
Abstract
Summary
We have sought the molecular diagnosis of OI in 38 Brazilian cases through targeted sequencing of 15 candidate genes. While 71% had type 1 collagen-related OI, defects in FKBP10, PLOD2 and SERPINF1, and a potential digenic P3H1/WNT1 interaction were prominent causes of OI in this underrepresented population.
Introduction
Defects in type 1 collagen reportedly account for 85–90% of osteogenesis imperfecta (OI) cases, but most available molecular data has derived from Sanger sequencing-based approaches in developed countries. Massively parallel sequencing (MPS) allows for systematic and comprehensive analysis of OI genes simultaneously. Our objective was to obtain the molecular diagnosis of OI in a single Brazilian tertiary center cohort.
Methods
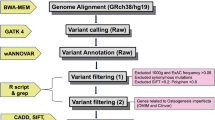
Forty-nine individuals (84% adults) with a clinical diagnosis of OI, corresponding to 30 sporadic and 8 familial cases, were studied. Sixty-three percent had moderate to severe OI, and consanguinity was common (26%). Coding regions and 25-bp boundaries of 15 OI genes (COL1A1, COL1A2, IFITM5 [plus 5′UTR], SERPINF1, CRTAP, P3H1, PPIB, SERPINH1, FKBP10, PLOD2, BMP1, SP7, TMEM38B, WNT1, CREB3L1) were analyzed by targeted MPS and variants of interest were confirmed by Sanger sequencing or SNP array.
Results
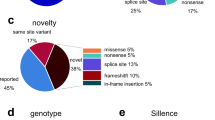
A molecular diagnosis was obtained in 97% of cases. COL1A1/COL1A2 variants were identified in 71%, whereas 26% had variants in other genes, predominantly FKBP10, PLOD2, and SERPINF1. A potential digenic interaction involving P3H1 and WNT1 was identified in one case. Phenotypic variability with collagen defects could not be explained by evident modifying variants. Four consanguineous cases were associated to heterozygous COL1A1/COL1A2 variants, and two nonconsanguineous cases had compound PLOD2 heterozygosity.
Conclusions
Novel disease-causing variants were identified in 29%, and a higher proportion of non-collagen defects was seen. Obtaining a precise diagnosis of OI in underrepresented populations allows expanding our understanding of its molecular landscape, potentially leading to improved personalized care in the future.


Similar content being viewed by others

References
Rauch F, Glorieux FH (2004) Osteogenesis imperfecta. Lancet 363:1377–1385
Marini JC, Forlino A, Bächinger HP et al (2017) Osteogenesis imperfecta. Nat Rev Dis Primers 3:17052
Forlino A, Cabral WA, Barnes AM, Marini JC (2011) New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol 7:540–557
Cho TJ, Lee KE, Lee SK, Song SJ, Kim KJ, Jeon D, Lee G, Kim HN, Lee HR, Eom HH, Lee ZH, Kim OH, Park WY, Park SS, Ikegawa S, Yoo WJ, Choi IH, Kim JW (2012) A single recurrent mutation in the 5'-UTR of IFITM5 causes osteogenesis imperfecta type V. Am J Hum Genet 91:343–348
Semler O, Garbes L, Keupp K, Swan D, Zimmermann K, Becker J, Iden S, Wirth B, Eysel P, Koerber F, Schoenau E, Bohlander SK, Wollnik B, Netzer C (2012) A mutation in the 5'-UTR of IFITM5 creates an in-frame start codon and causes autosomal-dominant osteogenesis imperfecta type V with hyperplastic callus. Am J Hum Genet 91:349–357
Dalgleish R (1997) The human type I collagen mutation database. Nucleic Acids Res 25:181–187
Dalgleish R (1998) The human collagen mutation database 1998. Nucleic Acids Res 26:253–255
Patel RM, Nagamani SC, Cuthbertson D et al (2015) A cross-sectional multicenter study of osteogenesis imperfecta in North America - results from the linked clinical research centers. Clin Genet 87:133–140
Lindahl K, Astrom E, Rubin CJ, Grigelioniene G, Malmgren B, Ljunggren O, Kindmark A (2015) Genetic epidemiology, prevalence, and genotype-phenotype correlations in the Swedish population with osteogenesis imperfecta. Eur J Hum Genet 23:1042–1050
Bardai G, Moffatt P, Glorieux FH, Rauch F (2016) DNA sequence analysis in 598 individuals with a clinical diagnosis of osteogenesis imperfecta: diagnostic yield and mutation spectrum. Osteoporos Int 27:3607–3613
Glorieux FH, Rauch F, Plotkin H, Ward L, Travers R, Roughley P, Lalic L, Glorieux DF, Fassier F, Bishop NJ (2000) Type V osteogenesis imperfecta: a new form of brittle bone disease. J Bone Miner Res 15:1650–1658
Liu Y, Asan MD et al (2017) Gene mutation spectrum and genotype-phenotype correlation in a cohort of Chinese osteogenesis imperfecta patients revealed by targeted next generation sequencing. Osteoporos Int 28:2985–2995
Caparros-Martin JA, Aglan MS, Temtamy S, Otaify GA, Valencia M, Nevado J, Vallespin E, del Pozo A, Prior de Castro C, Calatrava-Ferreras L, Gutierrez P, Bueno AM, Sagastizabal B, Guillen-Navarro E, Ballesta-Martinez M, Gonzalez V, Basaran SY, Buyukoglan R, Sarikepe B, Espinoza-Valdez C, Cammarata-Scalisi F, Martinez-Glez V, Heath KE, Lapunzina P, Ruiz-Perez VL (2017) Molecular spectrum and differential diagnosis in patients referred with sporadic or autosomal recessive osteogenesis imperfecta. Mol Genet Genomic Med 5:28–39
Mrosk J, Bhavani GS, Shah H, Hecht J, Krüger U, Shukla A, Kornak U, Girisha KM (2018) Diagnostic strategies and genotype-phenotype correlation in a large Indian cohort of osteogenesis imperfecta. Bone 110:368–377
Mohd Nawawi N, Selveindran NM, Rasat R, Chow YP, Abdul Latiff Z, Syed Zakaria SZ, Jamal R, Abdul Murad NA, Abd Aziz BB (2018) Genotype-phenotype correlation among Malaysian patients with osteogenesis imperfecta. Clin Chim Acta 484:141–147
Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy M, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur D, Exome Aggregation Consortium (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature 536:285–291
Naslavsky MS, Yamamoto GL, de Almeida TF, Ezquina SAM, Sunaga DY, Pho N, Bozoklian D, Sandberg TOM, Brito LA, Lazar M, Bernardo DV, Amaro E Jr, Duarte YAO, Lebrão ML, Passos-Bueno MR, Zatz M (2017) Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum Mutat 38:751–763
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1081
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 76:7.20.1–7.20.41
Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315
Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, Ryland GL, Tothill RW, Halgamuge SK, Campbell IG, Gorringe KL (2012) CONTRA: copy number analysis for targeted resequencing. Bioinformatics 28:1307–1313
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, ACMG Laboratory Quality Assurance Committee (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Minillo RM, Sobreira N, de Faria Soares Mde F, Jurgens J, Ling H, Hetrick KN, Doheny KF, Valle D, Brunoni D, Perez AB (2014) Novel deletion of SERPINF1 causes autosomal recessive osteogenesis imperfecta type VI in two Brazilian families. Mol Syndromol 5:268–275
Cabral WA, Chang W, Barnes AM, Weis M, Scott MA, Leikin S, Makareeva E, Kuznetsova NV, Rosenbaum KN, Tifft CJ, Bulas DI, Kozma C, Smith PA, Eyre DR, Marini JC (2007) Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat Genet 39:359–365
Rubinato E, Morgan A, D'Eustacchio A, Pecile V, Gortani G, Gasparini P, Faletra F (2014) A novel deletion mutation involving TMEM38B in a patient with autosomal recessive osteogenesis imperfecta. Gene 545:290–292
Rauch F, Lalic L, Roughley P, Glorieux FH (2010) Relationship between genotype and skeletal phenotype in children and adolescents with osteogenesis imperfecta. J Bone Miner Res 25:1367–1374
Liascovich R, Rittler M, Castilla EE (2001) Consanguinity in South America: demographic aspects. Hum Hered 51:27–34
Santos S, Kok F, Weller M, de Paiva FR, Otto PA (2010) Inbreeding levels in Northeast Brazil: strategies for the prospecting of new genetic disorders. Genet Mol Biol 33:220–223
Costa-Motta FM, Bender F, Acosta A et al (2014) A community-based study of mucopolysaccharidosis type VI in Brazil: the influence of founder effect, endogamy and consanguinity. Hum Hered 77:189–196
McInerney-Leo AM, Marshall MS, Gardiner B et al (2013) Whole exome sequencing is an efficient, sensitive and specific method of mutation detection in osteogenesis imperfecta and Marfan syndrome. Bonekey Rep 2:456
Trejo P, Rauch F (2016) Osteogenesis imperfecta in children and adolescents-new developments in diagnosis and treatment. Osteoporos Int 27:3427–3437
Tucker T, Marra M, Friedman JM (2009) Massively parallel sequencing: the next big thing in genetic medicine. Am J Hum Genet 85:142–154
Forlino A, Marini JC (2016) Osteogenesis imperfecta. Lancet 387:1657–1671
Mendoza-Londono R, Fahiminiya S, Majewski J, Care4Rare Canada Consortium, Tétreault M, Nadaf J, Kannu P, Sochett E, Howard A, Stimec J, Dupuis L, Roschger P, Klaushofer K, Palomo T, Ouellet J, al-Jallad H, Mort JS, Moffatt P, Boudko S, Bächinger HP, Rauch F (2015) Recessive osteogenesis imperfecta caused by missense mutations in SPARC. Am J Hum Genet 96:979–985
Lindert U, Cabral WA, Ausavarat S et al (2016) MBTPS2 mutations cause defective regulated intramembrane proteolysis in X-linked osteogenesis imperfecta. Nat Commun 7:11920
Li L, Mao B, Li S, Xiao J, Wang H, Zhang J, Ren X, Wang Y, Wu Y, Cao Y, Lu C, Gao J, You Y, Zhao F, Geng X, Xiao Y, Jiang C, Ye Y, Yang T, Zhao X, Zhang X (2019) Genotypic and phenotypic characterization of Chinese patients with osteogenesis imperfecta. Hum Mutat 40:588–600
Maioli M, Gnoli M, Boarini M et al (2019) Genotype-phenotype correlation study in 364 osteogenesis imperfecta Italian patients. Eur J Hum Genet 27:1090–1100
Acknowledgments
We are grateful to Amanda Narcizo for technical assistance with massively parallel sequencing.
Funding
This work was supported by the São Paulo Research Foundation (FAPESP, Multiusuário grant 2013/02162-8). BFdS also acknowledges support from FAPESP through a Young Investigator grant (2011/12696-4).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Adriana Fernandes, Manuela Rocha-Braz, Monica França, Antonio Lerario, Vivian Simões, Evelin Zanardo, Leslie Kulikowski, Regina Martin, Berenice Mendonca and Bruno Ferraz-de-Souza declare that they have no conflict of interest that could be perceived as prejudicing the impartiality of this study. Manuela Rocha-Braz has received lecture fees from EMS and Amgen Brazil. Bruno Ferraz-de-Souza has received consulting fees from Sandoz Brazil, UCB Brazil and Hypera Brazil, and lecture fees from Amgen Brazil, Sandoz Brazil, Sanofi Brazil and Hypera Brazil.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fernandes, A., Rocha-Braz, M., França, M. et al. The molecular landscape of osteogenesis imperfecta in a Brazilian tertiary service cohort. Osteoporos Int 31, 1341–1352 (2020). https://doi.org/10.1007/s00198-020-05366-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-020-05366-4