contains the conserved YEG motif (Fig. 5). Its proposed mechanism proceeds in two sequential steps: first, the substrate is oxidized by deprotonation at C2 (via the conserved Glu370) and hydride abstraction from C3 via the FAD cofactor, then the transient enzyme-bound glutaconyl-CoA intermediate is decarboxylated with re-donation of the proton from Glu370 (Fig. 7A; Rao et al. 2006). In contrast, benzylmalonyl-CoA dehydrogenase does not contain the conserved YEG motif of the active site, which is replaced by GGG. We propose a mechanism of IaaF that does not involve abstraction of a proton from C-2, but achieves the same effect by directly eliminating the carboxy group from C-2 as CO2 (Fig. 7B). The missing Tyr side chain may allow for the necessary space to accommodate the α-carboxy group of benzylmalonyl-CoA, whereas the missing Glu side chain prevents unwanted deprotonation of C-2, while direct decarboxylation should be stimulated by the expected cavity formed by the three consecutive small and apolar Gly residues of the active site (Fig. 7B).

Proposed mechanisms of decarboxylating acyl-CoA dehydratases. A Two-step mechanism of oxidation and γ-carboxy group elimination by glutaryl-CoA dehydrogenase. B One-step process of simultaneous α-carboxy group elimination and oxidation by benzylmalonyl-CoA dehydrogenase. Note the proposed proton transfer from C2 to C4 of the substrate exerted by Glu370 of glutaryl-CoA dehydrogenase, which is replaced by Gly in benzylmalonyl-CoA dehydrogenase
References
Aliverti A, Curti B, Vanoni MA (1999) Identifying and quantitating FAD and FMN in simple and in iron-sulfur-containing flavoproteins. Methods Mol Biol 131:9–23
Battailem KP, Nguyen TV, Vockley J, Kim JJ (2004) Structures of isobutyryl-CoA dehydrogenase and enzyme-product complex: comparison with isovaleryl- and short-chain acyl-CoA dehydrogenases. J Biol Chem 279:16526–16534
Blake-Hedges JM, Pereira JH, Cruz-Morales P, Thompson MG, Barajas JF, Chen J, Krishna RN, Chan LJG, Nimlos D, Alonso-Martinez C, Baidoo EEK, Chen Y, Gin JW, Katz L, Petzold CJ, Adams PD, Keasling JD (2020) Structural mechanism of regioselectivity in an unusual bacterial acyl-CoA dehydrogenase. J Amer Chem Soc 142:835–846
Chan YA, Podevels AM, Kevany BM, Thomas MG (2009) Biosynthesis of polyketide synthase extender units. Nat Prod Rep 26:90–114
Chang C, Huang R, Yan Y, Ma H, Dai Z, Zhang B, Deng Z, Liu W, Qu X (2015) Uncovering the formation and selection of benzylmalonyl-CoA from the biosynthesis of splenocin and enterocin reveals a versatile way to introduce amino acids into polyketide carbon scaffolds. J Amer Chem Soc 137:4183–4190
Chen YL, Zhao J, Liu W, Gao JF, Tao LM, Pan HX, Tang GL (2012) Identification of phoslactomycin biosynthetic gene clusters from Streptomyces platensis SAM-0654 and characterization of PnR1 and PnR2 as positive transcriptional regulators. Gene 509:195–200
Coligan JE, Dunn BM, Ploegh HL, Speicher DW, Wingfield PT (1995) Current protocols in protein science. Wiley, New York, NY
Ebenau-Jehle C, Thomas M, Scharf G, Kockelkorn D, Knapp B, Schühle K, Heider J, Fuchs G (2012) Anaerobic metabolism of indoleacetate. J Bacteriol 194:2894–2903
Erb TJ (2011) Carboxylases in natural and synthetic microbial pathways. Appl Environ Microbiol 77:8466–8477
Erb TJ, Berg IA, Brecht V, Müller M, Fuchs G, Alber B (2007) Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: The ethylmalonyl-CoA pathway. Proc Natl Acad Sci USA 104:10631–10636
Erb TJ, Rétey J, Fuchs G, Alber BE (2008) Ethylmalonyl-CoA mutase from Rhodobacter sphaeroides defines a new subclade of coenzyme B12-dependent acyl-CoA mutases. J Biol Chem 283:32283–32293
Foster DW (2012) Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest 122:1958–1959
Frerman FE (1987) Reaction of electron-transfer flavoprotein ubiquinone oxidoreductase with the mitochondrial respiratory chain. Biochim Biophys Acta 893:161–169
Fu Z, Wang M, Paschke R, Rao KS, Frerman FE, Kim JP (2004) Crystal structures of human glutaryl-CoA dehydrogenase with and without an alternate substrate: structural bases of dehydrogenation and decarboxylation reactions. Biochem 43:9674–9684
Fuchs G, Boll M, Heider J (2011) Microbial degradation of aromatic compounds—from one strategy to four. Nature Rev Microbiol 9:803–816
Gago G, Kurth D, Diacovich L, Tsai SC, Gramajo H (2006) Biochemical and structural characterization of an essential acyl coenzyme A carboxylase from Mycobacterium tuberculosis. J Bacteriol 188:477–486
Ghisla S, Thorpe C (2004) Acyl-CoA dehydrogenases: a mechanistic overview. Eur J Biochem 271:494–508
Johns N (1974) Synthesis and characterisation of acyl coenzyme A derivatives of aromatic carboxylic acids. Zeitsch Naturforsch Sect C 29:469–474
Kim J-J, Miura R (2004) Acyl-CoA dehydrogenases and acyl-CoA oxidases. Structural basis for mechanistic similarities and differences. Eur J Biochem 271:483–493
Leuthner B, Heider J (2000) Anaerobic toluene catabolism of Thauera aromatica: the bbs operon codes for enzymes of beta oxidation of the intermediate benzylsuccinate. J Bacteriol 182:272–277
Leutwein C, Heider J (2002) (R)-Benzylsuccinyl-CoA dehydrogenase of Thauera aromatica, an enzyme of the anaerobic toluene catabolic pathway. Arch Microbiol 178:517–524
Peter DM, Schada von Borzyskowski L, Kiefer P, Vorholt JA, Erb TJ (2015) Screening and engineering the synthetic potential of carboxylating reductases from central metabolism and polyketide biosynthesis. Angew Chemie Int Ed 54:13457–13461
Peter DM, Vögeli B, Cortina NS, Erb TJ (2016) A chemo-enzymatic road map to the synthesis of CoA esters. Molecules 21:1–11
Rabus R, Kube M, Heider J, Beck A, Heitmann K, Widdel F, Reinhard R (2005) The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch Microbiol 183:27–36
Rao KS, Albro M, Dwyer TM, Frerman FE (2006) Kinetic mechanism of glutaryl-CoA dehydrogenase. Biochem 45:15853–15861
Rosenthal RG, Vögeli B, Quade N, Capitani G, Kiefer P, Vorholt JA, Ebert MO, Erb TJ (2015) The use of ene adducts to study and engineer enoyl-thioester reductases. Nat Chem Biol 11:398–402
Schachter D, Taggart JV (1976) Benzoyl coenzyme A and hippurate synthesis. J Biol Chem 203:923–933
Schühle K, Nies J, Heider J (2016) An indoleacetate-CoA ligase and a phenylsuccinyl-CoA transferase involved in anaerobic metabolism of auxin. Environ Microbiol 18:3120–3132
Schürmann M, Meijers R, Schneider TR, Steinbüchel A, Cianci M (2015) 2-Sulfinopropionyl-CoA desulfinase from Adveniella mimigardefordensis DPN7. crystal structure and function of a desulfinase with an acyl-CoA dehydrogenase fold. Acta Cryst D71:1360–1372
Schwander T, von Borzyskowski LS, Burgener S, Cortina NS, Erb TJ (2016) A synthetic pathway for the fixation of carbon dioxide in vitro. Science 354:900–904
Staunton J, Wilkinson B (1997) Biosynthesis of erythromycin and rapamycin. Chem Rev 97:2611–2629
Tiffany KA, Roberts DL, Wang M, Paschke R, Mohsen AW, Vockley J, Kim JJ (1997) Structure of human isovaleryl-CoA dehydrogenase at 2.6 Å resolution: structural basis for substrate specificity. Biochem 36:8455–8464
Tohge T, Yonekura-Sakakibara K, Niida R, Watanabe-Takahashi A, Saito K (2007) Phytochemical genomics in Arabidopsis thaliana: A case study for functional identification of flavonoid biosynthesis genes. Pure Appl Chem 79:811–823
Toogood HS, van Thiel A, Basran J, Sutcliffe MJ, Scrutton NS, Leys D (2004) Extensive domain motion and electron transfer in the human electron transferring flavoprotein:medium chain acyl-CoA dehydrogenase complex. J Biol Chem 279:32904–32912
Vögeli B, Geyer K, Gerlinger PD, Benkstein S, Cortina NS, Erb TJ (2018) Combining promiscuous acyl-CoA oxidase and enoyl-CoA carboxylase/reductases for atypical polyketide extender unit biosynthesis. Cell Chem Biol 25:833-839.e4
Vogt MS, Schühle K, Kölzer S, Peschke P, Chowdhury NP, Kleinsorge D, Buckel W, Essen LO, Heider J (2019) Structural and functional characterization of an electron transfer flavoprotein Involved in toluene degradation in strictly anaerobic bacteria. J Bacteriol 201:e00326-e419
Weichler MT, Kurteva-Yaneva N, Przybylski D, Schuster J, Müller RH, Harms H, Rohwerder T (2015) Thermophilic coenzyme B12-dependent Acyl coenzyme A (CoA) mutase from Kyrpidia tusciae DSM 2912 preferentially catalyzes isomerization of (R)-3-hydroxybutyryl-CoA and 2-hydroxyisobutyryl-CoA. Appl Environ Microbiol 81:4564–4572
Wilkes H, Rabus R, Fischer T, Armstroff A, Behrends A, Widdel F (2002) Anaerobic degradation of n-hexane in a denitrifying bacterium: further degradation of the initial intermediate (1-methylpentyl)succinate via C-skeleton rearrangement. Arch Microbiol 177:235–243
Yagi K, Oishi N (1971) Separating determination of flavins using thin layer Chromatography. J Vitaminol 17:49–55
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schühle, K., Saft, M., Vögeli, B. et al. Benzylmalonyl-CoA dehydrogenase, an enzyme involved in bacterial auxin degradation. Arch Microbiol 203, 4149–4159 (2021). https://doi.org/10.1007/s00203-021-02406-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-021-02406-3