
Abstract
The appropriate utilisation, storage and conversion of nutrients in peripheral tissues, referred to as nutrient partitioning, is a fundamental process to adapt to nutritional and metabolic challenges and is thus critical for the maintenance of a healthy energy balance. Alterations in this process during nutrient excess can have deleterious effects on glucose and lipid homeostasis and contribute to the development of obesity and type 2 diabetes. Nutrient partitioning is a complex integrated process under the control of hormonal and neural signals. Neural control relies on the capacity of the brain to sense circulating metabolic signals and mount adaptive neuroendocrine and autonomic responses. This review aims to discuss the hypothalamic neurocircuits and molecular mechanisms controlling nutrient partitioning and their potential contribution to metabolic maladaptation and disease.
Similar content being viewed by others


Introduction
Obesity and associated complications, including type 2 diabetes, represent a huge health burden worldwide. Obesity is a complex multifactorial disease resulting from the interaction of genes, biology and the environment [1]. Our understanding of the aetiology of obesity is incomplete but it is clear that the dramatic increase in obesity rates are largely due to elevated intake of energy-dense food and, to a lesser extent, an inactive lifestyle. Despite the straightforward energy balance equation explaining fat mass gain, the processes and pathways underlying hyperphagia and the manner by which excess energy is handled in different tissues remain elusive. At the genetic level, BMI is strongly associated with genetic loci linked to central nervous system (CNS) processes (e.g. synaptic function and neurotransmitter signalling) that may increase susceptibility to overfeeding and body fat gain [2]. In addition to fat mass, fat distribution (central/upper vs lower body) is an important determinant of cardiometabolic complications. Upper body fat distribution (assessed by waist-to-hip ratio and adjusted for BMI) is associated with genes involved in lipid metabolism, adipocyte biology and differentiation [3]. Together, these studies suggest a complex polygenic architecture of fat mass gain and distribution involving both the CNS and the periphery. In addition to genetic factors, the type of ingested energy and the metabolic fate of nutrients, including their utilisation, storage and conversion (e.g. fatty acid synthesis from glucose), in peripheral tissues can affect fat distribution, mass and ectopic fat deposition [4]. This process, referred to as nutrient partitioning, occurs both at the whole body level, which relies on a complex neural and hormone-regulated interplay between organs and tissues, and at the cellular level, with coordinated coupling between nutrient metabolic pathways. Alterations in nutrient partitioning, often referred to as metabolic inflexibility, during nutrient surfeit can lead to the hyperglycaemia, dyslipidaemia and ectopic fat deposition that are major hallmarks of obesity and type 2 diabetes [5]. Although the direct action of hormones in peripheral tissues is a strong driver of nutrient metabolism, the CNS plays a key role in the regulation of nutrient fluxes and partitioning [6]. This control relies on the ability of the brain to sense and integrate circulating hormones and nutrients to, in turn, mount adaptive behavioural, autonomic and neuroendocrine responses to maintain energy homeostasis. These central mechanisms were likely selected throughout the evolution to adapt to periods of nutrient scarcity or starvation. In the current food-rich environment, they may favour energy intake and storage. The aim of this review is to discuss the neurocircuits and molecular mechanisms that regulate nutrient partitioning and their contribution to metabolic diseases.
Hypothalamic control of nutrient partitioning
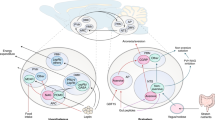
Within the CNS, it is well known that the hypothalamus integrates circulating metabolic signals and afferent autonomic inputs to regulate not only feeding and energy expenditure but also nutrient partitioning (Fig. 1). This control relies on the autonomic nervous system (ANS), which innervates every metabolic tissue and endocrine organ, and the hypothalamic–pituitary axis, which controls the secretion of metabolic hormones (e.g. cortisol, growth hormone). Decades ago, studies established that electrical stimulation of the lateral hypothalamus (LH) and the ventromedial hypothalamus (VMH), or electrical activation of the sympathetic or parasympathetic branch of the ANS had robust effects on glucose and fatty acid metabolism, pancreatic hormones and catecholamine secretion (reviewed in [7]) (Fig. 1). Electrical stimulation of the VMH or sympathetic nerves rapidly reduces insulin secretion and enhances glucagon secretion, hepatic glycogenolysis, lipolysis in white adipose tissue (WAT) and thermogenesis in brown adipose tissue (BAT) [7]. Conversely, stimulation of the LH or parasympathetic nervous system (PNS) increases insulin secretion and hepatic glycogenesis. These rapid metabolic effects in the liver and adipose tissues are initiated by autonomic inputs, while pancreatic hormones and adrenaline (epinephrine) secretion amplify and maintain the responses [7]. This line of work led to the notion that the VMH and LH act reciprocally in terms of the hormone secretion and metabolic functions stimulated, with the VMH promoting catabolic pathways via the sympathetic nervous system (SNS), and the LH and the PNS favouring energy storage. However, this binary model was challenged by the discovery of leptin in 1994 coupled with major methodological developments (e.g. Cre–lox system, opto- and chemogenetics), which recharged the field and led to the identification of novel neuronal pathways and mechanisms.

Central control of peripheral nutrient partitioning. In the hypothalamus, the activity of Agouti-related peptide (AgRP) and proopiomelanocortin (POMC) arcuate (ARC) neurons is regulated by circulating metabolic signals. In turn, the release of alpha-melanocyte stimulating hormone (α-MSH) and AgRP modulates the activity of post-synaptic melanocortin 4 receptor (MC4R) neurons in the paraventricular nucleus (PVN) projecting to the brainstem and regulating the activity of the autonomic nervous system. The autonomic nervous system innervates all metabolic tissues and endocrine organs via the parasympathetic and sympathetic branches. These autonomic inputs orchestrate the utilisation (transport and oxidation), storage (glycogen and triacylglycerol) and conversion (gluconeogenesis and fatty acid synthesis from glucose) of glucose and fatty acids (FA), and secretion of pancreatic hormones and adrenaline, which, in turn, contribute to nutrient partitioning. In addition, the hypothalamic–pituitary–adrenal axis regulates corticosterone secretion. Hormones in red stimulate glucose production while those in green stimulate glucose uptake and utilisation; the role of ghrelin is currently debated and so this is shown in black. GLP-1, glucagon-like peptide 1; GHSR, growth hormone secretagogue receptor (ghrelin receptor); IR, insulin receptor, LepR, Leptin receptor, TAG, triacylglycerol; TCA, tricarboxylic acid cycle. Created in Biorender.com. This figure is available as part of a downloadable slideset
The leptin era and the characterisation of hypothalamic neurocircuits
Leptin is a hormone secreted by the WAT with circulating levels proportional to fat mass. Leptin-deficient rodents and humans develop severe obesity, while leptin administration to leptin-deficient humans and mice lowers food intake and stimulates energy expenditure to ultimately reduce fat stores and promote leanness [8]. As such, leptin was initially proposed to act as an energy surfeit signal, elevated levels of which in states of positive energy balance stimulate energy expenditure and reduce energy intake. However, the current model rather suggests that the hormone is an energy deficiency signal, reduced levels of which during an energy deficit stimulates feeding and reduces energy expenditure. Although still debated, increased leptin levels during overfeeding may contribute to ‘leptin resistance’ and thus favour body weight gain [9]. Consistent with this, partial reduction or neutralisation of leptin action protects against diet-induced obesity in mice by increasing central leptin sensitivity [10]. Although studies in rodents suggest that leptin acts peripherally to regulate metabolism [11,12,13], the main targets of the hormone in energy balance regulation are localised in several brain regions, including the hypothalamus.
In addition to controlling feeding behaviour and energy expenditure, central administration of leptin was found to regulate glucose homeostasis. Intracerebroventricular or intra-VMH leptin administration acutely increases glucose utilisation in skeletal muscles, heart and BAT, while increasing hepatic glucose output via the SNS [14, 15]. Notably, leptin did not affect blood glucose and plasma insulin, suggesting that hepatic glucose production was counterbalanced by glucose utilisation. In line with this, central manipulations of leptin or leptin receptor (LepR) in pathological rodent models have beneficial effects on glucose metabolism. The rescue of LepR specifically in the arcuate nucleus of LepR-deficient obese rodents normalises hyperglycaemia [16, 17]. Notably, administration of leptin in the brain robustly improves hyperglycaemia in rodent models of insulin-deficient diabetes [18, 19], though the magnitude of the effects may be dependent on leptin dosage and treatment duration as well as residual insulin in a mouse model of streptozotocin-induced diabetes [20]. The glucose-lowering action of leptin is mediated by a reduction in glucagon and corticosterone levels, leading to reduced hepatic glucose production and increased glucose utilisation in the soleus muscle and BAT. Besides glucose, leptin stimulates lipid mobilisation in WAT [21] and fatty acid oxidation in skeletal muscles via the SNS [11] and reduces hepatic [22] and heart triacylglycerol content [23]. Thus, central administration of leptin stimulates glucose and fatty acid utilisation, thereby preventing ectopic lipid deposition.
The discovery of leptin led to the identification of leptin-responsive neuronal circuits that control nutrient partitioning, including the hypothalamic melanocortin pathway originating in the arcuate nucleus (ARC) [24]. The ARC contains two functionally opposing neuronal populations: the agouti-related peptide (AgRP)–Neuropeptide Y (NPY) neurons and proopiomelanocortin (POMC) neurons (Fig. 1). AgRP and POMC neurons project to similar post-synaptic targets located in the paraventricular nucleus (PVN), VMH, dorsomedial hypothalamus and LH. When activated by signals of energy sufficiency (e.g. leptin, insulin), POMC neurons release α-melanocyte-stimulating hormone (α-MSH), which activates the melanocortin-4 receptor (MC4R) in post-synaptic neurons projecting to the brainstem and downstream anorectic and catabolic responses via the ANS (Fig. 1). In contrast, signals of energy deprivation (e.g. ghrelin, hypoglycaemia) activate AgRP neurons and the release of AgRP, an endogenous MC4R antagonist, and stimulation of feeding and anabolic processes in peripheral tissues. In addition, AgRP neurons send inhibitory GABAergic inputs into neighbouring POMC neurons, thereby exerting a dual inhibition on POMC signalling (Fig. 1). The importance of this system in the aetiology of obesity is underscored by findings showing that MC4R or POMC deficiency leads to obesity in rodents and that mutations in the gene coding for MC4R are the most frequent form of monogenic human obesity [25].
There is mounting evidence that metabolic hormones (e.g. leptin, insulin, ghrelin, incretins) and nutrients directly influence the activity of POMC and AgRP neurons to regulate food intake. Alteration of hormonal signalling or nutrient sensing in neurons of the ARC and other nuclei can have a profound impact on feeding and body weight regulation. Summarising this stream of work is beyond the scope of this review and has been extensively discussed by others [26]. We will focus here on the neuronal and metabolic pathways in the mediobasal hypothalamus (MBH) that regulate peripheral nutrient partitioning.
Neuronal control of nutrient partitioning
Evidence of a role for AgRP neurons in nutrient partitioning surfaced from studies of AgRP neuronal ablation in neonatal mice. AgRP-ablated mice develop late-onset obesity when fed a standard diet [27]. Obesity was not the result of an increased energy intake but, rather, enhanced feed efficiency resulting from increased hepatic triacylglycerol synthesis from carbohydrates and a shift in nutrient oxidation coordinated by the ANS that favoured lipid vs carbohydrate utilisation. The enhanced lipid utilisation in muscles and adipose tissues of AgRP-ablated mice was a metabolic advantage during high-fat feeding by reducing glucose intolerance and diet-induced obesity [27]. Consistent with these findings, the ablation of AgRP neurons in adult mice subjected to enteral feeding (to avoid starvation and promote weight gain) reduces fat mass gain vs control mice receiving the same amount of calories enterally [28]. It is important to mention that AgRP neuron ablation also induces NPY and GABA deficiency that may contribute to the observed phenotypes. In contrast, chemo- or opto-genetic activation of AgRP neurons in the absence of food or under pair-fed conditions increases glucose utilisation in mice while decreasing lipid utilisation via the ANS thereby promoting lipogenesis and adiposity [28]. Consistent with the antagonistic properties of AgRP on MC4R, disruption or antagonism of MC4R reduces lipid utilisation while it increases triacylglycerol synthesis and fat accumulation in WAT (in normal and pair-fed conditions) [29] as well as HDL-cholesterol by reducing hepatic uptake via the ANS [30]. In addition, inhibition of MC4R reduces glucose utilisation in muscles and BAT [29] and impairs BAT thermogenic activity [31]. Conversely, MC4R activation induces lipolysis in WAT and thermogenesis in BAT [32]. Importantly, these changes in nutrient metabolism occur before any change in adiposity and independently of food intake. Finally, the glucose-lowering effect of central leptin delivery in models of diabetes is in part dependent on inhibition of AgRP neurons and increased melanocortinergic tone [18, 33,34,35,36]. Together, these studies demonstrate that AgRP neurons exert strong control over nutrient partitioning by promoting lipid storage, while POMC and post-synaptic MC4R neurons favour nutrient mobilisation and utilisation. Importantly, recent studies suggest that the melanocortin system also regulates the plasticity of adipose tissue and its capacity to expand. Enhancing leptin and insulin signalling in POMC neurons promotes an SNS-dependent browning of WAT associated with improved glucose homeostasis [37, 38], while browning is prevented by activation of AgRP neurons [39] (Fig. 1). These results are consistent with the well-described SNS-induced proliferation of BAT precursors and inhibition of preadipocyte proliferation in WAT [40]. Finally, MC4R disruption promotes a PNS-dependent proliferation of WAT precursors that favours WAT expansion and fat mass gain [41]. Thus, the ARC melanocortin system exerts dual control of adipose tissue plasticity and metabolism that contributes to the appropriate storage and utilisation of nutrients.
The control of nutrient partitioning does not only rely on ARC neurons; there are other neuronal circuits that play a critical role in metabolic adaptations. Notably, VMH neurons are critical for the counterregulatory response to hypoglycaemia (CRRH), a neuroendocrine response involving glucocorticoids, glucagon and adrenaline secretion and increased sympathetic tone to the liver and WAT. These responses stimulate hepatic glucose production and the release of fatty acids acting as an alternative fuel to spare glucose for the brain. As such, the CRRH is a centrally orchestrated pathway relying on glucose-sensing neurons and appropriate nutrient partitioning [42]. In the VMH, steroidogenic factor-1 (SF1)-glutamatergic neurons play a key role in glucose homeostasis during fasting and hypoglycaemia [43]. Genetic disruption of glutamate release by SF1 neurons impairs glucagon secretion and gluconeogenesis during fasting and hypoglycaemia. Conversely, optogenetic activation of SF1 neurons leads to hyperglycaemia whereas their silencing prevents the normalisation of glycaemia during hypoglycaemia by impairing glucagon and corticosterone secretion [44]. Whether SF1 neurons are also implicated in hypoglycaemia-induced lipolysis and fatty acid utilisation remains to be determined. In line with these findings, it was shown that increased GABAergic tone in the VMH impairs the CRRH [45]. Importantly, GABAA agonism blunts neuroendocrine and sympathetic responses to hypoglycaemia and exercise in individuals with type 1 diabetes [46, 47]. These studies suggest that alteration of VMH neuron activity may be implicated in hypoglycaemia unawareness, a condition characterised by impaired CRRH which is induced by antecedent hypoglycaemic episodes in individuals with diabetes receiving insulin treatment.
Intracellular mechanisms governing peripheral nutrient partitioning
As discussed above, nutrient partitioning also occurs at the cellular level. Several key enzymes in peripheral tissues are responsible for the glucose-dependent partitioning of fatty acid metabolism between oxidation and esterification. Glucose metabolism inhibits the energy sensor 5′ AMP-activated protein kinase (AMPK) leading to the activation of acetyl-CoA carboxylase (ACC) and the concomitant generation of malonyl-CoA from glucose-derived acetyl-CoA. Malonyl-CoA reduces acyl-CoA mitochondrial oxidation via inhibition of carnitine palmitoyl transferase-1 (CPT-1). Importantly, malonyl-CoA is the substrate for fatty acid synthesis via fatty acid synthase and glycerol-3-phosphate derived from glycolysis constitutes the backbone for triacylglycerol generation. As such, glucose metabolism reduces fatty acid oxidation and favours the storage of glucose-derived carbons as lipids.
Several lines of evidence suggest that this metabolic coupling pathway operates in MBH cells to regulate peripheral nutrient partitioning. As described in peripheral tissues, the malonyl-CoA level in the MBH is tightly regulated by glucose via AMPK. Once activated, AMPK inactivates ACC, leading to a decrease in MBH malonyl-CoA levels [48] (Fig. 2). Inversely, inhibition of AMPK by glucose leads to activation of ACC and malonyl-CoA synthesis. In line with this, our laboratory showed that glucose-induced inhibition of AMPK reduces fatty acid oxidation while increasing fatty acid esterification into triacylglycerol in hypothalamic neurons and astrocytes [49]. Thus, malonyl-CoA acts as a metabolic signal modulating fatty acid partitioning between esterification and oxidation in the MBH. Importantly, malonyl-CoA accumulation in the MBH inhibits feeding and increases fatty acid oxidation in skeletal muscle via the SNS [50], while reduction of malonyl-CoA leads to hyperphagia and obesity [51, 52]. Finally, intracellular accumulation of acyl-CoA in the MBH triggers neural responses to suppress hepatic glucose production via the PNS [53, 54]. Together, these data suggest tight regulation of fatty acid oxidation by glucose in the MBH which controls peripheral glucose production and fatty acid utilisation. Although these studies provided the first evidence that metabolic coupling pathways operate in the MBH to regulate nutrient partitioning, most of these interventions did not target specific cell or neuronal population(s) in the MBH. As such, the neuronal populations involved in peripheral metabolic responses remain unclear.

Nutrient partitioning in hypothalamic neurons. Key enzymes are responsible for a coordinated coupling between glucose and fatty acid (FA) intracellular metabolism in hypothalamic neurons. Glucose and malonyl-CoA regulates the partitioning of FA metabolism between mitochondrial oxidation and esterification into glycerolipids. In the mitochondria, carnitine and carnitine acetyltransferase (CrAT) regulates the pool of acetyl-CoA derived from glucose and FA oxidation and its partitioning between oxidation in the tricarboxylic acid cycle (TCA) or mitochondrial efflux for malonyl-CoA synthesis. The autophagy-dependent hydrolysis of triacylglycerol (TAG) stored in lipid droplets generates FA for mitochondrial oxidation. The partitioning of nutrient is controlled by metabolic hormones acting through the energy sensor 5′ AMP-activated protein kinase (AMPK). Whether nutrient partitioning occurs and is regulated similarly in AgRP and POMC neurons, other neuronal populations or cell types (glia) remains an important unanswered question. The green arrows indicate stimulatory actions, the red arrows indicate inhibitory actions. FA, fatty acid; GABA, γ-aminobutyric acid; ACC, acetyl-CoA carboxylase; ATG7, autophagy related 7; CPT-1, Carnitine palmitoyltransferase 1; DAG, diacylglycerol; FAS, Fatty acid synthase; G3P, glycerol 3-phosphate; GHSR, growth hormone secretagogue receptor (ghrelin receptor); MAG, monoacylglycerol; OXPHOS, oxidative phosphorylation; PDH, pyruvate dehydrogenase. Created in Biorender.com. This figure is available as part of a downloadable slideset
New evidence supporting the role of neuronal nutrient partitioning in the regulation of peripheral nutrient partitioning has been provided by recent studies on the enzyme carnitine acetyltransferase (CrAT). CrAT regulates the mitochondrial pool of acetyl-CoA and thus oxidative metabolism. During periods of nutrient surfeit, CrAT catalyses the formation of acetylcarnitine from carnitine and acetyl-CoA generated by fatty acid and glucose oxidation (Fig. 2). The cellular efflux of acetylcarnitine prevents the inhibition of glycolytic enzymes and pyruvate dehydrogenase by acetyl-CoA, thereby favouring glucose oxidation [55]. During times of energy deficit (e.g. exercise, food restriction), circulating acetylcarnitine uptake and its conversion to acetyl-CoA by CrAT in skeletal muscle fuel the tricarboxylic acid (TCA) cycle and support oxidative metabolism [56]. As such, CrAT plays a key role in metabolic adaptation and flexibility in skeletal muscle. Importantly, the disruption of CrAT specifically in AgRP neurons increases peripheral fatty acid oxidation and reduces glucose utilisation under fasting conditions [57]. In addition, AgRP CrAT knockout mice have a blunted switch from fatty acid to glucose utilisation during the transition from fasting to refeeding. During chronic energy restriction, CrAT deficiency in AgRP neurons increases fatty acid utilisation leading to a greater fat loss [58]. Together, these studies suggest that neuronal carnitine and CrAT may be key regulators of acetyl-CoA metabolic fate to control peripheral nutrient partitioning [59].
Autophagy is another pathway that plays a key role in cellular energy homeostasis. During times of nutrient deficiency, activation of AMPK stimulates autophagy and the breakdown of cellular components that generate metabolic substrates to maintain cellular energy. Lipophagy is the autophagic degradation of intracellular triacylglycerol stored in lipid droplets, thereby releasing fatty acids that can be oxidised (Fig. 2). This pathway is inhibited by activation of the mTOR pathway when nutrients are plentiful. Interestingly, autophagy is observed in MBH neurons under normal conditions, and disruption of autophagy related 7 (ATG7), an essential protein for the induction of autophagy, in ARC neurons has profound effects on energy homeostasis. Deletion of ATG7 in POMC neurons leads to overweight and glucose intolerance in chow-fed mice [60,61,62]. This phenotype is associated with disruption of POMC axonal projections to the PVN (without affecting the number of POMC neurons) [60] and reduced sympathetic outflow to WAT and lipolysis [61]. In addition, ATG7 deficiency in POMC neurons impairs lipophagy in BAT and liver during cold exposure, thereby increasing triacylglycerol content [63]. Conversely, pharmacological activation of autophagy in the MBH induces lipophagy in BAT and liver and leads to a lean phenotype with increased SNS activity, WAT lipolysis and BAT thermogenesis [63, 64]. Importantly, inactivation of ATG7 in AgRP neurons promotes neuronal triacylglycerol accumulation and a lean phenotype associated with increased locomotor activity and lipolysis [65]. These findings thus suggest that autophagy in MBH neurons regulates peripheral autophagy and further support the notion that autophagy dysregulation may have a causal role in the development of obesity and type 2 diabetes [66].
Taken together, these findings strongly suggest that pathways regulating nutrient metabolism and partitioning in MBH neurons are key regulators of peripheral nutrient partitioning. Importantly, the activity of these neuronal pathways is controlled by metabolic hormones, and the central effects of several hormones are dependent on nutrient availability and neuronal metabolism (Fig. 2). For example, the anorectic action of leptin relies on hypothalamic AMPK inhibition [67], mTOR activation [68] and the generation of malonyl-CoA by ACC [69]. Conversely, stimulation of food intake by ghrelin requires AMPK activation and increased mitochondrial fatty acid oxidation [70, 71]. Notably, feeding responses to ghrelin and glucagon-like peptide-1 are dependent on central glucose levels and AMPK activity [72,73,74]. Overall, this suggests the intricate and underappreciated metabolic integration of both humoral and nutrient signals in hypothalamic neurons to mount appropriate behavioural and metabolic responses. Key challenging questions remain as to whether (1) nutrient partitioning occurs and is regulated similarly in different hypothalamic neuronal populations and glia, and (2) these bioenergetic adaptations are required for metabolic sensing itself and/or to generate the energy and building blocks necessary for neuronal activity and plasticity in response to hormonal signals.
Implications for human obesity and diabetes
Translating these basic research findings to human physiology and assessing the contribution of the neural control of nutrient partitioning in the pathophysiology of human obesity and diabetes remain unmet challenges. Functional brain imaging has helped tremendously, allowing the mapping of human brain regions that respond to metabolic signals and how their activity is altered in obesity. Although these observations do not establish a causal link between changes in brain region activity and nutrient partitioning, recent clinical interventions support the idea that the brain regulates nutrient partitioning in humans and that this control can be improved or corrected in pathological conditions. For example, mutations in MC4R in obese humans are associated with decreased lipid utilisation compared with that in obese individuals with a normal MC4R genotype [29]. Short-term administration of setmelanotide, a novel MC4R agonist, rapidly increases lipid utilisation in obese individuals without changes in energy intake [75], thereby suggesting that the central melanocortin system regulates lipid partitioning in humans, as previously shown in rodents [29]. During long-term treatment, setmelanotide substantially decreases body weight mostly by reducing energy intake to suggest that MC4R agonism is a promising strategy for the treatment of obese people with defects in the melanocortin system [76]. Ongoing clinical studies may help determine if and to what extent MC4R agonism improves nutrient partitioning during long-term treatment.
Studies have shown that leptin-replacement therapy leads to major reductions in body weight and fat and normalises neuroendocrine and metabolic abnormalities in leptin-deficient individuals [8]. In addition, leptin has emerged as a treatment for lipodystrophy, a disorder characterised by adipose tissue deficiency, leading to hypoleptinaemia, glucose intolerance, hypertriglyceridaemia and hepatic steatosis [8]. Although the beneficial effects of leptin treatment in individuals with lipodystrophy are mostly explained by a reduction in food intake, a recent study showed that leptin therapy reduces circulating and hepatic triacylglycerol independently of energy intake [77]. Based on rodent studies showing that central administration of leptin stimulates peripheral nutrient utilisation via the melanocortin system and ANS, it is likely that the benefits of leptin replacement on ectopic fat deposition may implicate both its anorectic action and ANS-dependent stimulation of lipid utilisation.
Conclusion
As part of the critical function of the CNS in feeding behaviour and body weight regulation, it has become clear that specific brain regions and neurocircuits significantly contribute towards the orchestration of metabolic fluxes between peripheral tissues in rodents and humans. Increasing evidence suggests that alterations in these processes can contribute to the development of obesity and type 2 diabetes [5]. This is further supported by the strong association between altered ANS activity, ectopic fat and the metabolic and cardiovascular complications of obesity [78]. Although the literature highlights the fundamental role of specific hypothalamic neuronal populations and some underlying molecular mechanisms, much work will be required to characterise the complex interplay between nutrient and hormone sensing in MBH neurons, the contribution of other non-hypothalamic neurocircuits and glia in nutrient partitioning. In addition, the list of metabolic signals regulating nutrient partitioning is growing with the discovery of novel regulators harbouring potent glucose-lowering properties, including fibroblast growth factors 1 and 19 [79,80,81]. Finally, the central control of nutrient partitioning may also rely on external food cues and the pre-absorptive anticipatory phase (cephalic phase), which prime endocrine and metabolic organs for nutrient intake via the ANS [82]. This is a growing and exciting field of research that will potentially open up novel therapeutic avenues for obesity and diabetes.
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- AgRP:
-
Agouti-related peptide
- AMPK:
-
5′ AMP-activated protein kinase
- ANS:
-
Autonomic nervous system
- ARC:
-
Arcuate nucleus
- BAT:
-
Brown adipose tissue
- CNS:
-
Central nervous system
- CrAT:
-
Carnitine acetyl transferase
- CRRH:
-
Counterregulatory response to hypoglycaemia
- LepR:
-
Leptin receptor
- LH:
-
Lateral hypothalamus
- MBH:
-
Mediobasal hypothalamus
- MC4R:
-
Melanocortin-4 receptor
- α-MSH:
-
α-Melanocyte-stimulating hormone
- mTOR:
-
Mammalian target of rapamycin
- NPY:
-
Neuropeptide Y
- PNS:
-
Parasympathetic nervous system
- POMC:
-
Proopiomelanocortin
- PVN:
-
Paraventricular nucleus
- SF1:
-
Steroidogenic factor-1
- SNS:
-
Sympathetic nervous system
- VMH:
-
Ventromedial hypothalamus
- WAT:
-
White adipose tissue
References
Ghosh S, Bouchard C (2017) Convergence between biological, behavioural and genetic determinants of obesity. Nat Rev Genet 18(12):731–748. https://doi.org/10.1038/nrg.2017.72
Locke AE, Kahali B, Berndt SI et al (2015) Genetic studies of body mass index yield new insights for obesity biology. Nature 518(7538):197–206. https://doi.org/10.1038/nature14177
Justice AE, Karaderi T, Highland HM et al (2019) Protein-coding variants implicate novel genes related to lipid homeostasis contributing to body-fat distribution. Nat Genet 51(3):452–469. https://doi.org/10.1038/s41588-018-0334-2
Hall KD, Guo J (2017) Obesity energetics: body weight regulation and the effects of diet composition. Gastroenterology 152(7):1718–1727 e1713. https://doi.org/10.1053/j.gastro.2017.01.052
Goodpaster BH, Sparks LM (2017) Metabolic flexibility in health and disease. Cell Metab 25(5):1027–1036. https://doi.org/10.1016/j.cmet.2017.04.015
Myers MG Jr, Olson DP (2012) Central nervous system control of metabolism. Nature 491(7424):357–363. https://doi.org/10.1038/nature11705
Shimazu T (1981) Central nervous system regulation of liver and adipose tissue metabolism. Diabetologia 20(1):343–356. https://doi.org/10.1007/BF00254502
Flier JS (2019) Starvation in the midst of plenty: reflections on the history and biology of insulin and leptin. Endocr Rev 40(1):1–16. https://doi.org/10.1210/er.2018-00179
Flier JS, Maratos-Flier E (2017) Leptin’s physiologic role: does the emperor of energy balance have no clothes? Cell Metab 26(1):24–26. https://doi.org/10.1016/j.cmet.2017.05.013
Zhao S, Zhu Y, Schultz RD et al (2019) Partial leptin reduction as an insulin sensitization and weight loss strategy. Cell Metab 30(4):706–719 e706. https://doi.org/10.1016/j.cmet.2019.08.005
Minokoshi Y, Kim Y-B, Peroni OD et al (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415(6869):339–343. https://doi.org/10.1038/415339a
Huynh FK, Levi J, Denroche HC et al (2010) Disruption of hepatic leptin signaling protects mice from age- and diet-related glucose intolerance. Diabetes 59(12):3032–3040. https://doi.org/10.2337/db10-0074
Pereira S, O’Dwyer SM, Webber TD et al (2019) Metabolic effects of leptin receptor knockdown or reconstitution in adipose tissues. Sci Rep 9(1):3307. https://doi.org/10.1038/s41598-019-39498-3
Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ (1997) Acute stimulation of glucose metabolism in mice by leptin treatment. Nature 389(6649):374–377. https://doi.org/10.1038/38717
Minokoshi Y, Haque MS, Shimazu T (1999) Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes 48(2):287–291. https://doi.org/10.2337/diabetes.48.2.287
Coppari R, Ichinose M, Lee CE et al (2005) The hypothalamic arcuate nucleus: a key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab 1(1):63–72. https://doi.org/10.1016/j.cmet.2004.12.004
Morton GJ, Gelling RW, Niswender KD, Morrison CD, Rhodes CJ, Schwartz MW (2005) Leptin regulates insulin sensitivity via phosphatidylinositol-3-OH kinase signaling in mediobasal hypothalamic neurons. Cell Metab 2(6):411–420. https://doi.org/10.1016/j.cmet.2005.10.009
Fujikawa T, Berglund ED, Patel VR et al (2013) Leptin engages a hypothalamic neurocircuitry to permit survival in the absence of insulin. Cell Metab 18(3):431–444. https://doi.org/10.1016/j.cmet.2013.08.004
Perry RJ, Zhang XM, Zhang D et al (2014) Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat Med 20(7):759–763. https://doi.org/10.1038/nm.3579
Neumann UH, Denroche HC, Mojibian M, Covey SD, Kieffer TJ (2016) Insulin knockout mice have extended survival but volatile blood glucose levels on leptin therapy. Endocrinology 157(3):1007–1012. https://doi.org/10.1210/en.2015-1890
Zeng W, Pirzgalska RM, Pereira MM et al (2015) Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell 163(1):84–94. https://doi.org/10.1016/j.cell.2015.08.055
Hackl MT, Fürnsinn C, Schuh CM et al (2019) Brain leptin reduces liver lipids by increasing hepatic triglyceride secretion and lowering lipogenesis. Nat Commun 10(1):2717. https://doi.org/10.1038/s41467-019-10684-1
Mora C, Pintado C, Rubio B et al (2018) Central leptin regulates heart lipid content by selectively increasing PPAR beta/delta expression. J Endocrinol 236(1):43–56. https://doi.org/10.1530/joe-17-0554
Caron A, Richard D (2017) Neuronal systems and circuits involved in the control of food intake and adaptive thermogenesis. Ann N Y Acad Sci 1391(1):35–53. https://doi.org/10.1111/nyas.13263
Krashes MJ, Lowell BB, Garfield AS (2016) Melanocortin-4 receptor-regulated energy homeostasis. Nat Neurosci 19(2):206–219. https://doi.org/10.1038/nn.4202
Kim KS, Seeley RJ, Sandoval DA (2018) Signalling from the periphery to the brain that regulates energy homeostasis. Nat Rev Neurosci 19(4):185–196. https://doi.org/10.1038/nrn.2018.8
Joly-Amado A, Denis RG, Castel J et al (2012) Hypothalamic AgRP-neurons control peripheral substrate utilization and nutrient partitioning. EMBO J 31(22):4276–4288. https://doi.org/10.1038/emboj.2012.250
Cavalcanti-de-Albuquerque JP, Bober J, Zimmer MR, Dietrich MO (2019) Regulation of substrate utilization and adiposity by Agrp neurons. Nat Commun 10(1):311. https://doi.org/10.1038/s41467-018-08239-x
Nogueiras R, Wiedmer P, Perez-Tilve D et al (2007) The central melanocortin system directly controls peripheral lipid metabolism. J Clin Invest 117(11):3475–3488. https://doi.org/10.1172/JCI31743
Perez-Tilve D, Hofmann SM, Basford J et al (2010) Melanocortin signaling in the CNS directly regulates circulating cholesterol. Nat Neurosci 13(7):877–882. https://doi.org/10.1038/nn.2569
Kooijman S, Boon MR, Parlevliet ET et al (2014) Inhibition of the central melanocortin system decreases brown adipose tissue activity. J Lipid Res 55(10):2022–2032. https://doi.org/10.1194/jlr.M045989
Brito MN, Brito NA, Baro DJ, Song CK, Bartness TJ (2007) Differential activation of the sympathetic innervation of adipose tissues by melanocortin receptor stimulation. Endocrinology 148(11):5339–5347. https://doi.org/10.1210/en.2007-0621
da Silva AA, do Carmo JM, Freeman JN, Tallam LS, Hall JE (2009) A functional melanocortin system may be required for chronic CNS-mediated antidiabetic and cardiovascular actions of leptin. Diabetes 58(8):1749–1756. https://doi.org/10.2337/db08-1221
Goncalves GH, Li W, Garcia AV, Figueiredo MS, Bjorbaek C (2014) Hypothalamic agouti-related peptide neurons and the central melanocortin system are crucial mediators of leptin's antidiabetic actions. Cell Rep 7(4):1093–1103. https://doi.org/10.1016/j.celrep.2014.04.010
Xu J, Bartolome CL, Low CS et al (2018) Genetic identification of leptin neural circuits in energy and glucose homeostases. Nature 556(7702):505–509. https://doi.org/10.1038/s41586-018-0049-7
Singha AK, Yamaguchi J, Gonzalez NS, Ahmed N, Toney GM, Fujikawa T (2019) Glucose-lowering by leptin in the absence of insulin does not fully rely on the central melanocortin system in male mice. Endocrinology 160(3):651–663. https://doi.org/10.1210/en.2018-00907
Williams KW, Liu T, Kong X et al (2014) Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab 20(3):471–482. https://doi.org/10.1016/j.cmet.2014.06.002
Dodd GT, Decherf S, Loh K et al (2015) Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 160(1–2):88–104. https://doi.org/10.1016/j.cell.2014.12.022
Ruan HB, Dietrich MO, Liu ZW et al (2014) O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell 159(2):306–317. https://doi.org/10.1016/j.cell.2014.09.010
Schneider MK, Xue B, Shi H (2018) Activation of the sympathetic nervous system suppresses mouse white adipose tissue hyperplasia through the β1 adrenergic receptor. Physiol Rep 6(7):e13645. https://doi.org/10.14814/phy2.13645
Holland J, Sorrell J, Yates E et al (2019) A brain-melanocortin-vagus axis mediates adipose tissue expansion independently of energy intake. Cell Rep 27(8):2399–2410.e2396. https://doi.org/10.1016/j.celrep.2019.04.089
Verberne AJM, Sabetghadam A, Korim WS (2014) Neural pathways that control the glucose counterregulatory response. Front Neurosci 8:38–38. https://doi.org/10.3389/fnins.2014.00038
Tong Q, Ye C, McCrimmon RJ et al (2007) Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab 5(5):383–393. https://doi.org/10.1016/j.cmet.2007.04.001
Meek TH, Nelson JT, Matsen ME et al (2016) Functional identification of a neurocircuit regulating blood glucose. Proc Natl Acad Sci U S A 113(14):E2073–E2082. https://doi.org/10.1073/pnas.1521160113
Chan O, Cheng H, Herzog R et al (2008) Increased GABAergic tone in the ventromedial hypothalamus contributes to suppression of counterregulatory responses after antecedent hypoglycemia. Diabetes 57(5):1363–1370. https://doi.org/10.2337/db07-1559
Hedrington MS, Farmerie S, Ertl AC, Wang Z, Tate DB, Davis SN (2010) Effects of antecedent GABAA activation with alprazolam on counterregulatory responses to hypoglycemia in healthy humans. Diabetes 59(4):1074–1081. https://doi.org/10.2337/db09-1520
Hedrington MS, Mikeladze M, Tate DB, Younk LM, Davis I, Davis SN (2016) Effects of γ-aminobutyric acid a receptor activation on counterregulatory responses to subsequent exercise in individuals with type 1 diabetes. Diabetes 65(9):2754–2759. https://doi.org/10.2337/db16-0207
Wolfgang MJ, Cha SH, Sidhaye A et al (2007) Regulation of hypothalamic malonyl-CoA by central glucose and leptin. Proc Natl Acad Sci U S A 104(49):19285–19290. https://doi.org/10.1073/pnas.0709778104
Taib B, Bouyakdan K, Hryhorczuk C, Rodaros D, Fulton S, Alquier T (2013) Glucose regulates hypothalamic long-chain fatty acid metabolism via AMP-activated kinase (AMPK) in neurons and astrocytes. J Biol Chem 288(52):37216–37229. https://doi.org/10.1074/jbc.M113.506238
Cha SH, Hu Z, Chohnan S, Lane MD (2005) Inhibition of hypothalamic fatty acid synthase triggers rapid activation of fatty acid oxidation in skeletal muscle. Proc Natl Acad Sci U S A 102(41):14557–14562. https://doi.org/10.1073/pnas.0507300102
Hu Z, Dai Y, Prentki M, Chohnan S, Lane MD (2005) A role for hypothalamic malonyl-CoA in the control of food intake. J Biol Chem 280(48):39681–39683. https://doi.org/10.1074/jbc.C500398200
He W, Lam TKT, Obici S, Rossetti L (2006) Molecular disruption of hypothalamic nutrient sensing induces obesity. Nat Neurosci 9(2):227–233. https://doi.org/10.1038/nn1626
Obici S, Feng Z, Arduini A, Conti R, Rossetti L (2003) Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med 9(6):756–761. https://doi.org/10.1038/nm873
Lam TK, Pocai A, Gutierrez-Juarez R et al (2005) Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med 11(3):320–327. https://doi.org/10.1038/nm1201
Muoio DM, Noland RC, Kovalik JP et al (2012) Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab 15(5):764–777. https://doi.org/10.1016/j.cmet.2012.04.005
Seiler SE, Koves TR, Gooding JR et al (2015) Carnitine acetyltransferase mitigates metabolic inertia and muscle fatigue during exercise. Cell Metab 22(1):65–76. https://doi.org/10.1016/j.cmet.2015.06.003
Reichenbach A, Stark R, Mequinion M et al (2018) AgRP neurons require carnitine acetyltransferase to regulate metabolic flexibility and peripheral nutrient partitioning. Cell Rep 22(7):1745–1759. https://doi.org/10.1016/j.celrep.2018.01.067
Reichenbach A, Stark R, Mequinion M, et al (2018) Carnitine acetyltransferase (Crat) in hunger-sensing AgRP neurons permits adaptation to calorie restriction. FASEB J: fj201800634R. https://doi.org/10.1096/fj.201800634R
Stark R, Reichenbach A, Andrews ZB (2015) Hypothalamic carnitine metabolism integrates nutrient and hormonal feedback to regulate energy homeostasis. Mol Cell Endocrinol 418(Pt 1):9–16. https://doi.org/10.1016/j.mce.2015.08.002
Coupe B, Ishii Y, Dietrich MO, Komatsu M, Horvath TL, Bouret SG (2012) Loss of autophagy in pro-opiomelanocortin neurons perturbs axon growth and causes metabolic dysregulation. Cell Metab 15(2):247–255. https://doi.org/10.1016/j.cmet.2011.12.016
Kaushik S, Arias E, Kwon H et al (2012) Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep 13(3):258–265. https://doi.org/10.1038/embor.2011.260
Quan W, Kim HK, Moon EY et al (2012) Role of hypothalamic proopiomelanocortin neuron autophagy in the control of appetite and leptin response. Endocrinology 153(4):1817–1826. https://doi.org/10.1210/en.2011-1882
Martinez-Lopez N, Garcia-Macia M, Sahu S et al (2016) Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab 23(1):113–127. https://doi.org/10.1016/j.cmet.2015.10.008
Xiao Y, Deng Y, Yuan F et al (2017) An ATF4-ATG5 signaling in hypothalamic POMC neurons regulates obesity. Autophagy 13(6):1088–1089. https://doi.org/10.1080/15548627.2017.1307488
Kaushik S, Rodriguez-Navarro JA, Arias E et al (2011) Autophagy in hypothalamic AgRP neurons regulates food intake and energy balance. Cell Metab 14(2):173–183. https://doi.org/10.1016/j.cmet.2011.06.008
Zhang Y, Sowers JR, Ren J (2018) Targeting autophagy in obesity: from pathophysiology to management. Nat Rev Endocrinol 14(6):356–376. https://doi.org/10.1038/s41574-018-0009-1
Minokoshi Y, Alquier T, Furukawa N et al (2004) AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428(6982):569–574. https://doi.org/10.1038/nature02440
Cota D, Proulx K, Smith KA et al (2006) Hypothalamic mTOR signaling regulates food intake. Science 312(5775):927–930. https://doi.org/10.1126/science.1124147
Gao S, Kinzig KP, Aja S et al (2007) Leptin activates hypothalamic acetyl-CoA carboxylase to inhibit food intake. Proc Natl Acad Sci U S A 104(44):17358–17363. https://doi.org/10.1073/pnas.0708385104
Andrews ZB, Liu ZW, Walllingford N et al (2008) UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 454(7206):846–851. https://doi.org/10.1038/nature07181
Lopez M, Lage R, Saha AK et al (2008) Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab 7(5):389–399. https://doi.org/10.1016/j.cmet.2008.03.006
Sandoval D, Barrera JG, Stefater MA et al (2012) The anorectic effect of GLP-1 in rats is nutrient dependent. PLoS One 7(12):e51870. https://doi.org/10.1371/journal.pone.0051870
Burmeister MA, Ayala J, Drucker DJ, Ayala JE (2013) Central glucagon-like peptide 1 receptor-induced anorexia requires glucose metabolism-mediated suppression of AMPK and is impaired by central fructose. Am J Physiol Endocrinol Metab 304(7):E677–E685. https://doi.org/10.1152/ajpendo.00446.2012
Lockie SH, Stark R, Mequinion M et al (2018) Glucose availability predicts the feeding response to ghrelin in male mice, an effect dependent on AMPK in AgRP neurons. Endocrinology 159(11):3605–3614. https://doi.org/10.1210/en.2018-00536
Chen KY, Muniyappa R, Abel BS et al (2015) RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab 100(4):1639–1645. https://doi.org/10.1210/jc.2014-4024
Kuhnen P, Krude H, Biebermann H (2019) Melanocortin-4 receptor signalling: importance for weight regulation and obesity treatment. Trends Mol Med 25(2):136–148. https://doi.org/10.1016/j.molmed.2018.12.002
Brown RJ, Valencia A, Startzell M et al (2018) Metreleptin-mediated improvements in insulin sensitivity are independent of food intake in humans with lipodystrophy. J Clin Invest 128(8):3504–3516. https://doi.org/10.1172/JCI95476
Guarino D, Nannipieri M, Iervasi G, Taddei S, Bruno RM (2017) The role of the autonomic nervous system in the pathophysiology of obesity. Front Physiol 8:665. https://doi.org/10.3389/fphys.2017.00665
Morton GJ, Matsen ME, Bracy DP et al (2013) FGF19 action in the brain induces insulin-independent glucose lowering. J Clin Invest 123(11):4799–4808. https://doi.org/10.1172/jci70710
Marcelin G, Jo YH, Li X et al (2014) Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Mol Metab 3(1):19–28. https://doi.org/10.1016/j.molmet.2013.10.002
Scarlett JM, Rojas JM, Matsen ME et al (2016) Central injection of fibroblast growth factor 1 induces sustained remission of diabetic hyperglycemia in rodents. Nat Med 22(7):800–806. https://doi.org/10.1038/nm.4101
Brandt C, Nolte H, Henschke S et al (2018) Food perception primes hepatic ER homeostasis via melanocortin-dependent control of mTOR activation. Cell 175(5):1321–1335.e1320. https://doi.org/10.1016/j.cell.2018.10.015
Acknowledgements
The authors apologise to all colleagues whose studies could not be cited due to space constraints. Figures created with BioRender.com.
Funding
Work in TA’s laboratory is supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC RGPIN/04798) and the Canadian Institutes of Health Research (CIHR MOP115042), and a salary award from Fonds de Recherche Québec-Santé to TA. RM and DM are supported by PhD fellowships from the Neuroscience Department.
Author information
Authors and Affiliations
Contributions
The authors wrote and revised the manuscript and approved the version to be published.
Corresponding author
Ethics declarations
The authors have no duality of interest associated with this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Slideset of figures
(PPTX 571 kb)
Rights and permissions
About this article

Cite this article
Manceau, R., Majeur, D. & Alquier, T. Neuronal control of peripheral nutrient partitioning. Diabetologia 63, 673–682 (2020). https://doi.org/10.1007/s00125-020-05104-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-020-05104-9