
Abstract
Unusual hydrothermal native As-sulfide ± native Ag ± arsenide ± antimonide ± sulfosalt ore shoots and their co-genetic sulfide-fluorite-barite-quartz host veins, which are common in the region and in whole Central Europe, were investigated at three localities in the Schwarzwald, SW Germany, to understand the physico-chemical processes governing the change from a normal (= common) hydrothermal to an exceptional ore shoot regime. Based on fluid inclusions, the formation of the gangue minerals is the result of binary mixing between a NaCl-rich brine and a CaCl2-rich brine (both ~ 20 wt% NaCl aq.). This mixing correlation, major and minor fluid composition, formation temperature (~ 150 °C), and δ34S signature are identical (within error) in ore shoots and host veins. Thermodynamic modeling indicates that ore shoot formation must have resulted from a change in redox conditions by a local influx of a volumetrically minor reducing agent, probably hydrocarbons. The elemental content and the mineralogy of each ore shoot locality (Ag-As-rich: Münstertal; Ag–Ni-As-rich: Urberg; Ag–Ni-As-Sb-rich: Wieden) reflect the metal content of the binary mixed fluid, while mineral textures, successions, and assemblages are thermodynamically and, regarding sulfur, kinetically controlled. The formation of vein and ore shoot sulfides requires an addition of sulfide, most probably from the sulfide-bearing host rocks, because thermodynamic and kinetic reasons suggest that the two major vein-forming and metal-bearing fluids are not the source of the sulfur. The final ore shoot textures are influenced by later hydrothermal remobilization processes of As and Ag. This results in a number of sulfosalts, mostly proustite-pyrargyrite. Interestingly, the greater thermodynamic stability of Sb-endmember sulfosalts enables them to form even in As-dominated fluid systems.
Similar content being viewed by others

Introduction
In many hydrothermal deposits, the ratio of gangue to ore minerals varies significantly within a specific deposit (e.g., Lindgren 1913; Guilbert and Park 2007). Those parts which are or have been economically mineable are commonly referred to as ore shoot or—in the case of economically extremely valuable ore shoots—as bonanzas (e.g., Lindgren 1913; Guilbert and Park 2007; Squire et al. 2008).
The formation of ore shoots, in general, is closely linked to the origin of each deposit and the spatial variability of chemical and physical conditions during deposition (Hise 1900; Penrose 1910). Thus, reasons for the formation of local ore concentrates are numerous, including:
-
Physical segregation and accumulation (e.g., Penrose 1910; Ewers and Hudson 1972; Tomkins 2010);
-
Structure and intersection of fissures as well as the physical character of the wall rock that may provide preferred pathways, offer advantageous space, or trap the metal-bearing solution/magma into favorable positions (e.g., Hulin 1929; Gough 1965; Stone and Archibald 2004; Páez et al. 2016);
-
Local mixing or influx of a further gaseous or liquid fluid into or phase separation of the ore-bearing solution/magma (e.g., Penrose 1910; Hulin 1929; Shepherd et al. 1991; Markl et al. 2016);
-
Chemical character of the wall rock (e.g., Hulin 1929; Shepherd et al. 1991; Squire et al. 2008; Kreissl et al. 2018);
-
Post-depositional remobilization by alteration and/or weathering (e.g., Tomkins et al. 2004; Collins et al. 2012; Fougerouse et al. 2016; Páez et al. 2016).
Most ore shoots are structurally controlled, for example directly or indirectly by preferred fluid pathways, but structure alone does not promote ore formation (Penrose 1910). Thus, not only one, but a combination of the above-named and further factors leads to the formation of ore shoots.
In this study, three ore shoot-bearing unconformity-related hydrothermal fluorite-quartz-barite vein systems in the Schwarzwald, SW Germany, are investigated to understand the formation of such ore shoots and the transition from normal barite-fluorite-sulfide veins to these native element- and arsenide-dominated ore shoots. In the following, we chose to label the fluorite-barite-carbonate vein type with galena, sphalerite, and chalcopyrite, which is very common in the Schwarzwald region and over the whole of Europe, as “normal,” knowing that there are many other vein types which could be equally designated like this. The ore shoots investigated here differ from many other ore shoots worldwide in that they show not only enrichment in ore minerals but also a significant change in ore mineralogy relative to the larger, hosting, hydrothermal vein system. Specifically, ore mineralogy changes from a common galena-sphalerite-chalcopyrite assemblage to an unusual native element + arsenide + sulfarsenide association, while the gangue minerals vary only in relative mineral abundance (Otto 1967; Brill et al. 2018; Steen 2020).
Such strongly contrasting mineral assemblages in hydrothermal veins are easily interpreted either to represent two different mineralization stages operating at different times or to represent a change in fluid composition and/or source in combination with a change in parameters such as pH, fO2, or temperature. As in some cases most of the economic value of a specific vein lies in bonanza-style ore shoots (Lindgren 1913; Guilbert and Park 2007; Gloyn-Jones and Kisters 2019), it is vital to understand the formation of these features in relation to the larger hydrothermal vein system.
Hydrothermal veins generally form by either a change in pressure, a change in temperature (mostly cooling), fluid-host rock reaction, fluid mixing, effervescence, boiling, or a combination of those (Barnes 1997). For the ore shoot-bearing main hydrothermal stage in the localities investigated in this study, Scharrer et al. (2021) concluded the veins to have formed by fluid mixing which agrees with many precursor studies on similar veins in the region (e.g., Baatartsogt et al. 2007; Fusswinkel et al. 2013; Walter et al. 2016).
The ore shoot assemblage studied strongly resembles typical features of hydrothermal native element-arsenide assemblages (Kissin 1992). These consist of dendritic native Ag and/or native Bi; zoned Ni-, Co-, and Fe-arsenides and sulfarsenides; and late-stage native As (Bastin 1939; Kissin 1988, 1992; Markl et al. 2016; Scharrer et al. 2019; Guilcher et al. submitted). The importance of the redox state during the formation of native element-arsenide associations has been assumed before (Robinson and Ohmoto 1973; Kissin 1993; Ondrus et al. 2003a), but Markl et al. (2016) first proposed an integrated model explaining all textural, mineralogical, and geochemical features of five-element associations. Several studies showed their formation to be directly linked to the process of reduction (Naumov et al. 1971a; Markl et al. 2016; Burisch et al. 2017a; Kreissl et al. 2018; Scharrer et al. 2019). Scharrer et al. (2019) proposed (but could not prove unequivocally) that the sole process of reduction suffices to account for the formation of native element-arsenide associations and that the rest of the fluid chemistry could remain unchanged. This hypothesis was based on the fact that parameters such as elemental abundance in the ore, formation temperature, salinity, and host rock can greatly vary between all native element-arsenide associations worldwide, while their conspicuous dendritic textures are ubiquitous and the typical mineralogical sequence of native Ag ± native Bi → Ni-arsenides → Co-arsenides → Fe-arsenides → native As is the same at almost all localities. Furthermore, this mineral sequence can be thermodynamically modeled by simple reduction (Scharrer et al. 2019).
All these studies, however, could not show that the ore shoots formed from exactly the same fluid (or mixed fluid) as the rest of the vein. In this respect, we are lucky, because the main difference between our ore shoots and many other worldwide occurrences of native element-arsenide assemblages is that the latter typically form as an individual ore stage that is succeeded by a sulfide-bearing stage, while in our study area, the normal sulfide-bearing assemblage forms prior to, during, and subsequent to the native element-arsenide–bearing ore shoots. This fact renders them ideal candidates to investigate the difference in physico-chemical conditions between the normal and the native element-arsenide assemblages and the geological and/or geochemical process behind the change from one to the other.
Regional geology
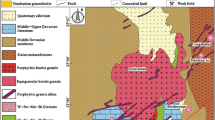
The three hydrothermal veins investigated in the present study include the former Teufelsgrund-Schindler-Herrenwald vein system in the Münstertal with Ag-As ore shoots, the former Gottesehre mine with associated parallel veins near Urberg with Ag–Ni-As ore shoots, and the Anton and Tannenboden veins near Wieden with Ag–Ni-As-Sb ore shoots. These three localities are the only ones in the Schwarzwald which show ore shoots rich in native arsenic and native silver and are described in detail in the subsequent chapter. The investigated vein systems are situated in the southern Schwarzwald, a Variscan basement area in SW Germany (Fig. 1a). The basement consists of mostly granitic and gneissic Variscan crystalline rocks which are discordantly overlain by a Mesozoic terrigenous to marine sedimentary cover (Geyer et al. 2011). This setting was disturbed during the opening of the Upper Rhine Graben with its normal fault-dominated regime (Geyer et al. 2011). Uplift of the rift flanks led to partial erosion of the sedimentary cover as well as part of the basement and resulted in the present-day topography: a mountain range with an elevation between 300 and 1500 m above sea level. Today’s outcropping surface in the southern Schwarzwald was originally situated about 2–3 km below the former basement-cover unconformity (Rupf and Nitsch 2008).

a Geological map of the Southern Schwarzwald in SW Germany with the investigated localities highlighted (modified from Scharrer et al. 2021; original geological map from LRGB GÜK300). 1: Teufelsgrund and Giftgrube mine, Münstertal; 2: Gottesehre mine, Urberg; 3: Anton and Tannenboden mine near Wieden. BLZ stands for Badenweiler-Lenzkirch zone, which represents an east–west striking Variscan suture zone filled with low-grade metamorphic clastic sediments. b Image of a typical banded vein by Matthias Zizelmann and c image of a granitic wall rock breccia which are typically cemented by sulfides and quartz. Both b and c are modified from Scharrer et al. (2021)
The preexisting geological, mineralogical, and geochemical literature on the Schwarzwald is vast and includes its geology (Geyer et al. 2011), stable and radiogenic isotope studies (e.g., Staude et al. 2011; Walter et al. 2015), mineralogical studies (e.g., Metz et al. 1957; Staude et al. 2007, 2010, 2012; Markl 2017a), geochronological studies (e.g., Pfaff et al. 2009; Walter et al. 2018b), fluid inclusion studies (e.g., Schwinn et al. 2006; Baatartsogt et al. 2007; Walter et al. 2016; Burisch et al. 2017b), and weathering studies (e.g., Keim et al. 2018). This solid geoscientific background helps in constraining the boundary conditions for the present study.
The region has a long-lasting history of hydrothermal activity which ranges from the Variscan Orogeny to recent times (Walter et al. 2018b). This hydrothermal activity produced more than 1000 unconformity-related, hydrothermal veins showing a large variability of mineral associations, of which some have been mined since pre-Roman times (Markl 2015, 2016, 2017a, 2017b). Walter et al. (2016) characterized five temporal maxima of hydrothermal mineralization based on the mineralogy, fluid characteristics, and age dating. Direct dating of individual minerals of the investigated vein systems by Walter et al. (2018b) showed that the ore shoot-bearing major hydrothermal stage lasted from the Triassic until the Early Cretaceous. This in combination with the fluid composition and vein mineralogy lets Scharrer et al. (2021) conclude that the investigated ore shoots and the associated main ore stage of these veins formed during the fourth of the five hydrothermal maxima in the Schwarzwald (see Walter et al. 2016, for the detailed terminology). Thus, this stage belongs to the Triassic-Cretaceous vein group (Walter et al. 2018b), commonly labeled Jurassic-Cretaceous fluorite-quartz-barite-carbonate vein group, which generally formed at temperatures between ~ 50 and 200 °C and salinities of 20–30 wt% NaCl + CaCl2 (Walter et al. 2016, 2017). Reactivation of these veins during the Upper Rhine Graben rifting produced other subsequent metal occurrences and deposits.
Location of the study
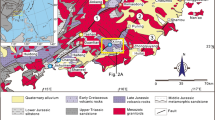
Each investigated vein system comprises several veins which are N-S and/or NE-SW striking and nearly vertically dipping. Individual veins are under 1 km, up to 2 km, and up to 5 km in length for Wieden (Zeschke 1959; Werner et al. 2020), Münstertal (Schürenberg 1950; Bliedtner 1978), and Urberg (Otto 1967; Brill et al. 2018), respectively. The veins consist of elongated, up to 100 m long lenses and vein sections with a highly variable width between 1 and 3.5 m (e.g., this study; Schürenberg 1950). The width is strongly dependent on the host rock, is wider in gneiss than in granite, and can locally decrease to only a few centimeters (this study; Metz et al. 1957; Otto 1964, 1967; Brill et al. 2018). On average, the veins consist of ~ 60 vol% fluorite, which, together with mainly quartz and barite, forms a banded structure (Fig. 1b) (Metz et al. 1957; Zeschke 1959; Otto 1964; Steen 2013; Markl 2017a; Brill et al. 2018). Sulfides occur as either local aggregates within the banded structure, or more commonly, are bound to host rock breccias (Fig. 1c). Each of the three vein systems hosted several ore shoots in the mined portions of the veins, at least three in the Münstertal, five at Wieden, and at least six at Urberg (Fig. 2). While the gangue mineralogy and mineral successions are fairly similar for the three vein systems (Metz et al. 1957), the ore shoots show an interesting variation from Ag-poor in the Münstertal to Sb-rich and Ni-bearing in Wieden and to Ag- and Ni-rich in Urberg. These ore shoots and/or vein sulfide accumulations are commonly spatially related to wider vein sections and brecciated zones and/or bound to the vein-host rock boundary (Fig. 1c) (Metz et al. 1957; Zeschke 1959; Grundmann 1971; Widemann 1988; Markl 2017a; Brill et al. 2018). Impregnations of the ore shoot mineralogy into the host rocks are very rare or absent (Zeschke 1959; Brill et al. 2018). The local host rocks of the veins at the three localities are mostly made up of gneisses with local enrichments of graphite and/or sulfides (Metz et al. 1957; Bliedtner 1978; Brill et al. 2018). Locally dikes of granitic rocks occur (Geyer et al. 2011). In addition to gneisses and granites, the region around Urberg also comprises local occurrences of diorites, amphibolites, and norites with associated magmatic Ni–Fe-sulfide lenses (Otto 1964; Brill et al. 2018).

a–c Maps and cross sections of the three investigated localities showing the ore shoot distribution. Ore shoot description shows mining date, characteristic mineralogy, and size. Depicted ore shoot information and location have been inferred from the literature (Grundmann 1971; Bliedtner 1978; Markl 2017a; Brill et al. 2018). Maps and ore shoot cross sections are modified from Brill et al. (2018) and Markl (2017a) and references therein
The veins in question were subject to various mining efforts dating back to medieval silver extraction and ending with fluorite extraction during the second half of the twentieth century (Markl 2004). In the twentieth century, the fluorite concentrate production totaled ~ 0.5 Mt and ~ 0.15 Mt for the Wieden and Urberg veins over a period of ~ 40 years and ~ 30 years, respectively (Markl 2017a). The calculated remaining reserves for Wieden were another 0.4 Mt at the time of mine closures in 1965 (Hauck 2020). Although fluorite concentrate was the main product, small amounts of barite concentrate and mixed sulfide concentrate were also recovered at Wieden in the twentieth century (Markl 2017a). The mixed sulfide concentrate was used to extract both silver and lead as minor byproducts, but the amounts were quite low (Markl 2017a). As an example, rough estimates indicate that the mined vein material without the ore shoots contained a total of ~ 20 t Ag at Urberg, while the ore shoots therein contained up to twice as much Ag.
All ore shoots have been mined out between 1960 and 1982 and are inaccessible today. Hence, the structural relations between vein and ore shoot can only be inferred from the literature (Schürenberg 1950; Otto 1967; Grundmann 1971; Bliedtner 1978; Brill et al. 2018; Steen 2020; Werner et al. 2020). Their mineralogical composition is based on collection material from the senior author and from the University of Tübingen, while additional microscopic images of samples from the Wieden vein system were provided by Günter Grundmann (1971) who had previously worked on the Wieden ore shoots. The hand specimens were carefully compared and inspected; representative samples of ore shoots and associated veins meters to tens of meters away from the ore shoots were selected for further investigations. In total, 38, 33, and 29 thick sections for microscopy and microprobe analytical work were prepared from Münstertal, Urberg, and Wieden, respectively.
Petrography of the mineralization
The paragenetic sequence of both the veins and the ore shoots is presented in Fig. 3 and augmented by literature data in Table 1.

Paragenetic sequence of the hydrothermal vein (a) and of the ore shoot mineralogy (b) with the corresponding schematic textural sketch provided in c. a & b Relative mineral abundance is indicated by line thickness, for which ore and gangue minerals are considered separately. Numbers in b and c represent the ore shoot sequences. The ages presented in a are taken from Walter et al. (2018b). Dotted arrows in b represent the typical replacement reactions observed in many samples
The hydrothermal veins
The schematic paragenetic sequence for each vein (Fig. 3a) was taken from Metz et al. (1957) and supplemented/revised according to own observations and to other authors, where reasonable (Schürenberg 1950; Zeschke 1959; Otto 1964, 1967; Bliedtner 1978; Steen 2013; Markl 2017a; Brill et al. 2018; Werner et al. 2020). The mineralization was initiated by a quartz-dominated stage (I) rich in Fe-sulfides and breccias. It shows initial pyrrhotite (not found at Urberg) which was subsequently replaced by pyrite and marcasite (Fig. 3a). The volumetrically most abundant stage is the fluorite-dominated second stage (II), which also contains variable amounts of quartz, barite, and carbonate. Barite content increases with time (Fig. 3a). Coarse-grained, massive carbonate is generally rare in stage II and only occurs in larger quantities in Urberg, where it formed prior to and during the transition to the subsequent fluorite-dominated substage of stage II. The fluorite-barite-quartz mixture forms a banded structure with variable proportions of the three minerals. The most abundant sulfide minerals within stage II of these veins are galena, sphalerite, and chalcopyrite, the last being the least common of the three (Metz et al. 1957; Steen 2013). The relative proportion of galena and sphalerite within individual thin sections ranges from solely galena to solely sphalerite and no typical sequence regarding the relative time of formation is visible, nor is there a systematic change from early to late within stage II. Tetrahedrite occurs only rarely and locally. Brecciated zones show an increasing abundance of sulfides and quartz—both minerals grow around clasts (this study; Metz et al. 1957; Zeschke 1959; Widemann 1988; Markl 2017a; Brill et al. 2018; Scharrer et al. 2021). There is no textural evidence for sulfide dissolution in the host rock during vein formation. This stage started > 200 Ma ago and lasted until about 75 Ma for the three localities studied here (Walter et al. 2018b). A less abundant, partial repetition of this stage with small variations forms stage III. The last stage (IV) is temporally and tectonically linked to the opening of the Upper Rhine Graben between ~ 22 and 8 Ma (Walter et al. 2018b). This stage is dominated by carbonates and forms discontinuously within the vein, mostly within vugs or small veinlets. In the later, generally fluorite-poorer, volumetrically minor hydrothermal stages III and IV, late-stage barite, quartz, and/or carbonates form together with rare galena, sphalerite, euhedral pyrite, and/or a colloform mixture of pyrite and marcasite.
As-(Ag)-ore shoots in the Münstertal
The Teufelsgrund-Schindler-Herrenwald vein system in the Münstertal comprises two ore shoot sequences which are distinctly different with respect to their gangue, but very similar concerning their ore minerals (Fig. 3b). The initial ore shoot (1) is characterized by the presence of abundant native As on (Fig. 4a) or within fluorite, commonly overgrown by fine-grained micro-crystalline quartz which successively increases in size and sometimes forms euhedral crystals in open spaces (Fig. 4a). The native As is also commonly associated with co-genetic galena (Fig. 4b, c). Less common is the association with sphalerite, which generally forms prior to native As. Fluorite co-precipitated with barite, quartz, galena, and native As (Fig. 4d, e), and the subhedral to euhedral barite was subsequently partially replaced by quartz (Fig. 4e). This texture is a common feature in many Schwarzwald veins (Burisch et al. 2017b). Rarely and locally confined sulfosalts such as minerals of the proustite-pyrargyrite series (Ag3AsS3-Ag3SbS3), freieslebenite (AgPbSbS3), argentopyrite (AgFe2S3), sternbergite (AgFe2S3), and boulangerite (Pb5Sb4S11) are present (Fig. 4c). These minerals generally form as reaction products from native As or galena either rimming galena, replacing native As aggregates (Fig. 4c) or as independent crystals on top of native As bands.

Textural images of ore shoot sequence 1 (a–g) and ore shoot sequence 2 (h & i). Reflected light images (a–f & i) and macro images (g & h). a Early cogenetic intergrowth of fluorite and galena with subsequent intergrowth of galena, native As, and fluorite. Late-stage quartz typically shows a coarsening from microcrystalline colloform quartz to palisade quartz. b Co-genetic intergrowth of galena and native As in which the latter has been dissolved during alteration. c Co-genetic intergrowths of commonly lath-shaped galena and native As in individual bands of the colloform native As. These textures occasionally show a replacement by Pb-sulfosalts such as boulangerite and freieslebenite. d Barite-rich samples show a co-genetic intergrowth between barite and native As, in which e barite is partially replaced by quartz during subsequent alteration. f & g Replacement of native As by quartz and rare barite. h & i Overgrowth and co-genetic growth of native As on carbonates and sulfides
The later ore shoot sequence (2) was identified in the Giftgrube mine ~ 1 km south of the Teufelsgrund mine (Fig. 2) (Bliedtner 1978) and in the Trudpert adit of the Teufelsgrund mine. It is characterized by the abundance of siderite. The common mineral sequence observed in these samples is early quartz with some fluorite overgrown by chalcopyrite, galena, and sphalerite, with the latter being most abundant. The base metal sulfides are, in turn, overgrown by siderite. During siderite formation, sphalerite and, less commonly, galena continued to form to a lesser extent. Co-genetic with these minerals, colloform native As (Fig. 4f, g) is abundantly present and can reach up to 60 vol% in some hand specimens (see also Bliedtner 1978). Later carbonates such as calcite and dolomite are not uncommon. Several repetitions of this mineral sequence have been observed even on a small scale.
As-Ag–Ni-Co-ore shoots at Urberg
The Gottesehre vein system near Urberg differs from the other two vein systems with respect to ore shoot abundance and the presence of an early pinkish calcite as one of the gangue minerals. Six different ore shoot pockets have been reported in the literature (Fig. 2) (Brill et al. 2018). The largest discovered ore shoot with dimensions of ~ 20 m × 20 m × 1 m was mined in 1962–1963 (Otto 1967). Five further (and smaller) ore shoots were discovered and mined in 1965, 1968, 1970, 1978, and 1982, respectively (Brill et al. 2018). The complete mineralogical ore shoot sequence (1–3; Fig. 3b) can only be found in samples of the first and largest ore shoot (this study; Otto 1967), and all later ones only show parts of the whole sequence. Individual samples commonly show a high variability in mineral abundance (this study; Brill et al. 2018). As an example, the 1968 ore shoot was mainly composed of native Ag, whereas the 1970 and 1978 ore shoots lacked native Ag and contained abundant arsenides (Brill et al. 2018). Furthermore, the 1970 and 1982 ore shoots showed signs of U enrichment recorded by the presence of secondary U-bearing minerals (Brill et al. 2018). The presence of U is also indicated by the characteristic dark purple color of fluorite of the 1982 ore shoot (Markl 2017a) which was in addition especially rich in niccolite. Although differences in modal amount and presence of minerals exist between ore shoots and samples, the overall mineral sequence (Fig. 3b) is identical in all of them.
The investigation of samples from all ore shoots shows three distinct mineral sequences. The earliest ore shoot sequence was initiated by dendritic aggregates of native Ag up to 10 cm in size and various textural forms of niccolite, ranging from subhedral or euhedral individual crystals to dendritic shapes. Interestingly, native Ag and niccolite are not intergrown, even in samples where they occur in close vicinity (< 1 mm). However, since native Ag is commonly isolated within the gangue minerals (Fig. 5a) and only sometimes overgrown by rammelsbergite (Fig. 5b), while niccolite is always overgrown by rammelsbergite (Fig. 5c), a slightly earlier formation of native Ag relative to niccolite may be inferred. The rammelsbergite is then overgrown by abundant skutterudite (Fig. 5d); at the transition, rammelsbergite and skutterudite zones alternate repetitively. Native Bi was only found as small flakes of less than 10 µm size included within arsenides of the first shoot sequence. These ore minerals are found in both colorless and dark purple fluorite and abundantly in fine- to medium-grained quartz. In these cases, the dark purple fluorite is almost exclusively associated with early-stage pinkish calcite. Skutterudite is rarely overgrown by native As (Fig. 5e) that can also incorporate relics of native Ag. Galena may be present at any point within this sequence but is generally more abundant during the second sequence (Fig. 5f) in which skutterudite is absent and fluorite is always colorless. Hydraulic brecciation (Jébrak 1997) of the first mineral sequence and cementation by subsequent minerals are typical.

Reflected light images (a–d & f) and macro image (e) of the Urberg locality. Native Ag is commonly found in dendritic form, either enclosed in the gangue mineral (a) or overgrown by arsenides (b). c & d The arsenide sequence is generally niccolite → rammelsbergite → skutterudite. d Skutterudite is commonly partially replaced by quartz. e The arsenides are, in turn, rarely overgrown by native As. Both native As and skutterudite are prone to remobilization (d & e). f Overgrowth of ore shoot 1 by ore shoot 2 and ore shoot 3, showing the change in mineralogy with time
In the second sequence, native Ag is overgrown by rammelsbergite with abundant galena (Fig. 5f).
In the third sequence, dendritic niccolite is overgrown by rammelsbergite and then by colloidal gersdorffite (Fig. 5f). These aggregates are occasionally overgrown by galena or colloform pyrite. Contemporaneous with and subsequent to the gersdorffite, barite could form, which, in turn, was commonly replaced by later quartz (Fig. 5f).
As-Ag-(Ni)-Sb-ore shoots at Wieden
A previous descriptive study (Grundmann 1971) reported the occurrence of two spatially separated ore shoots with two types of mineral assemblages in the Tannenboden vein near Wieden: an Sb-Ag-rich one mined in 1964/1965 and an As-Ag-rich one mined in 1966 (see Fig. 2c). Similar ore shoots were also found in the Anton, Spitzdobel, and Neue Hoffnung veins of the Wieden vein system (Steen 2020). Although the mineralogy varies between ore shoots and samples, the overall mineralogical sequence does not. Thus, for Wieden, each sample represents only a portion of the whole ore shoot mineralogical sequence. The samples investigated in this study, in combination with a revisited textural analysis of the original samples from the study of Grundmann (1971), let us conclude that the two ore shoot assemblages reported by Grundmann (1971) represent an earlier and a later stage within a continuous mineral sequence (see types 1 and 2 in Fig. 3b, c). It is important to note that many of the smaller ore shoots lacked the presence of arsenides, antimonides, and native antimony (Markl 2017a).
The Wieden vein system ore shoots are characterized by an abundance of native Ag, native As, and Ag-Sb-sulfosalts. The latter formed during the replacement of the primary ore shoot minerals. The initial and locally dominant ore mineral native Ag (Fig. 6a, b) occurs in a variety of dendritic shapes. Rarely it is overgrown by breithauptite (Fig. 6a, b), while rammelsbergite (Fig. 6c, d) is more common. The antimonide formed prior to the arsenide, but this sequence could be repeated several times (Fig. 6b). Intertwined intergrowth between native Ag and rammelsbergite occurs occasionally (Fig. 6e, f), especially in close spatial association with native As. Skutterudite was identified at this locality, but due to its rarity and due to the lack of suitable textures, its relative time of formation could not be ascertained. The replacement of native Ag by minerals of the pyrargyrite-proustite series and, to a lesser extent, also by other sulfosalts (see Table 1) is ubiquitous in all samples, and only the degree of replacement varies (Fig. 6d, f). Native As encloses intergrowths between rammelsbergite and minerals of the pyrargyrite-proustite series (originally native Ag ± native As) which implies a later formation of native As relative to rammelsbergite and native Ag (Fig. 6e, f). The native As is commonly intergrown with galena, showing a colloform-banded structure (Fig. 6g, h). Galena commonly forms elongated needles oriented perpendicular to the growth direction or monomineralic bands within the colloform structure. Sphalerite does not show these textures but more commonly forms euhedral crystals. Individual subhedral to euhedral crystals of native As are only visible in rare instances on the outermost zones of the native As aggregates. Not only quartz, but also native Sb rimmed by dyscrasite (As3Sb; Fig. 6g) can be found as primary cores within the native As aggregates. As in the Münstertal, native As and barite co-exist without a reaction boundary. The native As is overgrown by a large variety of different sulfides and sulfosalts, which include the base metal sulfides, tetrahedrite, minerals of the pyrargyrite-proustite series, xanthoconite, argentopyrite, sternbergite, and late-stage stephanite and polybasite. However, these sulfosalts more typically form as crystal aggregates together with quartz pseudomorphic after native Ag. They also replace individual layers or the cores of native As. The latter are interpreted as replacement textures of dyscrasite cores by sulfosalts (Fig. 6h, i).

Reflected light images (a–f) of the Wieden locality. a & b Native Ag is intergrown with and overgrown by breithauptite and galena, partial oscillatory repetition of these minerals with the addition of later rammelsbergite. c & d Common texture of dendritic native As being directly overgrown by rammelsbergite. e & f More intergrown dendritic native Ag with rammelsbergite, overgrown by native As. g–i Typical layered intergrowth between galena and native As with discrete sphalerite grains and seldom preserved cores of dyscrasite. Most cores and some native As–rich layers (h & i) as well as native Ag (d–f) are replaced by sulfosalts such as argentopyrite, minerals of the proustite-pyrargyrite series, and sometimes sphalerite or galena. These minerals are otherwise found as overgrowth on the primary ore shoot minerals (g & i). Images g and i have been kindly made available by Dr. Günter Grundmann on www.mindat.org (Photo ID: 1,038,791 and 881,932)
Supergene remobilization
A typical supergene alteration phenomenon in all of these ore shoots is the gradual dissolution of the native As along compositionally and texturally defined zones. Furthermore, the Ag-bearing sulfosalts are successively replaced by Ag-richer sulfides and finally by plates or wires of native Ag. The latter are also found in vugs on carbonates or quartz as well as in negative imprints of native As, produced by dissolution. These reactions are typically accompanied by the formation of other secondary minerals such as oxides, arsenates, and carbonates. They are not considered here further.
Methods
Mineral analyses
Scanning electron microscope and electron microprobe
The Hitachi TM3030 SEM Plus Tabletop Microscope in combination with reflected light microscopy at the University of Tübingen was used as a qualitative mineral identification and textural visualization tool. For quantitative mineral analysis in 31 thin sections, the JEOL SuperProbe JXA—8900RL at the University of Tübingen was used. To avoid the tarnishing of arsenic, the thin sections were re-polished, cleaned with ethanol, and dried at room temperature in a dry box for half a day before being sputtered with carbon. Analytical condition details and reference materials are described in ESM 1.
Special notice was given to the Lα line used for the analysis of As. Using the ZAF or φ(ρz) correction method yields vastly different results (up to ~ 10 wt% difference in As content). The latter provides acceptable results for As contents > 70 wt%, while the ZAF correction yields acceptable results for As contents < 55 wt%. For comparability, monoarsenide, diarsenide, and sulfarsenide were corrected using the ZAF correction method, while the triarsenides and native As were corrected using the φ(ρz) correction method. To forego this problem in general, we suggest using the Kα line instead.
Sulfur isotopes
Sulfur isotope samples were carefully handpicked and separated into monomineralic, preferably co-genetic samples of sphalerite, galena, chalcopyrite, pyrite, gersdorffite, proustite, and barite. The S isotope compositions were measured with He carrier gas and a Carlo Erba (CE 1100) elemental analyzer linked to a Thermo Fischer Delta V mass spectrometer at Lausanne University, Switzerland. Samples were reacted at 1050 °C in a stream of He carrier gas spiked with oxygen gas. External reproducibility of standards was better than 0.15‰, and samples were calibrated against IAEA standards S1 and S3 (Ag2S) and NBS-127 (BaSO4) with accepted values of − 0.3‰, − 32.1‰, and 20.3‰ δ34S (V-CDT), respectively.
Thermodynamic modeling
Thermodynamic modeling was done using the Geochemist’s Workbench 14 software bundle (Bethke 2007). Reaction path modeling was performed using the application react and predominance, and stability diagrams were produced using the application Phase2. The Thermoddem thermodynamic database (version June 2017) (Blanc et al. 2012) was used, and our mineral additions to it are presented in Table 2. Minerals for which Δf H0, S0, and cp(T) are known or estimated were implemented by the Thermoddem team to keep internal consistency as high as possible. Mineral additions include trechmannite, proustite, mirargyrite, pyrargyrite, rammelsbergite, dyscrasite, allargentum, and gersdorffite. In addition to minerals, the database was also extended to include Ni-chloride complexes and more aqueous Sb species. NiCl+, NiCl2 (aq), and NiCl3− from Liu et al. (2012) were implemented using the Ryzhenko parameters and the Thermoddem database temperature-dependent dissociation of H2O to further increase internal consistency. The added aqueous Sb species were taken from Obolensky et al. (2007) and augmented by the compilation of Bessinger and Apps (2003).
Results
Mineral analysis
Scanning electron microscope and electron microprobe
Representative analyses can be found in Table 3, and the entire dataset is presented in ESM 2. In the following, selected compositional features of each mineral will be discussed.
Native As has a variable composition, which can differ distinctly between localities, samples, and individual bands within colloform arsenic aggregates (ESM 3). These differences mainly concern the elements Ag, Pb, Sb, S, and Se which reach contents of up to 7.27 wt%, 7.49 wt%, 7.89 wt%, 3.04 wt%, and 0.56 wt% with an average of 1.32 wt%, 1.14 wt%, 1.87 wt%, 0.98 wt%, and 0.35 wt%, respectively. There is a negative correlation between As and these elements, generated by a typical gradual trend of decreasing elemental content within individual bands from the center to the outer margin of colloform arsenic aggregates (ESM 3). In some bands, inhomogeneities on the µm scale are visible. These could represent co-genetic or exsolved micrograins of Ag–S-rich minerals (ESM 3). Based on the Ag–S ratio (ESM 3), the presence of Sb, and the absence of other elements, the microinclusions seem to represent either Ag3Sb4S6 (aramayoit) or Ag3(As,Sb)S3 (e.g., proustite-pyrargyrite). However, these are only present in some individual bands, commonly linked to lower Sb contents in the surrounding native As, and it is not suggested that microinclusions are the reason for the high contents of minor elements in native arsenic in the continuously banded regions (ESM 3). Rather, these elements appear to represent real minor elements substituted into the crystal structure of native arsenic. Similar contents have been reported from other localities by George et al. (2015).
Native Ag shows a clear compositional distinction between primary dendritic silver formed co-genetically with the ore shoot minerals and remobilized platy or wire silver. The latter has significantly lower trace element contents, commonly below the detection limit of the electron microprobe. Average trace element contents of the primary silver are 0.25 wt% Sb, 0.36 wt% As, 0.10 wt% S, and 1.25 wt% Hg (ESM 3). In rare patchy zonations of native Ag of the first ore shoot sequence at Urberg, Hg may locally reach up to 14 wt% (ESM 3).
Due to the small size of native Bi, mixed analyses with the surrounding rammelsbergite cannot be excluded (Table 3). However, the analyzed compositional variation among the six analyses is fairly low and the molar Ni/As ratio is about 8 and not 0.5 as in rammelsbergite. The Ni and As contents in native Bi are 1.7 ± 0.3 wt% and 0.27 ± 0.04 wt%, respectively.
The arsenides, sulfarsenides, antimonides, and sulfantimonides are mostly Ni-dominated with average molar Ni/(Ni + Co + Fe) ratios of 0.79 for skutterudite, 0.80 for diarsenides, 0.995 for niccolite, 0.92 for gersdorffite, 0.993 for breithauptite, and 0.997 for ullmannite (Fig. 7a). Thus, the diarsenides are almost exclusively of rammelsbergite composition. This excludes one outlier sample from Wieden that contains diarsenides of the safflorite-loellingite series. These are associated only with quartz and therefore cannot be texturally correlated with the other minerals. The arsenides and antimonides incorporate sulfur to various degrees (Fig. 7b). This is especially visible for gersdorffite, which ranges from 0.31 to 0.95 atom per formula unit sulfur. Furthermore, the degree of As-S exchange varies with time at the Urberg locality, and thus, an increase in sulfur relative to arsenic is visible with progressing ore shoot sequences (Fig. 7b). Bismuth was detected in niccolite and shows an average content of ~ 1.4 wt%. Selenium is present in all arsenides and antimonides and reaches up to ~ 0.4 wt%. The arsenides and sulfarsenides typically show some degree of Sb-As exchange. For example, arsenides of the Wieden locality, which is also host to co-genetic antimonides, show the highest contents of antimony, reaching up to ~ 10 wt% (Fig. 7b).

Compositional variations of the arsenides and antimonides. a Most arsenides and antimonides are Ni-dominated, with the monoarsenides and monoantimonides showing the least spread with respect to Ni, Co, and Fe. b Compositional distribution with respect to sulfur content and As/(As + Sb) ratio within all samples and between different ore shoot sequences of Urberg. The distribution between the As- and Sb-endmembers shows the sole presence of arsenides for Urberg and the presence of both with a partial solid solution for Wieden. Arsenopyrite is excluded in this figure
Galena is the most common base metal sulfide, contains up to 2 wt% of silver, and has an average formula of Pb0.99Ag0.01Sb0.01S0.99. Sphalerite is more variable in composition, and the molar Fe/(Fe + Zn) ratio ranges from ~ 0.001 to 0.13. It is manganese-poor and the cadmium content is below 1 wt% with an average of ~ 0.3 wt%. Analyzed pyrite reveals a high arsenic content of on average 4.5 wt% and a trace elemental composition of on average ~ 0.5 wt% Ag, 0.3 wt% Sb, and 0.06 wt% Co. The colloform pyrite variety also contains up to 2.5 wt% Pb, which may be related to microinclusions of galena that could not be resolved.
Although the primary mineralogy of the ore shoots is dominated by arsenic-rich endmembers, most sulfosalts that form during remobilizing processes are, in fact, Sb-dominated (ESM 4). This includes diaphorite, freieslebenite, tetrahedrite, and bournonite with molar As/(As + Sb) ratios of less than 0.4. Minerals of the proustite-pyrargyrite solid solution series show the presence of both As- and Sb-rich minerals, ranging from the pure As-endmember to a molar As/(As + Sb) ratio of ~ 0.1. The Sb-rich pyrargyrite forms independent of texture, even if it directly replaces native As and no primary Sb-minerals are present. The only As-dominated sulfosalt besides proustite is argentopyrite with an average formula of Ag0.94Fe2.02As0.01S3.02 and an average molar As/(As + Sb) ratio of 0.997.
Sulfur isotopes
The minerals selected for sulfur isotope compositional analyses were chosen to cover (I) all localities, (II) all ore shoot mineral assemblages, (III) co-genetic barite and sulfides or sulfarsenides at each locality, and (IV) sulfides and barite from the hydrothermal veins far away from the ore shoots. All analyzed samples and respective values are presented in ESM 5 and, together with calculated equilibrium temperatures for co-genetic sulfide-barite pairs, in Fig. 8.

Isotopic signature of vein and ore shoot sulfates, sulfides, and sulfosalts. Texturally classified co-genetic sulfate-sulfide pairs are linked by a dashed line and the respective calculated equilibrium temperatures are given in °C. Possible regional sulfate and sulfide source, with the present-day aquifer isotopic range depicted by blue arrows and sulfate-/sulfide-mineral isotopic ranges depicted by black arrows. Literature data from Müller et al. (1966), Siewers (1977), and Rick (1990) of possible sulfate and sulfide sources reveal the large overlap between the sedimentary sulfate for sulfate and the basement sulfides for the sulfides
The δ34S (V-CDT) of barite ranges from ~ 10 to 20‰, whereas the sulfides show more negative values, below 0‰ δ34S (V-CDT). Only the late-stage colloform pyrite commonly associated with barite from Münstertal and Wieden reaches strongly depleted δ34S values of − 27.8 to − 36.2‰ (ESM 5), while the other sulfides are in the range from 0 to − 10‰ δ34S (V-CDT). The weighted δ34S (V-CDT) average including all localities is − 6.8‰ for galena, − 3.6‰ for sphalerite, and − 5.3‰ for chalcopyrite. Proustite and gersdorffite lie in the range of these sulfides.
Considering variations between the samples, there is no recognizable difference between the ore shoots and the rest of the hydrothermal system (Fig. 8): S isotope values of sulfides in the ore shoots and in the veins, and of barite in the ore shoots and in the veins are very similar to each other. The only exception is one barite analysis of the first ore shoot sequence of the Münstertal, which is more than 5‰ lower than barite from the vein.
In samples with co-genetic sulfide and sulfate, sulfate-sulfide equilibrium temperatures were calculated. These are presented in Fig. 8 and result in temperatures between 270 and 480 °C (excluding pyrite) for all three localities and independent of ore shoot or vein. In view of the fluid inclusion constraints (see below), these temperatures are considered unrealistic and the S-bearing phases were obviously not in isotopic equilibrium. This will be discussed below. The late-stage colloform pyrite, which generally occurs as layers within or on top of barite, indicates an equilibrium temperature of 110 °C.
Fluid constraints
Fluid composition and temperature
Extensive fluid inclusion investigations of more than 1600 individual fluid inclusions from both the ore shoots and the veins of all three localities are presented by Scharrer et al. (2021). The most important aspects of this work will be repeated here with a specific focus on the variations between the ore shoots and the vein and the range of trace element composition. The fluid composition was determined by microthermometry (Fig. 9) and LA-ICP-MS (Fig. 10) on individual fluid inclusions in fluorite, quartz, and carbonate. Fluid inclusion assemblages were characterized as primary, clusters, and secondary according to the classification of Goldstein and Reynolds (1994) and Walter et al. (2015). In addition, fluid inclusion assemblages were grouped into those co-genetic with the primary minerals of the ore shoots and those co-genetic with the common vein association. The dataset by Scharrer et al. (2021) includes ~ 1650 fluid inclusions analyzed by microthermometry that were chosen based on both spatial and temporal coverage of the main hydrothermal stage of the investigated veins. Furthermore, if available, fluid inclusion assemblages in fluorite that formed contemporaneously with minerals, in which fluid inclusions could not be measured, were preferentially analyzed (Scharrer et al. 2021). This includes early calcite for the Urberg samples and sulfides, arsenides, and native elements for all samples.

The binary fluid mixing of fluid A and fluid B is visible for both the ore shoot (colored) and the vein-hosted fluid inclusions (gray) for each of the three investigated localities. Furthermore, no change in homogenization temperature is visible between vein and ore shoot assemblage. Data from Scharrer et al. (2021)

Selected transition metals and As, Sb, and S trace element composition of fluid inclusions of both the vein and the ore shoots, as measured by LA-ICP-MS. Blue fields are estimated realistic ranges of the fluid composition. These estimations are based on detected elemental abundance (circle symbols), detection limit of those analyses for which the analysis was below the detection limit and thus quantification was not possible (line symbols). The number of analyses (in brackets) does not always reflect the number of symbols, as some elements could not be quantified for some analyses. Data from Scharrer et al. (2021). No significant difference between vein and ore shoot could be identified
Both major (Fig. 9) and some minor elements (Scharrer et al. 2021) in fluid inclusions show a linear correlation in samples from all three localities, suggesting a mixing process between two endmember fluids. These endmember fluids both showed a high salinity (20–25 wt% NaCl aq.); fluid A was rich in CaCl2, whereas fluid B was NaCl-dominated and richer in KCl (Table 4). The approximate position of endmember A’ in the NaCl-CaCl2 space presented in Fig. 9 is estimated by extrapolation of the KCl content to near 0. There is no statistical difference between fluids A and B regarding homogenization temperature and most trace elements, such as As, Bi, Sb, and S, and the transition metals. The homogenization temperature (in °C) varies between localities and is 90–150 (average: 122 ± 8), 100–170 (average: 131 ± 14), and 80–150 (average: 117 ± 12) for the Münstertal, Urberg, and Wieden vein systems, respectively (Fig. 9). Pressure-corrected, this results in formation temperatures (in °C) of 110–170 (average: 140), 110–180 (average: 140), and 100–170 (average: 140), respectively. These values perfectly agree with many earlier values in similar veins from the region reported in, e.g., Baatartsogt et al. (2007) and Walter et al. (2017).
The trace elemental abundance typically shows a large spread of commonly one or more orders of magnitude (Fig. 10). This can be attributed to either inhomogeneities in the primary fluid composition or, more likely, evidence for prior mineral precipitation (Scharrer et al. 2021). Due to the high detection limit and uncertainties of LA-ICP-MS fluid inclusion analyses in fluorite (Scharrer et al. 2021), it is vital to consider both the analyzed content and the detection limit for those analyses below the detection limit (Fig. 10).
For subsequent thermodynamic modeling, rounded maximum analyzed concentrations from LA-ICP-MS analyses were used. The Pb content was rounded up to 100 mg/kg to represent both Pb (up to 40 mg/kg) and Zn (up to 80 mg/kg). Such a simplification is possible due to the similar behavior of the two elements (e.g., Barrett and Anderson 1988). To reduce an obscuring complexity in the modeled diagrams, the number of components was reduced during modeling. To better depict the differences in mineralogy, Ni was assumed to be 0 for the Münstertal and Sb to be 0 for the Münstertal and Urberg localities. The summary of the analyzed fluid compositions and the fluid composition used for thermodynamic modeling is given in Table 4.
Aqueous species
The implementation of all essential and available aqueous thermodynamic species is a vital step for thermodynamic modeling. The aqueous species calculated for the established fluid composition (see section above and Table 4) at the temperature of vein formation (ca. 150 °C based on fluid inclusion data of Scharrer et al. 2021) are depicted in ESM 6. Although the Cl content of the fluid is high, the non-metal Cl complexes for arsenic and antimony are mostly confined to an acidic pH. Therefore, the most prominent species are hydroxide complexes. For As and for Sb, the H3AsO3 and the H3SbO3 complexes are the most prominent under hydrothermal, moderately reduced conditions, and a roughly neutral pH, which reflect a crustal environment. This is in accordance with Marini and Accornero (2007) for As. At more oxidizing conditions, arsenite complexes become more prominent. Unfortunately, the necessary thermodynamic data for some antimonite complexes are still lacking. The metal species show the importance of Cl complexes that are stable under a wide pH range, up to a pH of ~ 8–9.
Discussion
Gangue mineral formation (simple binary fluid mixing)
To understand the formation of the ore shoots themselves, it is crucial to first understand the formation of the normal hydrothermal veins that host them. Their petrogenesis in the Schwarzwald region was the subject of several previous contributions (e.g., Schwinn et al. 2006; Baatartsogt et al. 2007; Staude et al. 2011; Bons et al. 2014; Walter et al. 2016, 2018b, 2019). Furthermore, a recent study (Scharrer et al. 2021) focused specifically on the three vein systems of the current contribution. The previous studies showed that the vein stage hosting the ore shoots under consideration here belongs to a large group of so-called Jurassic-Cretaceous fluorite-quartz-barite-carbonate veins that formed from the late Triassic to the early Tertiary (Walter et al. 2016, 2018b; Scharrer et al. 2021). Gangue mineral precipitation occurred due to mixing of fluid A, a CaCl2-dominated, KCl-poor, deeper-seated, modified bittern brine with a salinity of ~ 25 wt% CaCl2 + NaCl, and fluid B, a NaCl-dominated and KCl-richer bittern or halite dissolution brine situated at shallower depths in the crystalline basement with salinities of ~ 22 wt% NaCl + CaCl2 (Scharrer et al. 2021). Simple binary fluid mixing, likely in combination with various degrees of fluid cooling, has resulted in the precipitation of gangue minerals (e.g., Schwinn et al. 2006; Baatartsogt et al. 2007; Walter et al. 2017; Scharrer et al. 2021). The sulfides in this normal vein type consist mainly of galena, sphalerite, and chalcopyrite. Their formation will be discussed in more below, together with the formation of sulfides in the arsenide-dominated ore shoots.
Formation of the main ore shoot mineralogy without the sulfides by reduction
The native As–bearing ore shoot mineral assemblage is quite different from the mineral association in the normal hydrothermal veins. The ore shoots can be regarded as different entities within the same vein structure that formed contemporaneously, but locally and temporally confined within the vein.
Formation mechanism
In general, the stabilities of hydrothermal mineral assemblages are defined by parameters such as pressure, temperature, fluid composition, redox, and pH at the time of formation. Hence, a change in one of these parameters should govern the change from the normal vein assemblage to an ore shoot assemblage.
As veins and ore shoots formed contemporaneously, a pressure change can be ruled out. Furthermore, our comprehensive fluid inclusion study showed no involvement of fluid boiling either in the veins or in the ore shoots (Scharrer et al. 2021). In addition, fluid inclusion homogenization temperatures of both the ore shoots and the rest of the vein cover the same range (Fig. 9) and, hence, also exclude changes in temperature to be responsible for the change from vein to ore shoot precipitation. The fluid inclusion work by Scharrer et al. (2021) also shows no systematic difference between the fluid mixture which formed the normal vein and that which formed the ore shoots with respect to their major (Fig. 9) or minor (Fig. 10) element composition. Furthermore, Fig. 9 provides clear evidence that both fluid A and fluid B, whose mixing formed the normal vein assemblage, were present not only during vein but also during ore shoot formation. A change in fluid composition or the involvement of a volumetrically major and compositionally different third fluid involved in the formation of the ore shoots can, thus, be excluded.
Based on the line of arguments above, the process of fluid mixing was still present during ore shoot formation. The fact that the normal vein assemblage is also present in the ore shoots corroborates this statement. On the other hand, native elements and/or arsenides exclusively formed in the ore shoots which call for a change in redox and or pH. Although pH is crucial in defining which aqueous and mineral species are stable, a change in pH does not suffice to explain the formation of these ore shoots (Fig. 11). For example, an increase or decrease in pH from roughly neutral starting conditions does not result in the formation of native As, in the consecutive formation of niccolite and rammelsbergite, or of native Ag followed by native As (see Figs. 12, 13, and 14) which are so strikingly present in our samples (Figs. 3, 4, 5 and 6). Based on these arguments, we conclude that normal vein mineralization and ore shoots formed from the same mixing process of the same two fluids, but that an additional reducing agent must have taken part in ore shoot formation. This is in agreement with earlier work of, e.g., Markl et al. (2016), Burisch et al. (2017a), Kreissl et al. (2018), and Scharrer et al. (2019), but it is a significant and important step forward, as for the first time, it is shown that identical fluids can produce normal veins and five-element ore shoots, just by adding a reducing agent.

Thermodynamic model of the redox-pH relationship of various minerals. For the sake of clarity, the stability region of native Ag is not completely filled. Its shaded area is only depicted as a rim. a The solubility of sulfur is depicted in an equilibrium system considering the rounded typical Ba and Pb content of the vein fluids. b Simplified mineral relations in the Ni-Ag-Pb-As-S system considering the typical fluid composition of Urberg as an example. Both native Ag and Ni-minerals (such as Ni-arsenides, Ni-sulfarsenide, Ni-sulfides, and native Ni) form at more oxidizing environments than galena. c Comparison between fluid inclusion based formation temperatures and calculated sulfide-sulfate isotopic equilibrium temperatures

Thermodynamic modeling of (Ag)-As ore shoots of the Münstertal locality. For the sake of clarity, the stability region of native Ag is not completely filled. Their shaded area, by diagonal and horizontal lines, respectively, is only depicted as a rim. Fluid composition is taken from Table 4, and a represents a sulfur-free system, b represents a sulfur poor system with sulfate-sulfide equilibrium, and c represents a sulfur-poor system with sulfate-sulfide disequilibrium as is the case at hand

Thermodynamic modeling of Ag–Ni-As ore shoots of the Urberg locality. For the sake of clarity, the stability region of native Ag is not completely filled. Their shaded area, by diagonal and horizontal lines, respectively, is only depicted as a rim. Fluid composition is taken from Table 4, and a represents a sulfur-free system, b represents a sulfur poor system with sulfate-sulfide equilibrium, and c represents a sulfur-poor system with sulfate-sulfide disequilibrium as is the case at hand

Thermodynamic modeling of Ag–Ni-Sb-As ore shoots of the Wieden locality. For the sake of clarity, the stability region of native Ag is not completely filled. Their shaded area, by diagonal and horizontal lines, respectively, is only depicted as a rim. Fluid composition is taken from Table 4, and a represents a sulfur-free system, b represents a sulfur poor system with sulfate-sulfide equilibrium, and c represents a sulfur-poor system with sulfate-sulfide disequilibrium as is the case at hand
Type and source of the reducing agent
In the following, possible reducing agents for the investigated ore shoot assemblages will be discussed. Solid reducing agents such as Fe2+-bearing minerals or graphite have been proposed as reducing agents for native element-arsenide associations (Robinson and Ohmoto 1973; Kissin 1993; Ondrus et al. 2003a; Kreissl et al. 2018). However, they are unlikely to have been involved here, because neither relics of such phases nor reaction textures involving such phases were observed in our samples. Instead, both graphite and Fe2+ minerals like biotite are present in the host rocks and there is no spatial correlation between them and the ore shoots. Furthermore, they are distributed throughout most of the host rocks (graphite only in the gneiss) which would not explain the very localized occurrence of the ore shoots.
This leaves as possible agents mobile species such as gases, liquids, or aqueous compounds, and specifically mobile hydrocarbons or possibly H2. Scharrer et al. (2021) did indeed find H2 and rarely minor amounts of hydrocarbons in the gaseous phase of the fluid inclusions by Raman spectroscopy at room temperature. The small amounts of such gases are not surprising, because as long as the reducing agent is the limiting factor, it will be completely consumed, while the oxidant, e.g., arsenite or arsenate, would only be partially consumed. Thus, it is very rare to find mobile reducing agents as they typically completely react as long as oxidized aqueous arsenic species are present. Kerrich et al. (1986), Essarraj et al. (2005), Levresse et al. (2016), and Burisch et al. (2017a) report such findings, which proves that hydrocarbons are viable reducing agents to produce native element-arsenide assemblages.
The source of such hydrocarbons, however, remains somewhat enigmatic. One potential source of hydrocarbons are rocks of the paleo subduction zone of the Badenweiler-Lenzkirch (BLZ) suture, which today outcrops ~ 5 km south of the Münstertal and Wieden localities and which dips towards the north (Huth and Zedler 2019). The rocks of this suture zone comprise, among others, greywackes and marine clay stones metamorphosed only at very low grades (Huth and Zedler 2019) which contain small coal occurrences (Geyer et al. 2011) and, hence, potentially also hydrocarbons. On the other hand, the Urberg vein system is south of the BLZ and, hence, cannot be related to it.
Other hydrocarbon-bearing lithologies in the region are, for example, organic-rich shales in the (partially eroded) sedimentary cover. If the sedimentary cover units acted as a hydrocarbon source, these buoyant substances must have migrated into the underlying basement to depths of several kilometers. Burisch et al. (2017a) described a reasonable scenario based on the model of Bons et al. (2014) and how sedimentary brines are sucked into the crust (Stober and Bucher 2004) and thus can transport hydrocarbons dissolved in aqueous fluids to greater depths. This process could explain the presence of hydrocarbons at all three localities.
The overlying sedimentary cover being the origin of the reducing agent requires the hydrocarbons to be present in an aqueous solution; gaseous or liquid hydrocarbons would be too buoyant. A mass balance calculation shows that in order to precipitate all of the 100–1000 mg/kg aqueous As as native As according to.
As(OH)3 (aq) + 0.375 CH4 (aq) ↔ As (s) + 2.25 H2O + 0.375 CO2 (aq) (1).
A total amount of only 0.5–5.0 mmol CH4 (aq) per kg fluid would be required. This amount is in agreement with the influx of ~ 1.5–1.9 mmol/l CH4 (aq) determined for the ore shoots of the Wittichen locality in the central Schwarzwald by Markl et al. (2016). An aqueous fluid at 150 °C at a lithostatic formation pressure of 250–550 bar (Scharrer et al. 2021) can transport up to ~ 60–100 mmol/kg CH4 (aq) (Duan and Mao 2006). The near-linear mixing between fluids A and B during ore shoot formation (Fig. 9) limits the relative amount of the reducing agent–bearing aqueous fluid which could take part in the mixing process without disturbing the linear binary mixing trend. If the reducing fluid was less than 5% of (fluid A + fluid B), a maximum of 3–5 mmol of CH4 (aq) would have been available for reduction of 1 kg (fluid A + fluid B), which is in good agreement with what is required.
Mineralogical variability and fluid composition
The mineralogical differences between the three investigated localities are attributed to a difference in the respective fluid’s metal content. This hypothesis is supported by LA-ICP-MS fluid inclusion analyses which show variations between the localities (Fig. 10) (Scharrer et al. 2021). The different ore shoot mineral assemblages at Münstertal (Fig. 12), Urberg (Fig. 13), and Wieden (Fig. 14) can be nicely modeled using these different metal contents (Table 4).
Before discussing the calculated diagrams, it is important to note that these thermodynamic models also show the importance of pH in defining the mineralogy. Hence, pH and other minor, local fluid variations are sufficient to explain the differences in mineralogy between various ore shoots at one specific locality.
In the simple Ag-As fluid system, as is present at the Münstertal, the only two minerals stable at moderate to reduced epithermal conditions are native Ag and native As. Under all but lower than slightly acidic pH conditions, two pH units below the neutral pH, they form at different redox states (Fig. 12a). During a fluid reduction of a neutral fluid, native Ag would precipitate first and would therefore form inclusions in native As (Fig. 12a). This is in accordance with our observations and with many observations worldwide: dendritic native Ag occurs within typically collomorphic native As (e.g., Hösel 2003; Ondrus et al. 2003b; Hiller and Schuppan 2008; Pekov et al. 2010).
In the case of Urberg, the availability of Ni in addition to As and Ag stabilizes Ni-arsenides. Due to the abundance of As relative to Ni, the reduction sequence at neutral pH is as follows: native Ag → niccolite → rammelsbergite → skutterudite → native As (Fig. 13a). This finding is congruent with that of Scharrer et al. (2019). Due to the lack of thermodynamic data, the stability field of NiAs3 could not be empirically modeled. However, a predicted NiAs2-NiAs3 stability boundary field is shown in Fig. 13a. The exact position of this and, thus, the formation of either NiAs2 or NiAs3 are strongly dependent on the As/Ni ratio of the fluid, the pH, and the redox conditions. At higher As/Ni ratios, lower pH, and more reducing conditions, the triarsenide is more stable than the diarsenide. One of these factors is most likely the reason for the abundance of triarsenides in ore shoot sequence 1 and the increased abundance of diarsenide in ore shoot sequence 2 at Urberg. The abundance of Ni in the Urberg fluids can be explained by the spatially close occurrence of mafic rocks and magmatic Ni–sulfide deposits in the region around Urberg (Otto 1964; Geyer et al. 2011; Markl 2017a; Brill et al. 2018); these rocks are very likely the source of the Ni in the Urberg fluids.
The Wieden ore shoots show Sb-bearing phases in addition to As phases. Based on both textural and thermodynamic evidence, the Sb-equivalent of niccolite, breithauptite, becomes thermodynamically stable over niccolite in the presence of additional aqueous Sb (Fig. 14a). This even occurs at fluid compositions, in which As is more abundant than Sb (see Wieden fluid composition; Fig. 10). In addition, allargentum, dyscrasite, and native Sb form over a large pH range at a redox state similar to that of native As (Fig. 14a). Their stability is strongly dependent on both the availability of Sb and Ag. During reduction of a fluid with a composition of, for example, the Ag–Ni-As-Sb ore shoot at Wieden (Table 4), allargentum and dyscrasite form after breithauptite and rammelsbergite, but prior to native As during fluid reduction. This is in accordance with the paragenetic sequence.
Formation of sulfides in normal vein and ore shoot assemblages
As shown above, gangue minerals form by simple binary fluid mixing, the typical native element-arsenide assemblage of the ore shoots by the same fluid mixing process accompanied by reduction. However, the presence of clustered and disseminated base metal sulfides (mainly galena and sphalerite) in the vein as well as the presence of sulfides in some and the absence of sulfides in other ore shoots have not been discussed and explained yet. Based on the lack of statistically significant differences between their sulfur isotope signatures (Fig. 8), sulfides in the ore shoots and sulfides in the normal veins are believed to have the same source and the same formation mechanism. We propose that the sulfide was added to the fluid discontinuously and locally during the mixing process, most likely by interaction with the surrounding host rock. The line of arguments is as follows:
-
(1)
Textures: The sulfides are not evenly distributed throughout the veins, but they occur as local accumulations such as bands or directly overgrowing host rock clasts in breccias (e.g., Fig. 1c; Metz et al. 1957; Markl 2017a). The brecciated clasts are typically overgrown by sulfide- and quartz-rich material which implies a formation process different from (or at least: additional to) the other normal vein minerals.
-
(2)
Fluid thermodynamics: Fluids A and B contain some metals such as Ba, Pb, and Zn (Fusswinkel et al. 2013; Scharrer et al. 2021) and thus cannot transport significant amounts of sulfur at the temperature range in question (e.g., Allison et al. 1991; Ball and Nordstrom 1991). Depending on redox conditions, thermodynamic modeling predicts that a maximum of 10 mg/kg of sulfate or a maximum of 0.05 mg/kg of sulfide can be transported at a neutral pH (Fig. 11a) for the fluid compositions of the three localities (Table 4). LA-ICP-MS measurements of Scharrer et al. (2021) showed detectable sulfur only in two fluid inclusions, namely 20 ± 10 mg/kg and 50 ± 25 mg/kg, which are in the same order of magnitude as values of thermodynamically possible dissolved sulfate. This implies that the sulfur in the fluid A or B (or both) was likely present as sulfate species.
-
(3)
Mineral thermodynamics: Based on the disagreement between thermodynamic phase stability fields and the observed mineralogy, a thermodynamic disequilibrium between a few minerals must be assumed both in the veins and in the ore shoots. The most striking example is the apparently stable assemblage of oxidized and reduced minerals such as barite and native As. Under equilibrium conditions, this assemblage would not form, as the sulfate in barite would be reduced to sulfide under the redox state of native As stability. Furthermore, fluids containing Ni, As, Pb, and Zn, as the ones investigated here (Fig. 10) cannot precipitate the base metal sulfides galena and sphalerite without the contemporaneous formation of Ni-sulfides and/or arsenides under equilibrium conditions (Fig. 11b). This is not only at odds with the observations from the three veins investigated here, but with thousands of hydrothermal veins in the Schwarzwald (Markl 2015, 2016, 2017a, 2017b), in Central Europe and worldwide (e.g., Baumann et al. 2000; Sangster 2009), where Ni-minerals are rather rare.
-
(4)
Kinetics: Experimentally determined kinetic rates of sulfate-sulfide equilibration (Ohmoto and Lasaga (1982) are insufficiently slow to allow sulfate reduction during short-lived mixing processes in hydrothermal systems below 200 °C. The formation of individual growth zones during such mixing processes takes less than minutes (Walter et al. 2018a), while the minimum time required for the attainment of a 90% equilibrium between aqueous sulfide and sulfate requires ~ 4000 years at 150 °C at roughly neutral conditions (Ohmoto and Lasaga 1982).
All these arguments imply that sulfide was not transported in fluid A or fluid B in sufficient quantities, but that it had to be added from outside. Also sulfate reduction can be ruled out for kinetic reasons.
Sulfate-sulfide disequilibrium based on δ 34 S
The kinetic disequilibrium between sulfate and sulfide discussed in the preceding chapter is corroborated by the δ34S signature of both sulfates and sulfides from ore shoots and normal vein assemblages (Fig. 8). The calculated equilibrium temperatures between sulfides and co-genetic sulfates from the three investigated localities seem to record temperatures of formation between 270 and 470 °C (Fig. 12c), while the pressure-corrected fluid inclusions record temperatures of about 150 °C (Scharrer et al. 2021) which are in agreement with many other fluid inclusion studies in the region (see above). Hence, the apparently co-genetic sulfate-sulfide textures do not prove sulfate-sulfide isotope equilibrium; rather, sulfides and sulfates are in textural equilibrium, but in sulfur isotope disequilibrium. This agrees with a sulfur isotope study from the whole Schwarzwald showing unrealistic equilibrium temperatures ranging from ~ 200 to ~ 500 °C (Schwinn et al. 2006). There is no evidence for a post-formational isotopic equilibration of barite or the sulfides with each other or a further sulfur source. This is because there is no correlation between the barite, galena, and sphalerite δ34S value and the sampled crystal size or mineral paragenesis. Furthermore, the sulfides and barite from this Jurassic-Cretaceous hydrothermal stage from all over the Schwarzwald show a similar disequilibrium isotopic range (Gehlen et al. 1962; Schwinn et al. 2006; Staude et al. 2011). A full equilibration of all sulfides or sulfates with a subsequent fluid is disregarded since these are systems governed by localized fluid pathways.
In summary, we interpret the sulfur isotope values of barite and sulfides to reflect a sulfate-sulfide isotopic disequilibrium during vein formation. Therefore, sulfate and sulfide must have originated from different sources, and for sulfide, this source was not fluid A or fluid B. This finding is in sharp contrast to the results and interpretations of previous studies (Walter et al. 2016, 2018a, 2020).
Sulfide source
This chapter discusses the potential sources of the externally derived sulfide. The regional geology permits the sulfide to be sourced either from the surrounding host rock or from the (at the time of ore formation) overlying sedimentary lithologies. The latter would require sulfide from the organic-rich and/or sulfide-rich layers in the sedimentary cover penetrating the basement during downward fluid flow as was, e.g., suggested by Bons et al. (2014). However, sedimentary, mostly organically or diagenetically sourced, sulfide generally shows strongly negative δ34S values, mostly between − 5 and − 50 (Marowsky 1969; Maynard 1980; Gautier 1987) as does the most likely sedimentary sulfide source candidate in the region, the Lower Jurassic Posidonian shale (Geyer et al. 2011), namely δ34S values between − 15 and − 40 (Brumsack 1991; Raiswell et al. 1993). This is significantly lighter than our analyzed sulfides (Fig. 8). Thus, a major organic sulfide source component can, in all probability, be excluded.
The most likely sulfide source is, hence, the surrounding basement rock sulfides. Remobilization of this sulfide during hydraulic fracturing is a viable mechanism of sulfide addition to the mixing hydrothermal fluids. It is well known that the Schwarzwald basement rocks, specifically the gneisses, bear high amounts of sulfides, including pyrite, pyrrhotite, and arsenopyrite (e.g., Wager 1935; Otto 1964; Hofmann 1989; Geyer et al. 2011; Staude et al. 2011; Drüppel et al. 2020). We also observed them in our host rock samples. The importance of the host rock characteristics on the occurrence of sulfides in Schwarzwald hydrothermal veins has also been described by Walter et al. (2019) who found that gneiss-hosted veins contain a higher modal abundance of sulfides compared to granite-hosted veins. Furthermore, galena and sphalerite are mostly abundant in hydraulic breccia zones (this study; Metz et al. 1957; Widemann 1988; Markl 2017a), where interactions between the hydrothermal fluid and the fresh, non-leached host rock are strongest. As the new fractures typically occur in the center of a vein or at the marginal selvages, which are covered and thereby sealed by clay minerals (Metz et al. 1957; Burisch et al. 2016; Walter et al. 2016), repetitive opening of the same fractures nicely explains the typical banded gangue textures (e.g., Keim et al. 2019) and the general rarity of sulfides. The latter are distributed unevenly reflecting the rare addition of sulfide to the hydrothermal fluid. This is related to the preexisting vein being a structurally inherited weak zone (Virgo et al. 2014) which is a typical feature in many hydrothermal systems (e.g., Cathelineau et al. 2017). The sulfide isotope signature of both ore shoots and vein sulfides (δ34S ~ 0 to − 12) overlaps with the range of the basement sulfides (δ34S ~ + 2 to − 9‰; Siewers 1977) which corroborates our inference that sulfide was derived from the gneissic host rocks.
This concludes the sulfide, which is the prerequisite for sulfide formation, and the reducing agent, which is required for the ore shoot formation, having two separate origins. Which of these fluids, a sulfide-bearing or a reducing agent–bearing fluid, is tapped, results in the formation of the normal vein sulfides or an ore shoot assemblage, respectively.
The mineralogical consequences of sulfide addition and sulfate-sulfide disequilibrium on the ore shoot assemblage
The ore shoots show a highly variable sulfide content, from absent to abundant, which implies that the availability of sulfide and reducing agent may have been variable between ore shoots. Thus, it is vital to consider the effect sulfide has on the ore shoot mineralogy. In the following, the consequences of sulfide addition under equilibrium (Figs. 12b, 13b and 14b) and under sulfate-sulfide disequilibrium (Figs. 12c, 13c and 14c) conditions on the ore shoot mineralogy will be discussed.
In a simplified As-S system and under equilibrium conditions, realgar and orpiment would form at acidic to neutral conditions. Their stability field is increased to more oxidized environments under sulfate-sulfide disequilibrium conditions. Only when the available sulfide is used up, native arsenic would form. However, realgar and orpiment were not found in any of the investigated samples. This is attributed to their stability strongly depending on the availability of reduced sulfur, which, in turn, is also dependent on the stability of other sulfides and, thus, on the availability of, for example, Pb and Zn in the fluid (Scharrer et al. 2020). As Pb and Zn are very abundant in the mixture of fluid A with fluid B, nearly all LA-ICP-MS analyses show them in a similar range of 1 to 100 ppm, and as galena and sphalerite have very low solubilities, their formation uses up the reduced aqueous sulfide which is the ultimate reason for the stability of native As over realgar and orpiment.
During reduction of a sulfide-bearing Ag-As-Pb fluid under equilibrium conditions and at neutral pH, galena precipitation is succeeded by native As formation (Fig. 12b). In our samples, although galena does also form prior and subsequent to native As, it also forms simultaneously to native As (e.g., Fig. 4b). As an example, in an equilibrium system with the fluid condition at Münstertal (Table 4), the amount of galena that can precipitate simultaneously with native As during reduction is several orders of magnitude less than native As. A co-precipitation is however possible, when sulfide influx occurs contemporaneous with the reduction, as is the case at hand. Thus, the simultaneous precipitation of these two minerals at a neutral pH reflects the sulfide influx during reduction under sulfate-sulfide disequilibrium conditions. At these conditions, the stability and formation of galena is independent of the redox state of the fluid (Fig. 12c) and whether galena or native As forms depends on the relative abundance of sulfide and reducing agent. Temporal variations in redox state and/or sulfide influx nicely explain the commonly observed banded onion-layer texture between native As and galena and the lath-shaped oriented galena crystals in native As (Fig. 6e).
In the presence of dissolved Ni, the thermodynamic modeling predicts the formation of gersdorffite under slightly basic to strongly acidic conditions (Fig. 13b). Under sulfate-sulfide disequilibrium conditions, the stability of gersdorffite increases, based on the availability of sulfide, to more oxidizing and more basic conditions (Fig. 13c). Due to the As-S exchange in the arsenides and sulfarsenides (Fig. 7b), we assume that S-rich rammelsbergite forms at low sulfide, As-rich gersdorffite at moderate sulfide, and stoichiometric gersdorffite at high sulfide availability. This would explain the mineralogical differences in the three ore shoot sequences at Urberg, in which a steady increase in sulfur content in the Ni(As, S)2 and NiAs3 minerals can be witnessed from ore shoot sequences 1 to 3 (Fig. 7b). It is important to note that under moderately reduced conditions, only a few mg/kg of sulfide are needed to increase the stability of even stoichiometric gersdorffite to form over a large pH range. The non-stoichiometric gersdorffite would form at even lower sulfide contents. The availability of sulfide is, however, not only constrained by the absolute sulfide content, but (as already described above) also by the content of other elements such as Pb or Zn that contest for the sulfide due to the low solubility of their respective sulfides (Scharrer et al. 2020). The fact that both Pb and Zn were detected in most fluid inclusions (i.e., in the mixed fluids after mineral precipitation) indicates a high metal/sulfide ratio.
The presence of Sb in addition to Ni, As, Pb, and sulfide in the fluid stabilizes some Sb-minerals at the expense of their respective As counterparts, even if the fluid is richer in As than in Sb (Fig. 14b). This explains the formation of breithauptite (Fig. 14b) and possibly ullmannite in As-dominated ore shoots, although the stability of the latter could not be modeled due to the lack of suitable thermodynamic data.
The mineralogical consequences of sulfate-sulfide disequilibrium on the normal vein assemblage
Although the sulfate-sulfide disequilibrium does influence the mineralogy of the ore shoots (Figs. 12c, 13c and 14c), with minor changes in mineral abundance and mineral succession, similar ore shoots could also form under sulfate-sulfide equilibrium conditions (Figs. 12b, 13b and 14b). However, the normal sulfide assemblage of the vein observed here could not have formed under equilibrium conditions. The sulfate-sulfide disequilibrium does not only stabilize mineral assemblages that would not form otherwise, but it also prevents the formation of some equilibrium assemblages or textural successions which would usually be expected to form. For example, under equilibrium conditions (Figs. 12b, 13b, and 14b), the base metal sulfides should always form after native Ag (Fig. 12c), arsenides (Fig. 13c), or antimonides (Fig. 14c), but the typical vein sulfide assemblage completely lacks native Ag, arsenides, and antimonides. This is identical in almost all other hydrothermal veins of the same hydrothermal stage in the Schwarzwald (Markl 2015, 2016, 2017a, 2017b) and can be explained by the disequilibrium conditions, which (1) prevent sulfide from acting as a reducing agent for, e.g., Ag+, aqueous arsenite, or aqueous antimonite and (2) increase the stability of sulfide-bearing minerals such as base metal sulfides to oxidizing conditions.
Finally, under sulfate-sulfide disequilibrium conditions, an influx of sulfide into a fluid of variable redox state invariably either co-precipitates galena and Ag-sulfides/Ag-sulfosalts (Figs. 12c, 13c and 14c) or Ag is incorporated into galena via coupled substitution. The latter could not be modeled due to the lack of thermodynamic miscibility data. However, extrapolation from higher temperature experiments to below 200 °C of the coupled substitution of Ag(As, Sb, Bi)S2 into galena reveals that up to ~ 3 mol% of ∑(Ag, As, Sb, Bi) can be incorporated into galena (Chutas et al. 2008; Renock and Becker 2011). Thus, the observed elevated contents of Ag, As, Sb, and Bi in galena which are both present in the ore shoots and the vein sulfide assemblages could only form due to the sulfate-sulfide disequilibrium, which prohibits native Ag to form. Hence, the main medieval Ag source, argentiferous galena, is a direct result of the sulfate-sulfide disequilibrium during the formation of these veins (Markl 2017a).
Alternative formation mechanisms for ore shoot and vein sulfides
The model presented here suggests that the vein formed by mixing of fluid A and fluid B, the vein sulfides precipitated due to an influx of host rock-sourced sulfide, and the ore shoot assemblages formed by a local influx of a mobile reducing agent, most likely hydrocarbons. Although we have provided abundant textural, mineralogical, geochemical, fluid inclusion, and modeling data to support the hypothesis of such a three fluid ± host rock interaction model, possible alternatives should also be mentioned. Alternative models to explain the presence of both vein sulfides and ore shoots are, for example, that the change from vein sulfides to ore shoots may be governed by (a) a different magnitude of external (rock) vs. internal (fluid) buffering and (b) differences in the magnitude of the net fluid flux.
We do not consider both alternatives as viable, because, concerning (a), the internal buffering of fluids A + B forms only the gangue minerals, while the external (rock) buffering is, in principle, the mechanism that we suggest forming the vein sulfides. A stronger interaction with the surrounding host rocks produces a higher sulfide ore content in the vein, but not an arsenide-bearing ore shoot. This statement is even texturally supported by the increased sulfide concentrations at places where host rock breccias are present in the banded vein (see vein sulfide formation chapter). Alternative (b) proposes that a different degree of reaction progress of the overall fluid system is responsible for the shift between the normal vein sulfide and the ore shoot assemblage. However, our modeling clearly shows that variations in magnitude of net fluid flux of fluids A + B or the sulfide- and reducing agent–bearing fluid do not suffice to explain the mineralogical difference between the vein sulfides and the ore shoots. A single fluid with a given sulfide/reducing agent ratio cannot thermodynamically result in the formation of sulfide-free ore shoots, sulfide-bearing ore shoots, and normal vein sulfides.
Hydrothermal overprint, sulfosalt assemblage, and worldwide comparison to other occurrences
The previous models nicely explained the formation of the native element-arsenide-sulfarsenide assemblages, with or without the base-metal sulfides, but they did not discuss the silver sulfides and sulfosalts, mainly proustite-pyrargyrite solid solutions, which are abundant. These minerals formed in a later process by hydrothermal remobilization of the primary ore assemblages. Similar processes have been described for other Ag-rich hydrothermal systems in the region (Keim et al. 2019). To understand their formation is of vital importance since remobilization processes can increase or decrease the economic value of many types of deposit dramatically (e.g., Hobbs 1987; Marshall et al. 2000; van Dongen et al. 2010; Hastie et al. 2020).
In the deposits investigated in the present contribution, the most common consequence of hydrothermal overprint of the primary ore shoot assemblages is the formation of minerals of the proustite (Ag3AsS3)-pyrargyrite (Ag3SbS3) solid solution series at the cost of mainly native Ag and sometimes native As. Depending on sample and locality, they form in association with other sulfosalts and sulfides (Table 1). As is the case in our samples, these Ag-sulfosalts generally form during a later stage or at the end of an ore stage in many Ag-rich hydrothermal associations worldwide (Ryall 1979; Ruvalcaba-Ruiz and Thompson 1988; Mladenova et al. 2004; Camprubí et al. 2006; Staude et al. 2007; Fanlo et al. 2010; Kallstrom 2012; Yesares et al. 2017; Epp et al. 2018). The formation of sulfosalts has been linked both to a primary hydrothermal fluid (Foley 1984), but more commonly to exsolution and/or remobilization processes (e.g., Boni and Koeppel 1985; Loucks and Petersen 1988; Mladenova et al. 2004; Takahashi et al. 2006; Epp et al. 2018).
The formation of proustite-pyrargyrite series minerals must have started after the ore shoot formation, but during the same hydrothermal stage II (Fig. 3a) since euhedral sulfosalts are found imbedded in gangue minerals of this stage. As the sulfosalts do not just replace, but also overgrow the primary minerals, an elemental flux at constant volume during remobilization cannot be calculated. However, the observation that native arsenic and native silver are replaced by proustite clearly shows that an influx of sulfide is a necessary prerequisite for their formation. It is suggested that the replacement processes involved batches of either fluid A or fluid B, both of which are not in equilibrium with and more oxidized than the ore shoot, plus sulfide.
The sulfide isotope signature of proustite ranges from the same as that of the primary ore shoot and main stage vein sulfides to significantly lower. The same isotopic signature as the rest of the vein sulfides suggests that the sulfide originated from the same sulfide fluid source or remobilization of sulfides within the vein. The significantly lower isotopic signature can be explained by a subsequent fluid, or sulfate reduction being involved. Such reaction processes are not temporally confined to a short process such as fluid mixing. Thus, sulfate reduction may play a role for these remobilization processes.
For the investigated samples at hand, the large variety of sulfosalts forming during the replacement process can be traced back to local, small-scale variations in preexisting mineralogy and, possibly, fluid composition. Although minerals of the proustite-pyrargyrite series (Ag3(As,Sb)S3) are abundantly present, minerals of the smithite-miargyrite series (Ag(As,Sb)S2) are rare or absent. This is a common feature at many Ag-sulfosalt–bearing localities worldwide (e.g., Gillerman and Whitebread 1953; Gemmell et al. 1989; Chutas and Sack 2004; Camprubí et al. 2006). The formation of either of these minerals is defined by the elemental abundance (Chutas and Sack 2004), specifically of Ag, Sb, As, and S, and by prevailing formation conditions, specifically pH. The proustite-pyrargyrite series preferably forms at more neutral to slightly basic conditions and at higher Ag and S activities relative to miargyrite (Fig. 14c). A high Ag activity was provided by the dissolution of native Ag. The favorable neutral to basic pH was given if the involved initial fluid was at roughly neutral pH, moderately reduced to moderately oxidized and As-bearing. Local variations in fluid composition and primary ore shoot mineralogy then determined, which sulfosalt formed during this hydrothermal overprint (see also Keim et al. 2019).
These required initial fluid conditions are met by fluids A and B. At initially roughly neutral pH and under moderately reduced to moderately oxidized conditions, both arsenite and arsenate are present in the fluid (ESM 6). During dissolution of native Ag or native As, arsenate is progressively reduced to arsenite (Scharrer et al. 2019), which, in turn, increases the fluids pH (Eqs. 2 and 3).
As (s) + 1.5 H+ + 1.5 H2O + 1.5 H2AsO4− ↔ 2.5 As(OH)3 (aq) (2).
Ag (s) + 1.5 H+ + 0.5 H2AsO4− + 2Cl− ↔ 0.5 H2O + 0.5 As(OH)3 (aq) + AgCl2− (3).
Hence, the sulfosalts form in the presence of a sulfide influx during ore shoot remobilization and the sulfosalt mineralogy is defined by the elemental availability and formation conditions. Interestingly, if fluids A and B react with the ore shoots in the absence of sulfide, the primary mineralogy is only dissolved and replaced by gangue minerals, without the formation of secondary ore minerals. This has also been observed in several samples, in which preferably native As and native Ag were dissolved, while their colloform and dendritic shapes are still preserved in the replacing gangue minerals (Fig. 4f, g).
In some samples from Wieden, minerals of the proustite-pyrargyrite series are occasionally replaced by acanthite (Ag2S) and, in turn, by native Ag. This formation of secondary native Ag by the dissolution of acanthite is a typical reaction which has been described from many localities worldwide (e.g., Fanlo et al. 2010; Arribas et al. 2020). The secondary native Ag commonly forms wires in open vugs. This process shows a progressive enrichment of Ag relative to As, Sb, and S. While the primary native Ag contains significant amounts of Sb, Hg, and As, the process of remobilization, first to proustite and then to secondary native silver, preferably removes Sb, Hg, and As and, hence, purifies the native silver. The whole process is most likely related to weathering fluids of different compositions than fluid A or B.
Conclusions
Although drastic mineralogical changes in a hydrothermal vein are commonly interpreted to indicate a new hydrothermal stage with the influx of a new fluid of different composition, this study shows that such changes can simply reflect a spatially and temporally restricted change in redox conditions. In this case, a local influx of a reducing agent can result in the formation of Ag- and As-rich ore shoots. The mineralogy of these ore shoots is strongly dependent on the presence of the metals in the fluid, as the mineralogy reflects the maximum elemental fluid composition analyzed by LA-ICP-MS in single-fluid inclusions. This, in turn, implies that the elemental composition of the ore, at least to some degree, reflects the source fluid and, thus, the source rock.
The mineralogy of the ore shoots is further influenced by the redox state, the pH, the availability of sulfide, and the already precipitated mineralogy. The presence of sulfide during reduction results in the formation of sulfide-bearing minerals such as base metal sulfides, sulfarsenides, and sulfantimonides. The influx of sulfide and lack of reduction of sulfate is due to a thermodynamic disequilibrium between sulfate and sulfide, which stem from two different sources and which could not equilibrate due to slow reaction kinetics (Ohmoto and Lasaga 1982). In the presence of a sulfide influx without a reducing agent, only normal base metal sulfides such as galena, sphalerite, or chalcopyrite form. This sulfide influx also promotes the incorporation of Ag, Sb, As, and Bi into galena. The most likely sulfide source is the sulfide-bearing gneisses (= host rocks), while the reducing agent was most probably represented by hydrocarbons from the sedimentary cover.
The final ore shoot textures found in the samples are strongly influenced by post-depositional remobilization processes. These include the typical hydrothermal dissolution of native Ag and native As and reprecipitation as sulfosalts as well as later reaction processes that re-remobilize the Ag-bearing sulfosalts and form secondary native Ag. Thermodynamic calculations show that the larger stability of Ag-Sb-sulfosalts such as pyrargyrite compared to their respective Ag-As-endmembers (in this case, proustite) enables their formation even in As-dominated hydrothermal systems.
Data availability
All new data provided in this manuscript, EPMA data and sulfur isotope data, are provided in the electronic supplement.
Change history
28 August 2022
Missing Open Access funding information has been added in the Funding Note.
References
Allison JD, Brown DS, Novo-Gradac KJ (1991) MINTEQA2/PRODEFA2, a geochemical assessment model for environmental systems: version 3.0 user’s manual. Environmental Research Laboratory, Office of Research and Development, US Environmental Protection Agency.
Arribas A, Mathur R, Megaw P, Arribas I (2020) The isotopic composition of silver in ore minerals. Geochem Geophys Geosyst 21.https://doi.org/10.1029/2020GC009097
Aspiala M, Tesfaye F, Taskinen P (2015) Electrochemical study on the Ag–Sb system by advanced experimental method. Electrochim Acta 173:649–655. https://doi.org/10.1016/j.electacta.2015.05.065
Aspiala M, Tesfaye F, Taskinen P (2016) Thermodynamic study in the Ag–Sb–S system by the EMF method. J Chem Thermodyn 98:361–366. https://doi.org/10.1016/j.jct.2016.03.009
Baatartsogt B, Schwinn G, Wagner T, Taubald H, Beitter T, Markl G (2007) Contrasting paleofluid systems in the continental basement: a fluid inclusion and stable isotope study of hydrothermal vein mineralization, Schwarzwald district, Germany. Geofluids 7:123–147. https://doi.org/10.1111/j.1468-8123.2007.00169.x
Ball JW, Nordstrom DK (1991) WATEQ4F--user’s manual with revised thermodynamic data base and test cases for calculating speciation of major, trace and redox elements in natural waters. Survey USG. Open-File Report 91–183.
Barin I (1995) Thermochemical data of pure substances. Wiley-VCH, New York
Barnes HL (1997) Geochemistry of hydrothermal ore deposits. John Wiley & Sons
Barrett TJ, Anderson GM (1988) The solubility of sphalerite and galena in 1–5 m NaCl solutions to 300 C. Geochim Cosmochim Ac 52:813–820. https://doi.org/10.1016/0016-7037(88)90353-5
Bastin ES (1939) The nickel-cobalt-native silver ore type. Econ Geol 34:40–79. https://doi.org/10.2113/gsecongeo.34.1.1
Baumann L, Kuschka E, Seifert T (2000) Lagerstätten des Erzgebirges. Enke, Stuttgart
Bessinger B, Apps JA (2003) The hydrothermal chemistry of gold, arsenic, antimony, mercury and silver. U.S. Department of Energy. pp 52.
Bethke CM (2007) Geochemical and biogeochemical reaction modeling. Cambridge University Press
Blanc P, Lassin A, Piantone P, Azaroual M, Jacquemet N, Fabbri A, Gaucher EC (2012) Thermoddem: a geochemical database focused on low temperature water/rock interactions and waste materials. Appl Geochem 27:2107–2116. https://doi.org/10.1016/j.apgeochem.2012.06.002
Bliedtner M (1978) Blei- und Zink-Prospektion im Gebiet Kaltwasser/Mulden-Untersmünstertal Südschwarzwald. Albert-Ludwigs-Universität Freiburg
Boni M, Koeppel V (1985) Ore-lead isotope pattern from the Iglesiente-Sulcis area (SW Sardinia) and the problem of remobilization of metals. Miner Deposita 20:185–193. https://doi.org/10.1007/BF00204563
Bons PD, Fusswinkel T, Gomez-Rivas E, Markl G, Wagner T, Walter B (2014) Fluid mixing from below in unconformity-related hydrothermal ore deposits. Geology 42:1035–1038. https://doi.org/10.1130/G35708.1
Brill RA, Falkenstein F, Gulich J (2018) Die Grube “Gottesehre” bei Urberg im Südschwarzwald. Erzgräber 32:118
Brumsack H-J (1991) Inorganic geochemistry of the German ‘Posidonia Shale’: palaeoenvironmental consequences. Geol Soc Lon, Spec Publ 58:353–362. https://doi.org/10.1144/GSL.SP.1991.058.01.22
Burisch M, Walter BF, Wälle M, Markl G (2016) Tracing fluid migration pathways in the root zone below unconformity-related hydrothermal veins: insights from trace element systematics of individual fluid inclusions. Chem Geol 429:44–60. https://doi.org/10.1016/j.chemgeo.2016.03.004
Burisch M, Gerdes A, Walter BF, Neumann U, Fettel M, Markl G (2017a) Methane and the origin of five-element veins: mineralogy, age, fluid inclusion chemistry and ore forming processes in the Odenwald, SW Germany. Ore Geol Rev 81:42–60. https://doi.org/10.1016/j.oregeorev.2016.10.033
Burisch M, Walter BF, Markl G (2017b) Silicification of hydrothermal gangue minerals in Pb-Zn-Cu-fluorite-quartz-baryte veins. Can Mineral 55:501–514. https://doi.org/10.3749/canmin.1700005
Camprubí A, González-Partida E, Iriondo A, Levresse G (2006) Mineralogy, fluid characteristics, and depositional environment of the paleocene epithermal Au-Ag deposits of the El Barqueno District, Jalisco, Mexico. Econ Geol 101:235–247. https://doi.org/10.2113/gsecongeo.101.1.235
Cathelineau M, Myagkiy A, Quesnel B, Boiron M-C, Gautier P, Boulvais P, Ulrich M, Truche L, Golfier F, Drouillet M (2017) Multistage crack seal vein and hydrothermal Ni enrichment in serpentinized ultramafic rocks (Koniambo Massif, New Caledonia). Miner Deposita 52:945–960. https://doi.org/10.1007/s00126-016-0695-3
Chutas NI, Sack RO (2004) Ore genesis at La Colorada Ag-Zn-Pb deposit in Zacatecas, Mexico. Mineral Mag 68:923–937. https://doi.org/10.1180/0026461046860231
Chutas NI, Kress VC, Ghiorso MS, Sack RO (2008) A solution model for high-temperature PbS-AgSbS2-AgBiS2 galena. Am Mineral 93:1630–1640. https://doi.org/10.2138/am.2008.2695
Collins JE, Barnes SJ, Hagemann SG, McCuaig TC, Frost K (2012) Postmagmatic variability in ore composition and mineralogy in the T4 and T5 ore shoots at the high-grade flying fox Ni-Cu-PGE deposit, Yilgarn craton, Western Australia. Econ Geol 107:859–879. https://doi.org/10.2113/econgeo.107.5.859
Drüppel K, Stober I, Grimmer JC, Mertz-Kraus R (2020) Experimental alteration of granitic rocks: implications for the evolution of geothermal brines in the Upper Rhine Graben. Germany Geothermics 88:101903. https://doi.org/10.1016/j.geothermics.2020.101903
Duan Z, Mao S (2006) A thermodynamic model for calculating methane solubility, density and gas phase composition of methane-bearing aqueous fluids from 273 to 523 K and from 1 to 2000 bar. Geochim Cosmochim Ac 70:3369–3386. https://doi.org/10.1016/j.gca.2006.03.018
Epp T, Walter B, Scharrer M, Lehmann G, Henze K, Heimgärtner C, Bach W, Markl G (2018) Quartz veins with associated Sb-Pb-Ag±Au mineralization in the Schwarzwald, SW Germany: a record of metamorphic cooling, tectonic rifting, and element remobilization processes in the Variscan belt. Miner Deposita 52:1–26. https://doi.org/10.1007/s00126-018-0855-8
Essarraj S, Boiron M-C, Cathelineau M, Banks DA, Benharref M (2005) Penetration of surface-evaporated brines into the Proterozoic basement and deposition of Co and Ag at Bou Azzer (Morocco): evidence from fluid inclusions. J African Earth Sci 41:25–39. https://doi.org/10.1016/j.jafrearsci.2005.03.001
Ewers W, Hudson D (1972) An interpretive study of a nickel-iron sulfide ore intersection, Lunnon Shoot, Kambalda, Western Australia. Econ Geol 67:1075–1092. https://doi.org/10.2113/gsecongeo.67.8.1075
Fanlo I, Subías I, Mateo E (2010) Supergene enrichment of primary Cu-Ag assemblages in Ag at the Padros deposit, Iberian range, Spain. Can Mineral 48:415–430. https://doi.org/10.3749/canmin.48.2.415
Foley NK (1984) Characteristics of some silver-, and base metal-bearing, epithermal deposits of Mexico and Peru. US Geological Survey.
Fougerouse D, Micklethwaite S, Tomkins AG, Mei Y, Kilburn M, Guagliardo P, Fisher LA, Halfpenny A, Gee M, Paterson D (2016) Gold remobilisation and formation of high grade ore shoots driven by dissolution-reprecipitation replacement and Ni substitution into auriferous arsenopyrite. Geochim Cosmochim Ac 178:143–159. https://doi.org/10.1016/j.gca.2016.01.040
Fusswinkel T, Wagner T, Wälle M, Wenzel T, Heinrich CA, Markl G (2013) Fluid mixing forms basement-hosted Pb-Zn deposits: insight from metal and halogen geochemistry of individual fluid inclusions. Geology 41:679–682. https://doi.org/10.1130/G34092.1
Gasanova Z, Mashadieva L, Zlomanov V, Babanly M (2014) Thermodynamic study of the Ag2S-As2S3-S system by EMF measurements with Ag4RbI5 as a solid electrolyte. Inorg Mater 50:6–9. https://doi.org/10.1134/S0020168514010075
Gautier DL (1987) Isotopic composition of pyrite: relationship to organic matter type and iron availability in some North American Cretaceous shales. Chem Geol: Iso Geo Sec 65:293–303. https://doi.org/10.1016/0168-9622(87)90009-1
Gehlen Kv, Nielsen H, Ricke W (1962) S-Isotopen-Verhältnisse in Baryt und Sulfiden aus hydrothermalen Gängen im Schwarzwald und Jängeren Barytgängen in Süddeutschland und ihre genetische Bedeutung. Geochim Cosmochim Ac 26:1189–1207. https://doi.org/10.1016/0016-7037(62)90051-0
Gemmell JB, Zantop H, Birnie RW (1989) Silver sulfosalts of the Santo Nino vein, Fresnillo District, Zacatecas, Mexico. Can Mineral 27:401–418
George L, Cook NJ, Ciobanu CL, Wade BP (2015) Trace and minor elements in galena: a reconnaissance LA-ICP-MS study. Am Mineral 100:548–569. https://doi.org/10.2138/am-2015-4862
Geyer OF, Gwinner MP, Geyer M, Nitsch E, Simon T (2011) Geologie von Baden-Württemberg. Schweizerbart.
Gillerman E, Whitebread DH (1953) The Uranium-bearing nickel-cobalt-native silver deposits in the Black Hawk district, Grant County, New Mexico. US Geological Survey. Bulletin pp 313.
Gloyn-Jones J, Kisters A (2019) Ore-shoot formation in the Main Reef Complex of the Fairview Mine—multiphase gold mineralization during regional folding, Barberton Greenstone Belt, South Africa. Miner Deposita 54:1157–1178. https://doi.org/10.1007/s00126-019-00865-9
Goldstein H, Reynolds T (1994) Systematics of fluid inclusions in diagenetic minerals. SEPM Short Course 31:199
Gough D (1965) Structural analysis of ore shoots at Greenside lead mine, Cumberland, England. Econ Geol 60:1459–1477. https://doi.org/10.2113/gsecongeo.60.7.1459
Grundmann G (1971) Neues über Silbererze vom Tannenbodengang bei Wieden (Sdschwarzwald). Aufschluss 22:195–200
Guilbert JM, Park CFJ (2007) The geology of ore deposits. Waveland Press
Guilcher M, Gerdes A, Albert R, Gutzmer J, Burisch M (submitted) Timing of native metal-arsenide (Bi-Co-Ni-As-Ag±U) veins in continental rift zones – new insights from LA-ICP-MS U-Pb geochronology of carbonates from the Erzgebirge/Krušné Hory province (Germany and the Czech Republic).
Hastie EC, Kontak DJ, Lafrance B (2020) Gold remobilization: insights from gold deposits in the Archean Swayze greenstone belt, Abitibi Subprovince, Canada. Econ Geol 115:241–277. https://doi.org/10.5382/econgeo.4709
Hauck M (2020) Bergbau- und Fördertechnik auf Wiedener Gruben, in Lagerstätten und Bergbau bei Wieden im Südschwarzwald. Unter besonderer Berücksichtigung der Grube Finstergrund. Erzgräber 35:117–136
Hiller A, Schuppan W (2008) Geologie und Uranbergbau im Revier Schlema-Alberoda. Landesamt für Umwelt, Landwirtschaft und Geologie. Bergbau in Sachsen pp 200.
Hise CV (1900) Some principles controlling the deposition of ores. J Geol 8:730–770
Hobbs BE (1987) Principles involved in mobilization and remobilization. Ore Geol Rev 2:37–45. https://doi.org/10.1016/0169-1368(87)90022-9
Hofmann B (1989) Genese, Alteration und rezentes Fliess-System der Uranlagerstätte Krunkelbach (Menzenschwand, Südschwarzwald). Dissertation, Universität Bern
Hösel G (2003) Die polymetallische Skarnlagerstätte Pöhla-Globenstein. Landesamt für Umwelt, Landwirtschaft und Geologie. Bergbau in Sachsen pp 147.
Hulin CD (1929) Structural control of ore deposition. Econ Geol 24:15–49. https://doi.org/10.2113/gsecongeo.24.1.15
Huth T, Zedler H (2019) Entlang der kontinentalen Schweißnaht im Südschwarzwald–die Badenweiler–Lenzkirch-Zone (BLZ). Schriftenreihe der Deutschen Gesellschaft für Geowissenschaften:166–192.
Jébrak M (1997) Hydrothermal breccias in vein-type ore deposits: a review of mechanisms, morphology and size distribution. Ore Geol Rev 12:111–134. https://doi.org/10.1016/S0169-1368(97)00009-7
Kallstrom MJ (2012) Fluid and metal sourcing for the native silver deposits in the Batopilas Mining District, Chihuahua, Mexico. Unpublished MSc Thesis, University of Texas at Austin
Keim MF, Staude S, Marquardt K, Bachmann K, Opitz J, Markl G (2018) Weathering of Bi-bearing tennantite. Chem Geol 499:1–25. https://doi.org/10.1016/j.chemgeo.2018.07.032
Keim MF, Walter BF, Neumann U, Kreissl S, Bayerl R, Markl G (2019) Polyphase enrichment and redistribution processes in silver-rich mineral associations of the hydrothermal fluorite-barite-(Ag-Cu) Clara deposit, SW Germany. Miner Deposita 54:155–174. https://doi.org/10.1007/s00126-018-0799-z
Kerrich R, Strong D, Andrews A, Owsiacki L (1986) The silver deposits at Cobalt and Gowganda, Ontario. III: Hydrothermal regimes and source reservoirs-evidence from H, O, C, and Sr isotopes and fluid inclusions. Can J Earth Sci 23:1519–1550. https://doi.org/10.1139/e86-145
Kissin SA (1988) Nickel-cobalt-native silver (five-element) veins: a riftrelated ore type. In: Kisvarsany G, Grant S (eds) North American Conference on tectonic control of ore deposits and the vertical and horizontal extent of ore systems. Univ, Missouri - Rolla, Rolla, pp 268–279
Kissin SA (1992) Five-element (Ni-Co-As-Ag-Bi) veins. Geosci Can 19:113–124
Kissin SA (1993) The geochemistry of transport and deposition in the formation of five-element (Ag-Ni-Co-As-Bi) veins. Schweizerbart’sche Verlagsbuchhandlung, Eight Quadrennial International Association on the Genesis of Ore Deposits Symposium, p 14
Kreissl S, Gerdes A, Walter B, Neumann U, Wenzel T, Markl G (2018) Reconstruction of a >200 Ma multi-stage “five element” Bi-Co-Ni-Fe-As-S system in the Penninic Alps, Switzerland. Ore Geol Rev 95:746–788. https://doi.org/10.1016/j.oregeorev.2018.02.008
Levresse G, Bouabdellah M, Cheilletz A, Gasquet D, Maacha L, Tritlla J, Banks D, Rachid ASM (2016) Degassing as the main ore-forming process at the Giant Imiter Ag–Hg vein deposit in the Anti-Atlas Mountains. Springer, Morocco Mineral Deposits of North Africa, p 22
Lindgren W (1913) Mineral deposits. McGraw-Hill Book Company, Incorporated
Liu W, Migdisov A, Williams-Jones A (2012) The stability of aqueous nickel(II) chloride complexes in hydrothermal solutions: results of UV–visible spectroscopic experiments. Geochim Cosmochim Ac 94:276–290. https://doi.org/10.1016/j.gca.2012.04.055
Loucks RR, Petersen U (1988) Polymetallic epithermal fissure vein mineralization, Topia, Durango, Mexico; part II, Silver mineral chemistry and high resolution patterns of chemical zoning in veins. Econ Geol 83:1529–1558. https://doi.org/10.2113/gsecongeo.83.8.1529
Marini L, Accornero M (2007) Prediction of the thermodynamic properties of metal–arsenate and metal–arsenite aqueous complexes to high temperatures and pressures and some geological consequences. Environ Geol 52:1343–1363. https://doi.org/10.1007/s00254-006-0578-5
Markl G (2004) Silber Kupfer Kobalt. Markstein Verlag für Kultur- und Wissenschaftsgeschichte, Filderstadt, Bergbau im Schwarzwald
Markl G (2015) Schwarzwald, vol 1. Bode Verlag, Lauenstein, Nordschwarzwald and Grube Clara
Markl G (2016) Schwarzwald, Vol. 2, Mittlerer Schwarzwald Teil 1. Bode Verlag, Lauenstein.
Markl G, Burisch M, Neumann U (2016) Natural fracking and the genesis of five-element veins. Miner Deposita 51:703–712. https://doi.org/10.1007/s00126-016-0662-z
Markl G (2017a) Schwarzwald, vol 4. Bode Verlag, Lauenstein, Südlicher Schwarzwald
Markl G (2017b) Schwarzwald, Vol. 3, Mittlerer Schwarzwald Teil 2. Bode Verlag, Lauenstein.
Marowsky G (1969) Schwefel-, Kohlenstoff-und Sauerstoff-Isotopenuntersuchungen am Kupferschiefer als Beitrag zur genetischen Deutung. Contrib Mineral Petr 22:290–334. https://doi.org/10.1007/BF00400127
Marshall B, Vokes F, Larocque A (2000) Regional metamorphic remobilization: upgrading and formation of ore deposits. Rev Econ Geol 11:19–38. https://doi.org/10.5382/Rev.11.02
Maynard JB (1980) Sulfur isotopes of iron sulfides in Devonian-Mississippian shales of the Appalachian Basin: control by rate of sedimentation. Am J Sci;(United States) 280. https://doi.org/10.2475/ajs.280.8.772.
Metz R, Richter M, Schürenberg (1957) Die Blei-Zink-Erzgänge des Schwarzwaldes. Amt für Bodenforschung, Hannover.
Mladenova V, Kerestedjian T, Dimitrova D (2004) The Balkan mountains Paleozoic gold deposits. Bull Geol Soc Greece 36:424–433. https://doi.org/10.12681/bgsg.16729.
Müller G, Nielsen H, Ricke W (1966) Schwefel-isotopen-verhältnisse in formationswässern und evaporiten nord-und süddeutschlands. Chem Geol 1:211–220. https://doi.org/10.1016/0009-2541(66)90017-9
Naumov GB, Motorina ZM, Naumov VB (1971a) Conditions of formation of carbonates in veins of the lead-cobalt-nickel-silver-uranium type (translation from Geokhimiya, No 8, pp. 938–948, 1971). Geochem Int+ 8:590–598.
Naumov GB, Ryzhenko BN, Khodakovsky IL (1971b) Handbook of thermodynamic data. Moscow Atomizdat, Moscow ((in Russian))
Obolensky A, Gushchina L, Borisenko A, Borovikov A, Pavlova G (2007) Antimony in hydrothermal processes: solubility, conditions of transfer, and metal-bearing capacity of solutions. Russ Geol Geophys 48:992–1001. https://doi.org/10.1016/j.rgg.2007.11.006
Ohmoto H, Lasaga AC (1982) Kinetics of reactions between aqueous sulfates and sulfides in hydrothermal systems. Geochim Cosmochim Ac 46:1727–1745. https://doi.org/10.1016/0016-7037(82)90113-2
Ondrus P, Veselovsky F, Gabasova A, Drabek M, Dobes P, Maly K, Hlousek J, Sejkora J (2003a) Ore-forming processes and mineral parageneses of the Jáchymov ore district. J GeoSci-Czech 48:157–192
Ondrus P, Veselovsky F, Gabasova A, Hlousek J, Srein V, Vavrin I, Skala R, Sejkora J, Drabek M (2003b) Primary minerals of the Jáchymov ore district. J GeoSci-Czech 48:19–146
Otto J (1964) Die Fluoritgrube “Gottesehre”. Unpublished Diplom Thesis, Albert Ludwigs Universität zu Freiburg
Otto J (1967) Der Fluoritgang der Grube “Gottesehre” bei Urberg Kr. Säckingen (Südschwarzwald). Jh Geolog Landesamt Baden-Württemberg 9:25–50
Páez GN, Ruiz R, Guido DM, Ríos FJ, Subias I, Recio C, Schalamuk I (2016) High-grade ore shoots at the Martha epithermal vein system, Deseado Massif, Argentina: the interplay of tectonic, hydrothermal and supergene processes in ore genesis. Ore Geol Rev 72:546–561. https://doi.org/10.1016/j.oregeorev.2015.07.026
Pekov IV, Levitskiy VV, Krivovichev VG (2010) Mineralogy of the Belorechenskoye deposit (Northern Caucasus, Russia). Mineral Almanac 15:1–96
Penrose RAF (1910) Some causes of ore shoots. Econ Geol 5:97–133. https://doi.org/10.2113/gsecongeo.5.2.97
Pfaff K, Romer RL, Markl G (2009) U-Pb ages of ferberite, chalcedony, agate, ‘U-mica’ and pitchblende: constraints on the mineralization history of the Schwarzwald ore district. Eur J Mineral 21:817–836. https://doi.org/10.1127/0935-1221/2009/0021-1944
Raiswell R, Bottrell SH, Al-Biatty HJ, Tan MM (1993) The influence of bottom water oxygenation and reactive content on sulfur incorporation into bitumen from jurassic marine shales. Am J Sci 293:569–596. https://doi.org/10.2475/ajs.293.6.569
Renock D, Becker U (2011) A first principles study of coupled substitution in galena. Ore Geol Rev 42:71–83. https://doi.org/10.1016/j.oregeorev.2011.04.001
Rick B (1990) Sulphur and oxygen isotopic composition of Swiss Gipskeuper (Upper Triassic). Chem Geol: Iso Geo Sec 80:243–250. https://doi.org/10.1016/0168-9622(90)90031-7
Robinson B, Ohmoto H (1973) Mineralogy, fluid inclusions, and stable isotopes of the Echo Bay U-Ni-Ag-Cu deposits, Northwest Territories, Canada. Econ Geol 68:635–656. https://doi.org/10.2113/gsecongeo.68.5.635
Rupf I, Nitsch E (2008) Das Geologische Landesmodell von Baden-Württemberg: Datengrund lagen, technische Umsetzung und erste geologische Ergebnisse. Regierungspräsidium Freiburg: Landesamt für Geologie RuB. Regierungspräsidium Freiburg: Landesamt für Geologie, Rohstoffe und Bergbau. LGRB-Informationen 21: pp 82.
Ruvalcaba-Ruiz DC, Thompson TB (1988) Ore deposits at the Fresnillo mine, Zacatecas, Mexico. Econ Geol 83:1583–1597. https://doi.org/10.2113/gsecongeo.83.8.1583
Ryall W (1979) Mercury in the Broken Hill (NSW, Australia) lead-zinc-silver lodes. J Geochem Explor 11:175–194. https://doi.org/10.1016/0375-6742(79)90022-0
Sangster D (2009) Geology of base metal deposits. Eolss Publishers Co., Ltd., Singapore
Scharrer M, Kreissl S, Markl G (2019) The mineralogical variability of hydrothermal native element-arsenide (five-element) mineralizations and the role of aqueous sulfide. Ore Geol Rev 113.https://doi.org/10.1016/j.oregeorev.2019.103025
Scharrer M, Sandritter K, Walter BF, Neumann U, Markl G (2020) Formation of native arsenic in hydrothermal base metal deposits and related supergene U6+ enrichment: The Michael vein near Lahr, SW Germany. Am Mineral 105:727–744. https://doi.org/10.2138/am-2020-7062
Scharrer M, Reich R, Fusswinkel T, Walter B, Markl G (2021) Physical and chemical aspects of the formation of unconformity-related hydrothermal vein type deposits in a continental basement setting (Schwarzwald, Germany) based on LA-ICP-MS analyses of single fluid inclusions in fluorite Chem Geol 575:120260. https://doi.org/10.1016/j.chemgeo.2021.120260.
Schürenberg H (1950) Die Erzgänge Teufelsgrund und Schindler im Untermünstertal und ihr quantitativer Mineralgehalt. Dissertation, Albert-Ludwigs-Universität Freiburg
Schwinn G, Wagner T, Baatartsogt B, Markl G (2006) Quantification of mixing processes in ore-forming hydrothermal systems by combination of stable isotope and fluid inclusion analyses. Geochim Cosmochim Ac 70:965–982. https://doi.org/10.1016/j.gca.2005.10.022
Shepherd T, Bottrell S, Miller M (1991) Fluid inclusion volatiles as an exploration guide to black shale-hosted gold deposits, Dolgellau gold belt, North Wales, UK. J Geochem Explor 42:5–24. https://doi.org/10.1016/0375-6742(91)90058-3
Siewers U (1977) Beitrag zur Häufigkeit und Isotopenzusammensetzung des Schwefels in magmatischen und metamorphen Gesteinen. PhD Thesis, University of Göttingen
Squire RJ, Robinson JA, Rawling TJ, Wilson CJ (2008) Controls on ore shoot locations and geometries at the Stawell Gold Mine, Southeastern Australia: contributions of the volcanosedimentary, alteration, and structural architecture. Econ Geol 103:1029–1041. https://doi.org/10.2113/gsecongeo.103.5.1029
Staude S, Wagner T, Markl G (2007) Mineralogy, mineral compositions and fluid evolution at the Wenzel hydrothermal deposit, Southern Germany: implications for the formation of Kongsberg-type silver deposits. Can Mineral 45:1147–1176. https://doi.org/10.2113/gscanmin.45.5.1147
Staude S, Mordhorst T, Neumann R, Prebeck W, Markl G (2010) Compositional variation of the tennantite–tetrahedrite solid-solution series in the Schwarzwald ore district (SW Germany): the role of mineralization processes and fluid source. Mineral Mag 74:309–339. https://doi.org/10.1180/minmag.2010.074.2.309
Staude S, Göb S, Pfaff K, Ströbele F, Premo WR, Markl G (2011) Deciphering fluid sources of hydrothermal systems: a combined Sr-and S-isotope study on barite (Schwarzwald, SW Germany). Chem Geol 286:1–20. https://doi.org/10.1016/j.chemgeo.2011.04.009
Staude S, Werner W, Mordhorst T, Wemmer K, Jacob DE, Markl G (2012) Multi-stage Ag–Bi–Co–Ni–U and Cu–Bi vein mineralization at Wittichen, Schwarzwald, SW Germany: geological setting, ore mineralogy, and fluid evolution. Miner Deposita 47:251–276. https://doi.org/10.1007/s00126-011-0365-4
Steen H (2013) Bergbau auf Lagerstätten des Südlichen Schwarzwalds. Ein Beitrag zur Bergbaugeschichte und Lagerstättenkunde zwischen Dreisamtal und Hochrhein. Books on Demand, Norderstedt.
Steen H (2020) Mineralien, in Lagerstätten und Bergbau bei Wieden im Südschwarzwald. Unter besonderer Berücksichtigung der Grube Finstergrund. Erzgräber 35:59–96
Stober I, Bucher K (2004) Fluid sinks within the earth’s crust. Geofluids 4:143–151. https://doi.org/10.1111/j.1468-8115.2004.00078.x
Stone W, Archibald N (2004) Structural controls on nickel sulphide ore shoots in Archaean komatiite, Kambalda, WA: the volcanic trough controversy revisited. J Struct Geol 26:1173–1194. https://doi.org/10.1016/j.jsg.2003.11.014
Takahashi R, Matsueda H, Okrugin VM, Ono S (2006) Polymetallic and Au-Ag mineralizations at the Mutnovskoe deposit in South Kamchatka, Russia. Resour Geol 56:141–156. https://doi.org/10.1111/j.1751-3928.2006.tb00275.x
Tomkins AG, Pattison DR, Zaleski E (2004) The Hemlo gold deposit, Ontario: an example of melting and mobilization of a precious metal-sulfosalt assemblage during amphibolite facies metamorphism and deformation. Econ Geol 99:1063–1084. https://doi.org/10.2113/gsecongeo.99.6.1063
Tomkins AG (2010) Wetting facilitates late-stage segregation of precious metal–enriched sulfosalt melt in magmatic sulfide systems. Geology 38:951–954. https://doi.org/10.1130/G31263.1
van Dongen M, Weinberg RF, Tomkins AG (2010) REE-Y, Ti, and P remobilization in magmatic rocks by hydrothermal alteration during Cu-Au deposit formation. Econ Geol 105:763–776. https://doi.org/10.2113/gsecongeo.105.4.763
Virgo S, Abe S, Urai JL (2014) The evolution of crack seal vein and fracture networks in an evolving stress field: insights from discrete element models of fracture sealing. J Geophys Res-Sol Ea 119:8708–8727. https://doi.org/10.1002/2014JB011520
Wager R (1935) Die Schwarzwälder Gneise Geol Rundsch 26:161–185. https://doi.org/10.1007/BF01807727
Walter BF, Immenhauser A, Geske A, Markl G (2015) Exploration of hydrothermal carbonate magnesium isotope signatures as tracers for continental fluid aquifers, Schwarzwald mining district, SW Germany. Chem Geol 400:87–105. https://doi.org/10.1016/j.chemgeo.2015.02.009
Walter BF, Burisch M, Markl G (2016) Long-term chemical evolution and modification of continental basement brines–a field study from the Schwarzwald, SW Germany. Geofluids 16:604–623. https://doi.org/10.1111/gfl.12167
Walter BF, Burisch M, Marks MA, Markl G (2017) Major element compositions of fluid inclusions from hydrothermal vein-type deposits record eroded sedimentary units in the Schwarzwald district, SW Germany. Miner Deposita 52:1191–1204. https://doi.org/10.1007/s00126-017-0719-7
Walter BF, Burisch M, Fusswinkel T, Marks MA, Steele-MacInnis M, Wälle M, Apukhtina OB, Markl G (2018a) Multi-reservoir fluid mixing processes in rift-related hydrothermal veins, Schwarzwald, SW-Germany. J Geochem Explor 186:158–186. https://doi.org/10.1016/j.gexplo.2017.12.004
Walter BF, Gerdes A, Kleinhanns IC, Dunkl I, von Eynatten H, Kreissl S, Markl G (2018b) The connection between hydrothermal fluids, mineralization, tectonics and magmatism in a continental rift setting: fluorite Sm-Nd and hematite and carbonates U-Pb geochronology from the Rhinegraben in SW Germany. Geochim Cosmochim Ac 240:11–42. https://doi.org/10.1016/j.gca.2018.08.012
Walter BF, Kortenbruck P, Scharrer M, Zeitvogel C, Wälle M, Mertz-Kraus R, Markl G (2019) Chemical evolution of ore-forming brines–basement leaching, metal provenance, and the redox link between barren and ore-bearing hydrothermal veins. A case study from the Schwarzwald mining district in SW-Germany. Chem Geol 506:126–148. https://doi.org/10.1016/j.chemgeo.2018.12.038
Walter BF, Jensen JL, Coutinho P, Laurent O Markl G, Steele-MacInnis M (2020) Formation of hydrothermal fluorite-hematite veins by mixing of continental basement brine and redbed-derived fluid: Schwarzwald mining district SW-Germany. J Geochem Explor: Article Nr 106512.https://doi.org/10.1016/j.gexplo.2020.106512
Werner W, Markl G, Steen H (2020) Lagerstätteninhalt und Entstehung der Gänge bei Wieden, in Lagerstätten und Bergbau bei Wieden im Südschwarzwald. Unter besonderer Berücksichtigung der Grube Finstergrund. Erzgräber 35:28–45
Widemann N (1988) Die neuen Gang- und Nebengesteinsaufschlüsse im Bereich der Grube ‘Gottesehre’ bei Urberg im Südschwarzwald mit Aufnahme der Gangmineralisation bei den ‘Erzlöchern.’ Diplomarbeit, Univ, Freiburg
Yesares L, Sáez R, De Almodóvar GR, Nieto JM, Gómez C, Ovejero G (2017) Mineralogical evolution of the Las Cruces gossan cap (Iberian Pyrite Belt): from subaerial to underground conditions. Ore Geol Rev 80:377–405. https://doi.org/10.1016/j.oregeorev.2016.05.018
Zeschke G (1959) Die Flusspatvorkommen der Gewerkschaft Finstergrund. pp 44.
Acknowledgements
First, we thank the Thermoddem team of the BRGM for the very friendly cooperation and the implementation of new species into the thermodynamic database. Furthermore, we thank Dr. Günter Grundmann for providing the microscopic images of his private samples of the Wieden ore shoots. We would like to acknowledge the Chief and Associate Editor and the reviewers for their constructive criticism and feedback, which led to an improved version of this manuscript. Last but not least, we thank S. Schafflick for the fast and reliable sample preparation.
Funding
Open Access funding enabled and organized by Projekt DEAL. This research was funded by a combination of the German Research Foundation (DFG), grant no. MA 2135/25–1, and by the German-American Fulbright Commission.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Editorial handling: D. Dolejs
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scharrer, M., Epp, T., Walter, B. et al. The formation of (Ni-Co-Sb)-Ag-As ore shoots in hydrothermal galena-sphalerite-fluorite veins. Miner Deposita 57, 853–885 (2022). https://doi.org/10.1007/s00126-021-01059-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00126-021-01059-y