
Abstract
Aims/hypothesis
The role of non-cardiomyocytes in diabetic cardiomyopathy has not been fully addressed. This study investigated whether endothelial cell calpain plays a role in myocardial endothelial injury and microvascular rarefaction in diabetes, thereby contributing to diabetic cardiomyopathy.
Methods
Endothelial cell-specific Capns1-knockout (KO) mice were generated. Conditions mimicking prediabetes and type 1 and type 2 diabetes were induced in these KO mice and their wild-type littermates. Myocardial function and coronary flow reserve were assessed by echocardiography. Histological analyses were performed to determine capillary density, cardiomyocyte size and fibrosis in the heart. Isolated aortas were assayed for neovascularisation. Cultured cardiac microvascular endothelial cells were stimulated with high palmitate. Angiogenesis and apoptosis were analysed.
Results
Endothelial cell-specific deletion of Capns1 disrupted calpain 1 and calpain 2 in endothelial cells, reduced cardiac fibrosis and hypertrophy, and alleviated myocardial dysfunction in mouse models of diabetes without significantly affecting systemic metabolic variables. These protective effects of calpain disruption in endothelial cells were associated with an increase in myocardial capillary density (wild-type vs Capns1-KO 3646.14 ± 423.51 vs 4708.7 ± 417.93 capillary number/high-power field in prediabetes, 2999.36 ± 854.77 vs 4579.22 ± 672.56 capillary number/high-power field in type 2 diabetes and 2364.87 ± 249.57 vs 3014.63 ± 215.46 capillary number/high-power field in type 1 diabetes) and coronary flow reserve. Ex vivo analysis of neovascularisation revealed more endothelial cell sprouts from aortic rings of prediabetic and diabetic Capns1-KO mice compared with their wild-type littermates. In cultured cardiac microvascular endothelial cells, inhibition of calpain improved angiogenesis and prevented apoptosis under metabolic stress. Mechanistically, deletion of Capns1 elevated the protein levels of β-catenin in endothelial cells of Capns1-KO mice and constitutive activity of calpain 2 suppressed β-catenin protein expression in cultured endothelial cells. Upregulation of β-catenin promoted angiogenesis and inhibited apoptosis whereas knockdown of β-catenin offset the protective effects of calpain inhibition in endothelial cells under metabolic stress.
Conclusions/interpretation
These results delineate a primary role of calpain in inducing cardiac endothelial cell injury and impairing neovascularisation via suppression of β-catenin, thereby promoting diabetic cardiomyopathy, and indicate that calpain is a promising therapeutic target to prevent diabetic cardiac complications.
Similar content being viewed by others


Introduction
By 2030, it is estimated that diabetes will be the seventh leading cause of death [1]. People with diabetes are at increased risk of developing cardiovascular diseases, which account for ~80% of diabetes-related deaths [2]. Both type 1 and type 2 diabetes can affect cardiac structure and function independent of vascular defects and hypertension, a condition termed diabetic cardiomyopathy [3]. Diabetic cardiomyopathy is characterised by early diastolic dysfunction and later systolic dysfunction and heart failure. The pathogenesis of diabetic cardiomyopathy remains incompletely understood and effective therapies are limited.
Calpains belong to a family of calcium-dependent cysteine proteases [4]. Among 15 family members, only calpain 1 and calpain 2 are expressed in the endothelial cells [5]. Calpain 1 and calpain 2 are heterodimers consisting of a distinct large 80 kDa catalytic subunit called CAPN1 or CAPN2, respectively (encoded by the genes CAPN1 and CAPN2), and a common small 28 kDa regulatory subunit (encoded by CAPNS1). The small subunit is indispensable for calpain 1 and calpain 2 stability and activity [6]. Both calpain 1 and calpain 2 activities are negatively regulated by calpastatin [4]. We have recently reported that cardiomyocyte-specific deletion of Capns1 reduces adverse cardiac changes in mouse models of impaired glucose tolerance (induced by feeding a high-fat diet [HFD]) and type 1 diabetes [7,8,9]. These findings indicate a critical role of cardiomyocyte calpain in diabetic cardiomyopathy.
Whereas dysfunction of cardiomyocytes plays a central role in diabetic cardiomyopathy, non-cardiomyocytes in the heart, such as coronary microvascular endothelial cells, are important in maintaining coronary vessel function, ventricular homeostasis and cardiac function [10]. Myocardial endothelial cell dysfunction, death and rarefaction contribute to diabetic cardiac complications [11]. Indeed, diabetes impairs the stability of myocardial microvascular vessels in both diabetic human myocardial explants and experimental diabetes [12], and microvascular endothelial dysfunction has been observed in impaired glucose tolerance, which may explain the increased risk of complications of microvascular origin in impaired glucose tolerance and early type 2 diabetes [13, 14]. However, the causes of coronary microvascular endothelial cell injury and dysfunction that thereby facilitate diabetic cardiomyopathy remain incompletely understood. Calpain activation has been implicated in endothelial dysfunction and inflammation under diabetic conditions [15,16,17,18,19]. This raises an intriguing possibility that calpain-mediated endothelial cell injury and dysfunction may contribute to diabetic cardiomyopathy.
In this study, we investigated the role of endothelial cell calpain in diabetic cardiomyopathy using mice with endothelial cell-specific deletion of Capns1.
Methods
Animals
The Guide for the care and use of laboratory animals, 8th edition (2011) (http://grants.nih.gov/grants/olaw/guide-for-the-care-and-use-of-laboratory-animals.pdf) was followed. All experimental procedures were approved by the Animal Use Subcommittee of Soochow University (Suzhou, China). C57BL/6 mice and mice with endothelial cell-specific expression of Cre recombinase under the control of Tie2 (TEK-CRE+/−) were purchased from the Jackson Laboratory (Sacramento, CA, USA). Mice bearing the targeted Capns1PZ allele containing loxP sites flanking essential coding exons (Capns1PZ+/+) were generated as previously described [6]. A breeding programme was implemented at Soochow University’s animal care facilities.
Mouse models of diabetes
Male mice were allocated to different experimental groups by matching body weight.
Prediabetes was induced by feeding male mice an HFD for 12 weeks starting at age 4 weeks [20]. This HFD contained 26.2% protein, 26.3% carbohydrate and 34.9% fat (% by weight) (Research Diets, New Brunswick, NJ, USA) [8].
A condition mimicking type 2 diabetes was induced in male mice (age 4 weeks) by feeding an HFD for 4 weeks, followed by injection of a single dose of streptozotocin (STZ, 100 mg/kg, i.p.). HFD was maintained for an additional 8 weeks after STZ injection. At 48 h after STZ injection, mice were considered diabetic only if they exhibited hyperglycaemia (≥15 mol/l) [21].
A condition mimicking type 1 diabetes was induced in adult male mice (2 months old) by consecutive peritoneal injections of STZ (50 mg kg−1 day−1) for 5 days [7]. At 2 months after induction of diabetes, mice were subjected to the experiments described below.
Isolation of cardiomyocytes, endothelial cells, fibroblasts and macrophages
Adult mice ventricular cardiomyocytes were isolated as previously described [22]. Endothelial cells were isolated from lung tissues using microbeads conjugated with CD31 antibody as previously described [23]. Cardiac fibroblasts were prepared and cultured as previously described [7]. Peritoneal macrophages were isolated as previously described [24].
Echocardiography
Animals were lightly anaesthetised with inhalant isoflurane (1–3%) and imaged using a 40 MHz linear array transducer attached to a preclinical ultrasound system (Vevo 2100, FUJIFILM VisualSonics, Toronto, ON, Canada) as previously described [9]. Changes in left ventricle (LV) end-systolic inner diameter, LV end-diastolic inner diameter, ejection fraction and fractional shortening were analysed. The pulsed wave Doppler measurements of maximal early and late transmitral velocities in diastole were obtained in the apical view with a cursor at mitral valve inflow. The coronary blood flow was measured as previously described [25]. Blinding was carried out for echocardiographic analysis.
IPGTT
An IPGTT was performed as previously described [8].
Determination of capillary density
Mice were anaesthetised with inhalant isoflurane (1–3%). A 50 μl amount of Griffonia (Bandeiraea) simplicifolia lectin 1 (BS1 lectin, 1 mg/ml, Vector Laboratories, Burlingame, CA, USA) was directly injected into the LV chamber. After 15 min, the heart tissues were harvested and fixed in 10% (vol./vol.) formalin and embedded and sectioned at 5 μm thickness. After processing, the sections were incubated with goat anti-BS1 lectin antibody (1:100; Vector Laboratories) and then stained with Alexa Fluor 594-conjugated rabbit anti-goat IgG (1:500, Invitrogen, Waltham, MA, USA). The fluorescent signals were quantified from at least eight random high-power fields for each section. The results were presented as capillary density per field. Blinding was carried out.
Blood lipid profiles
Total cholesterol, triacylglycerol, LDL-cholesterol and HDL-cholesterol were measured in blood as previously described [8].
Mouse aortic ring assay
Angiogenic assays of mouse aortic discs were carried out as previously described [23]. Briefly, mouse aortas were isolated and 1 mm discs placed in culture dishes and overlaid with 300 μl Matrigel (Corning, Corning, NY, USA) and endothelial growth medium. After 5 days of incubation, vessel outgrowths from aorta explants were counted.
Histological analyses
Total collagen content and cardiomyocyte cross-sectional areas of heart sections were assessed as previously described [7,8,9]. Collagen I and collagen III deposition was determined by polarisation microscopy. Blinding was carried out for these analyses.
Adenoviral infection and DNA transfection of cultured CMVECs
Cardiac microvascular endothelial cells (CMVECs) (within six passages) (CELLutions Biosystems, Toronto, ON, Canada) were infected with adenoviral vector containing rat calpastatin ([Ad-CAST] University of Buffalo, Buffalo, NW, USA) or β-galactosidase ([Ad-gal] Vector Biolabs) at a multiplicity of infection of 100 plaque-forming units/cell. All experiments were performed after 24 h of adenoviral infection.
CMVECs were transfected with pcDNA3-eGFP/β-catenin (Addgene), pcDNA3-eGFP, β-catenin CRISPR/Cas9 knockout (KO) plasmid (β-catenin Double Nickase Plasmid) or a control CRISPR/Cas9 KO plasmid from Santa Cruz Biotechnology, Dallas, TX, USA, using the jetPRIME DNA transfection reagent (Polyplus-Transfection, Illkirch, France) according to the manufacturer’s instructions.
Treatment with siRNA
CMVECs were transfected with small interfering (si)RNAs against mouse Capn1 (siCapn1) or Capn2 (siCapn2, Santa Cruz Biotechnology), or a scrambled control siRNA, as previously described [25].
In vitro two-dimensional CMVEC culture
In vitro angiogenesis of CMVECs was assessed using the Endothelial Tube Formation Assay (Cell Biolabs, San Diego, CA, USA) as previously described [25]. Tube formation was determined by measuring the tube length.
Scratch wound healing assay
CMVECs were wounded with a sterile pipette tip to remove cells by two perpendicular linear scrapes. After dislodged cells were washed away, the cells were refed with FBS-free media. Wound closure was photographed both immediately and 12 h post-wounding at the same spot. The extent of healing was defined as the difference between the original and remaining wound areas as a percentage of the original area [26].
Endothelial cell permeability
Endothelial cell permeability was assessed as previously described [27]. Briefly, CMVECs were infected with Ad-CAST or Ad-gal, and then were seeded to the insert in upper chamber of transwell (pore size 0.4 μm). Cells were cultured for 24 h to allow the growth of a confluent monolayer. After treatment, culture media in the upper chamber were replaced with 20 μl Evans Blue dye (0.5%). After 1 h, the liquid was collected from the lower chamber and spectrophotometric absorbance at 650 nm was determined.
Determination of oxidative damage
Protein carbonyl content and malondialdehyde (MDA) production in heart tissue lysates were measured using a protein carbonyl content assay kit (Sigma-Aldrich, St. Louis, MO, USA) and a lipid peroxidation MDA assay kit (Beyotime, Shanghai, China), respectively, according to the manufacturer’s instructions.
Calpain activity
Calpain activities were measured in CMVEC lysates using a fluorescence substrate N-succinyl-LLVY-AMC (Cedarlane Laboratories, Burlington, ON, Canada) as previously described [22].
Analysis of apoptosis
Apoptosis was determined by measuring caspase 3 activity and cellular DNA fragmentation as previously described [17].
Real-time RT-PCR
Real-time RT-PCR was performed to analyse mRNA expression of collagen I, collagen III, hypoxia-inducible factor (HIF)-1α, vascular endothelial growth factor (VEGF), β-catenin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [7,8,9].
Western blot analysis
The protein levels of CAPN1, CAPN2, calpastatin, β-catenin, histone H3, c-Myc and GAPDH were determined by western blot analysis using respective specific antibodies (1:1000 dilutions, Cell Signaling Technology, Danvers, MA, USA). The proteins recognised by their antibodies were determined by their molecular mass. Both CAPN1 and CAPN2 antibodies were validated using wild-type and Capns1 KO cells.
Statistical analysis
All data are presented as mean ± SD. One- or two-way ANOVA followed by Newman–Keuls tests were performed for multi-group comparisons, as appropriate. A Student’s t test was used for comparison between two groups. A value of p < 0.05 was considered statistically significant.
Results
Characterisation of endothelial cell-specific Capns1-KO mice
Mice with endothelial cell-specific Capns1 KO were generated by breeding Capns1PZ+/+ and TEK-CRE+/− mice (electronic supplementary material [ESM] Fig. 1a). As the regulatory subunit encoded by Capns1 is indispensable for the stability of calpain 1 and calpain 2 [4], the protein levels of CAPN1 and CAPN2 were decreased by about 90% and 75%, respectively, in microvascular endothelial cells isolated from Capns1-KO mice compared with wild-type littermates (Capns1PZ+/+), whereas their levels were not changed in fibroblasts, macrophages and cardiomyocytes from Capns1-KO mice and their wild-type littermates (ESM Fig. 1b–d), confirming endothelial cell-specific disruption of calpain in this Capns1-KO mouse model.
Myocardial function, heart weight, body weight, blood pressure, blood lipid profiles, blood glucose and glucose tolerance were similar between Capns1-KO mice and their wild-type littermates at age 3 months (Fig. 1, ESM Tables 1–4, ESM Fig. 2), though the peak glucose levels were higher in Capns1-KO mice compared with their wild-type littermates (ESM Fig. 2a). These observations suggest that endothelial cell-specific disruption of calpain is not deleterious to the heart and its impact on systemic metabolism is negligible in Capns1-KO mice.

Assessment of myocardial function. Prediabetes, type 2 diabetes and type 1 diabetes were induced in Capns1-KO mice and their wild-type littermates. Myocardial function was assessed by echocardiographic analysis. (a) LV fractional shortening. (b) E/A ratio. Data are mean ± SD, n = 5–8 mice in each group. *p < 0.05 vs wild-type, non-diabetic mice and †p < 0.05 vs wild-type mice in the same category (two-way ANOVA followed by Newman–Keuls test). LVFS, LV fractional shortening; ND, non-diabetic; preD, prediabetes; T1D, type 1 diabetes; T2D, type 2 diabetes; WT, wild-type
Deficiency of endothelial cell Capns1 protects myocardial function in diabetic mice
Both Capns1-KO mice and their wild-type littermates exhibited similar prediabetic changes [20], including obesity, hyperlipidaemia and impaired glucose tolerance (ESM Tables 1 and 3, ESM Fig. 2b). Echocardiographic analysis revealed a reduction in fractional shortening and the ratio of maximal early and late transmitral velocities (E/A ratio) in prediabetic wild-type mice, indicative of myocardial dysfunction. However, deletion of Capns1 attenuated myocardial dysfunction in prediabetic Capns1-KO mice (Fig. 1a, b and ESM Table 4) while not changing systemic metabolic variables or blood pressure (ESM Tables 1–3, ESM Fig. 2).
Myocardial function was decreased in wild-type type 1 and type 2 diabetic mice (Fig. 1a, b), but was relatively maintained in Capns1-KO diabetic mice without changes in systemic metabolic variables (ESM Tables 1 and 3, ESM Fig. 2). These results suggest endothelial cell calpain has a role in myocardial dysfunction in diabetic mice.
Deletion of endothelial cell Capns1 reduces myocardial hypertrophy and fibrosis in diabetic mice
Collagen deposition and cardiomyocyte cross-sectional area (i.e. cell size) were significantly increased in hearts of prediabetic and diabetic mice compared with non-diabetic mouse hearts, indicative of cardiomyocyte hypertrophy and fibrosis (Fig. 2a–c), two characteristic changes in diabetic cardiomyopathy [3]. However, deletion of Capns1 significantly reduced myocardial hypertrophy and fibrosis in prediabetes and diabetes. As additional evidence, deletion of Capns1 attenuated the deposition and mRNA levels of collagen I and collagen III in prediabetic mouse hearts (ESM Fig. 3a–e).

Determination of fibrosis and cardiac hypertrophy. (a) Representative Picrosirius Red staining for collagen deposition (red colour) from Capns1-KO mice and their wild-type littermates under non-diabetic or diabetic conditions (scale bar, 100 μm). (b) Quantitative data for total collagen deposition in hearts. (c) Cross-sectional cardiomyocyte area. Data are mean ± SD, n = 4–6 hearts in each group. *p < 0.05 vs wild-type, non-diabetic mice and †p < 0.05 vs wild-type mice in the same category (two-way ANOVA followed by Newman–Keuls test). ND, non-diabetic; preD, prediabetes; T1D, type 1 diabetes; T2D, type 2 diabetes; WT, wild-type
Coronary vascular dysfunction and microvascular rarefaction in diabetic mouse hearts are attenuated by endothelial cell-specific Capns1-KO
To determine coronary vascular function, we analysed coronary flow reserve (CFR) [28]. CFR was significantly decreased in mice with prediabetes and diabetes. This decrease was significantly less in Capns1-KO mice (Fig. 3a), implicating endothelial cell calpain in diabetic coronary vascular dysfunction.

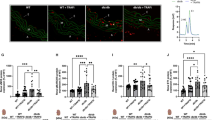
Measurement of CFR, cardiac capillary density and analysis of aortic sprouting. (a) CFR by echocardiographic analysis. (b) A representative micrograph for capillary density in hearts (red colour; scale bar, 50 μm). (c) Quantitative data for cardiac capillary density. Data are mean ± SD, n = 4–6 hearts in each group. (d) A representative micrograph of vessel sprouting from aortic rings (black scale bar, 200 μm; red scale bar, 100 μm). (e) Quantitative data for aortic sprouting. Data are mean ± SD, n = 3 different mice in each group (at least six aortic rings analysed for each mouse). *p < 0.05 vs wild-type, non-diabetic mice and †p < 0.05 vs wild-type mice in the same category (two-way ANOVA followed by Newman–Keuls test). H/B; hyperaemic/baseline; HPF, high-power field; ND, non-diabetic; preD, prediabetes; T1D, type 1 diabetes; T2D, type 2 diabetes; WT, wild-type
As coronary microvascular rarefaction occurs in diabetic hearts [29, 30], and can impair CFR [12], we examined capillary density in diabetic mouse hearts. Both prediabetes and diabetes decreased capillary density in wild-type mouse hearts (Fig. 3b, c); however, this phenotype was suppressed in Capns1-KO mice (wild-type vs Capns1-KO 3646.14 ± 423.51 vs 4708.7 ± 417.93 capillary number/high-power field in prediabetes, 2999.36 ± 854.77 vs 4579.22 ± 672.56 capillary number/high-power field in type 2 diabetes and 2364.87 ± 249.57 vs 3014.63 ± 215.46 capillary number/high-power field in type 1 diabetes). The reduction of coronary capillary density was associated with increases in protein carbonyl content and MDA production and the expression of HIF-1α and VEGF in prediabetic hearts, suggesting a hypoxic response; these effects were prevented by deletion of Capns1 (ESM Fig. 4a–d).
As impaired neovascularisation contributes to coronary microvascular rarefaction in diabetes [31, 32], we analysed neovascularisation in isolated aortic rings. Capillary-like outgrowth of endothelial sprouts was significantly impaired in aortic rings from wild-type prediabetic and type 2 diabetic mice (Fig. 3d, e), but it was relatively maintained in Capns1-KO mice with prediabetes and type 2 diabetes. A similar observation was made with aortic rings from non-diabetic mice on culturing with, to provide metabolic stress, high palmitate (100 μmol/l), high palmitate (50 μmol/l) plus high glucose (15 mmol/l) or high glucose (30 mmol/l) (ESM Fig. 5a, b). In the absence of endothelial Capns1, the extent of aortic sprouting was significantly greater when compared with tissues taken from wild-type littermates under the same metabolic stress, though considerably lower than in the control environment (incubation with oleate). These results indicate that deletion of endothelial cell Capns1 benefits aortic neovascularisation in diabetes.
Inhibition of calpain improves angiogenesis and prevents apoptosis in CMVECs under metabolic stress
To provide direct evidence to support the role of calpain in endothelial injury and angiogenesis, we infected CMVECs with Ad-CAST or Ad-gal, and then incubated them with high palmitate (to mimic metabolic stress) or oleate (100 μmol/l). High palmitate increased calpain activity (ESM Fig. 6), impaired tube formation (Fig. 4a, c), suppressed wound healing (Fig. 4b, d) and increased apoptosis in CMVECs (Fig. 4e, f). All these effects of palmitate were prevented by infection with Ad-CAST (Fig. 4a–f). A similar protective effect of calpain inhibition with Ad-CAST was made when CMVECs were incubated with high NEFA (50 μmol/l palmitate +50 μmol/l oleate) in combination with high glucose (15 mmol/l) (ESM Fig. 7). Consistently, incubation with calpain inhibitor III achieved similar protective effects in CMVECs under metabolic stress (ESM Fig. 8). Although high palmitate increased the permeability of endothelial cells, inhibition of calpain did not affect the permeability (ESM Fig. 9). These results demonstrate that inhibition of calpain promotes angiogenesis and protects endothelial cells under metabolic stress.

Effects of calpastatin upregulation on angiogenesis and apoptosis. Cultured CMVECs were infected with Ad-CAST or Ad-gal as a control in the presence of high palmitate or oleate. (a) A representative micrograph of endothelial cell tube formation (scale bar, 100 μm). (b) A representative micrograph of endothelial cell wound healing (scale bar, 200 μm). (c, d) Quantitative data for endothelial cell tube formation, determined by tube length (c), and percentage healing (d). (e) Caspase 3 activity. (f) DNA fragmentation. Data are mean ± SD, n = 5 independent cell batches. *p < 0.05 vs oleate + Ad-gal and †p < 0.05 vs palmitate + Ad-gal (two-way ANOVA followed by Newman–Keuls test)
Constitutive activities of calpain suppress β-catenin protein expression in endothelial cells
To investigate the mechanisms by which calpain impairs neovascularisation and induces endothelial injury, we focused on β-catenin as it has been implicated in angiogenesis and calpain induces its proteolysis [33, 34]. The protein levels of β-catenin were much higher in endothelial cells of Capns1-KO mice compared with their wild-type littermates (Fig. 5a), whereas the protein levels of β-catenin in whole-heart lysates did not differ between Capns1-KO and wild-type mice (Fig. 5b), suggesting that β-catenin protein may not increase in cardiomyocytes. To provide direct evidence, we showed that infection with Ad-CAST upregulated β-catenin protein in CMVECs (Fig. 5c, d), but it did not increase the mRNA levels of β-catenin (Fig. 5e). Although high palmitate (ESM Fig. 10a, b) and prediabetes (Fig. 5b) did not change the total protein levels of β-catenin, high palmitate significantly reduced the nuclear protein levels of β-catenin in endothelial cells (ESM Fig. 10c, d), indicative of its inactivation, which was prevented by calpain inhibition. In contrast, inhibition of calpain induced c-Myc protein expression in both oleate- and palmitate-stimulated endothelial cells (ESM Fig. 10e, f), suggesting that nuclear accumulation of β-catenin may not account for c-Myc induction in this scenario. Furthermore, knockdown of CAPN2 but not CAPN1 increased β-catenin protein (Fig. 5f–h). These results suggest that the constitutive activity of calpain 2 suppresses β-catenin protein expression in endothelial cells.

Measurement of calpain and β-catenin expression. (a) Microvascular endothelial cells were isolated from Capns1-KO mice and their wild-type littermates. A representative western blot for β-catenin and GAPDH from two of four different hearts in each group, together with quantification of β-catenin protein levels relative to GAPDH, showing that the protein levels of β-catenin are much higher in endothelial cells from KO compared with wild-type mice. (b) β-Catenin and GAPDH protein in whole-heart lysates. A representative western blot for β-catenin and GAPDH from two of four different hearts in each group, together with quantification of β-catenin protein levels relative to GAPDH. (c–e) Cultured CMVECs were infected with Ad-CAST or Ad-gal as a control. At 6, 12 and 24 h after infection, the protein levels of calpastatin, β-catenin and GAPDH were determined by western blot analysis. The mRNA levels of β-catenin and Gapdh were analysed by real-time RT-PCR. (c) Representative western blots for calpastatin, β-catenin and GAPDH protein. (d) β-catenin protein levels relative to GAPDH. (e) mRNA level of β-catenin relative to Gapdh. (f–h) Cultured CMVECs were transfected with siRNAs for Capn1 (siCapn1) and Capn2 (siCapn2) or scramble siRNA as a control. (f) Representative western blots for CAPN1, β-catenin and GAPDH. (g) Representative western blots for CAPN2, β-catenin and GAPDH. (h) β-Catenin protein levels relative to GAPDH. The same scramble siRNA was used in two experiments. Data are mean ± SD, n = 5 independent cell batches. *p < 0.05 vs WT for (a), Ad-gal for (d) and scramble for (h) (one-way ANOVA followed by Newman–Keuls tests for [d] and Student’s t test for [a] and [h]). CAST, calpastatin; ND, non-diabetic; preD, prediabetes; T1D, type 1 diabetes; T2D, type 2 diabetes; WT, wild-type
Upregulation of β-catenin promotes angiogenesis in CMVECS under metabolic stress
To investigate the role of β-catenin in angiogenesis and apoptosis, we introduced pcDNA3-eGFP/β-catenin into CMVECs to upregulate β-catenin protein. An empty plasmid pcDNA3-eGFP served as a control. After 24 h, we incubated these cells with palmitate or oleate (100 μmol/l). Palmitate decreased tube formation, suppressed wound healing and induced apoptosis in CMVECs (Fig. 6a–f). These effects of palmitate were not seen in cells with upregulated β-catenin (Fig. 6a–f, ESM Fig. 11a).

Effects of β-catenin upregulation on angiogenesis and apoptosis. Cultured CMVECs were transfected with pcDNA3-eGFP/β-catenin or pcDNA3-eGFP as a control plasmid, and then incubated with palmitate or oleate. (a) A representative micrograph of endothelial cell tube formation (scale bar, 100 μm). (b) A representative micrograph of wound healing (scale bar, 200 μm). (c, d) Quantitative data for endothelial cell tube formation, determined by tube length (c), and percentage healing (d). (e) Caspase 3 activity. (f) DNA fragmentation. Data are mean ± SD, n = 5 independent cell batches. *p < 0.05 vs oleate + pcDNA3-eGFP and †p < 0.05 vs palmitate + pcDNA3-eGFP (two-way ANOVA followed by Newman–Keuls test)
Knockout of β-catenin offsets the pro-angiogenic and anti-apoptotic effects of calpain inhibition in CMVECS under metabolic stress
To determine whether inhibition of calpain promotes angiogenesis and inhibits apoptosis through β-catenin signalling, we knocked out β-catenin in CMVECs using CRISPR/cas9 (ESM Fig. 11b) and then infected these cells with Ad-CAST or Ad-gal. At 24 h after infection, we exposed these cells to palmitate or oleate (100 μmol/l). Palmitate decreased tube formation, suppressed wound healing and induced apoptosis in CMVECs (Fig. 7a–d). These effects of palmitate were reduced by infection with Ad-CAST. However, knockdown of β-catenin offset the protective effects conferred by infection with Ad-CAST in palmitate-stimulated CMVECs (Fig. 7a–d).

Analyses of angiogenesis and apoptosis. CMVECs were transfected with β-catenin CRISPR/Cas9 plasmid or control CRISPR/Cas9 as a control, and then infected with Ad-CAST or Ad-gal as a control. These cells were incubated with high palmitate or oleate. (a) Quantitative data for endothelial cell tube formation, determined by tube length. (b) Quantitative data for wound healing. (c) Caspase 3 activity. (d) DNA fragmentation. Data are mean ± SD, n = 5 independent cell batches. *p < 0.05 vs oleate + control CRISPR/Cas9 + Ad-gal, †p < 0.05 vs palmitate + control CRISPR/Cas9 + Ad-gal, and ‡p < 0.05 vs palmitate + control CRISPR/Cas9 + Ad-CAST (two-way ANOVA followed by Newman–Keuls tests)
Discussion
The major finding of this study is that endothelial cell-specific disruption of calpain reduced diabetic cardiomyopathy in mice (Figs 1 and 2). The protective effect of endothelial cell-specific calpain disruption may be attributed to attenuated endothelial injury and improved angiogenesis in diabetic hearts and CMVECs under metabolic stress (Figs 3 and 4). Furthermore, the constitutive activity of calpain 2 suppressed β-catenin protein expression in endothelial cells (Fig. 5), and upregulation of β-catenin promoted angiogenesis and inhibited apoptosis; knockout of β-catenin offset the effects of calpain inhibition on angiogenesis and apoptosis in endothelial cells under metabolic stress (Figs 6 and 7). These findings highlight a critical role of endothelial cell calpain in coronary vascular injury and microvascular rarefaction, contributing to diabetic cardiomyopathy.
Calpain has been implicated in diabetic cardiomyopathy. Calpain is activated in the diabetic heart and in cultured cardiomyocytes under metabolic stress [7,8,9]. In cardiomyocytes, inhibition of calpain prevented apoptosis induced by high glucose and palmitate [7, 8]. In mouse models of prediabetes and type 1 diabetes, we recently demonstrated that cardiomyocyte-specific deletion of Capns1 reduced diabetic cardiomyopathy [7,8,9]. These earlier studies indicated a critical role of cardiomyocyte calpain in diabetic cardiomyopathy. The present study provides direct evidence for a primary role of endothelial cell calpain in diabetic cardiomyopathy (Figs 1 and 2). The role of endothelial cell calpain may be associated with coronary circulation dysfunction in the diabetic heart as deletion of Capns1 increased CFR [35] (Fig. 3a). Thus, our observations are consistent with a model whereby calpain activation compromises coronary circulation, which can directly impair myocardial function in diabetes. Compromised coronary circulation may induce a condition of sub-ischaemia in hearts, as supported by increases in oxidative damage, HIF-1α and VEGF expression (ESM Fig. 4), which initiates cardiomyocyte death and subsequent myocardial remodelling [36, 37], characteristic changes in diabetic cardiomyopathy [3]. This may partially explain how calpain-mediated endothelial cell injury and dysfunction contributes to myocardial hypertrophy and fibrosis in diabetic cardiomyopathy. However, other mechanisms merit future investigation. Although deleting endothelial cell calpain may have unknown systemic impacts that potentially confound the interpretation of our findings, our studies using cultured endothelial cells and aortic rings provide direct evidence supporting the role of calpain in endothelial injury and angiogenesis (Figs 3 and 4). Furthermore, deficiency of endothelial cell Capns1 did not affect systemic metabolic variables and blood pressure. Thus, the global effects of Capns1 deletion, if any, may be negligible in this scenario.
Certain pathophysiological conditions can influence CFR. Among them, coronary vascular dysfunction (e.g. mild to moderate coronary artery stenosis, myocardial vascular dysfunction and capillary rarefaction) is directly related to abnormalities of endothelial cells [38]. As myocardial capillary rarefaction and vascular dysfunction are observed in cases where no coronary artery stenosis has been reported in diabetes [12], reduced CFR is likely caused by capillary rarefaction and vascular dysfunction [35]. Indeed, capillary density was significantly decreased in diabetic mouse hearts (Fig. 3b, c); this phenotype was suppressed by endothelial cell-specific disruption of calpain. The detrimental role of calpain in endothelial/vascular dysfunction has also been demonstrated in diabetes. In cultured endothelial cells, calpain activation decreased NO production and promoted apoptosis under metabolic stress [18]. This was further confirmed in vasculature from mouse and rat models of type 1 and type 2 diabetes [15, 39]. Calpain was also reported to cleave prostacyclin synthase in mesenteric artery from type 1 diabetic mice; the consequent reduction of prostacyclin led to endothelial dysfunction [19]. In addition, previous studies have reported that calpain inhibition maintains the microcirculatory endothelial cell barrier in type 1 and type 2 diabetes [15,16,17], which is not supported by our study on endothelial cell permeability induced by palmitate (ESM Fig. 9). The cause of this discrepancy is unknown.
Although the exact mechanisms by which calpain disruption in endothelial cells improves capillary density in diabetic hearts are currently unknown, our data suggest that decreased endothelial injury and improved neovascularisation may be important in this scenario (Figs 3d, e and 4). Previous studies have also demonstrated that inhibition of calpain promotes angiogenesis in a variety of experimental models. In an in vitro study on pericyte regulation of nascent endothelial tubes, calpain activation mediated chemokine (C-X-C motif) receptor 3 (CXCR3)-induced repression of angiogenic vessel formation in human dermal microvascular endothelial cells [40]. More recently, it was reported that calpain 1 activation decreased circulating angiogenic cells in response to IFN-β, and thus inhibited vessel formation [41], and that thromboxane-induced calpain 2 activation inhibited endothelial cell differentiation of autologous adipose-derived stromal cells and angiogenesis in ischaemic mouse hindlimbs [34]. However, these reports, together with our present findings, contrast with some prior studies in non-cardiac endothelial cells induced by VEGF, IL-6 or shear stress, and in skin wound healing during diabetes [42,43,44]. These differences suggest that calpain may modulate angiogenesis via multiple mechanisms, depending on conditions and organs/tissues.
The present study demonstrates that inhibition of calpain promotes angiogenesis and prevents apoptosis through upregulation of β-catenin under metabolic stress (Figs 6 and 7). Silencing of Capn2 elevated β-catenin levels, whereas silencing of Capn1 did not alter the protein levels of β-catenin under normal conditions (Fig. 5g, h). Relevantly, deficiency of Capns1 increased β-catenin protein levels in endothelial cells of Capns1-KO mice (Fig. 5a). These data suggest that constitutive activity of calpain 2 suppresses β-catenin protein expression, which is consistent with a study in human osteosarcoma cells [33]. Several studies have also shown that calpain cleaves β-catenin protein [45, 46]. In line with these reports, our data show that inhibition of calpain increased the protein levels of β-catenin without changing its mRNA levels (Fig. 5a–e). Metabolic stress did not increase β-catenin expression in endothelial cells and heart tissues. However, calpain inhibits β-catenin activation under metabolic stress.
Studies have implicated β-catenin in diabetic cardiomyopathy. In the hearts from STZ-induced diabetic rats, β-catenin expression was increased [47], suggesting that β-catenin may play a role in diabetic cardiomyopathy. Indeed, glucagon-like peptide-mediated activation of β-catenin prevented apoptosis in palmitate-stimulated cardiomyocytes [48]. Although the present study shows increased β-catenin protein in endothelial cells from HFD-fed mice, its levels in whole-heart tissues do not differ between HFD-fed mice and those fed a normal diet. This discrepancy between rats with STZ-induced diabetes and HFD-fed mice suggests different responses to type 1 diabetes and HFD in terms of β-catenin induction. Deletion of Capns1 in endothelial cells provides similar cardiac protection in different diabetic models. This suggests that calpain activation may be a common mechanism contributing to adverse cardiac changes in different types of diabetes. Indeed, we show that inhibition of calpain prevents apoptosis and improves angiogenesis in endothelial cells under different metabolic stresses, including high glucose and high palmitate alone or in combination, and in diabetic hearts (Figs 3d, e and 4 and ESM Figs 5, 7 and 8). Furthermore, disruption of calpain in cardiomyocytes reduces adverse cardiac changes similarly in different type 1 diabetic and HFD-fed mice [7,8,9].
In summary, we have demonstrated a primary role for calpain-mediated coronary microvascular endothelial cell injury and dysfunction in the pathogenesis of diabetic cardiac complications. This effect of endothelial cell calpain may be mediated through impaired neovascularisation and endothelial injury in diabetic cardiomyopathy. Given that cardiomyocyte calpain also plays an important role in diabetic cardiomyopathy [6, 8] and that calpain activation induces beta cell death [49], peripheral vascular dysfunction [15,16,17] and retinopathy [50] during diabetes, pharmaceutical approaches targeting calpain may be a useful therapy for diabetes and its cardiovascular complications.
Data availability
The data are available on request from the authors.
Abbreviations
- Ad-CAST:
-
Adenoviral vector containing rat calpastatin
- Ad-gal:
-
Adenoviral vector containing β-galactosidase
- CFR:
-
Coronary flow reserve
- CMVEC:
-
Cardiac microvascular endothelial cell
- E/A ratio:
-
Ratio of maximal early and late transmitral velocities
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- HFD:
-
High-fat diet
- HIF:
-
Hypoxia-inducible factor
- KO:
-
Knockout
- LV:
-
Left ventricle
- MDA:
-
Malondialdehyde
- siRNA:
-
Small interfering RNA
- STZ:
-
Streptozotocin
- VEGF:
-
Vascular endothelial growth factor
References
Mathers CD, Loncar D (2006) Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3(11):e442. https://doi.org/10.1371/journal.pmed.0030442
Bertoni AG, Hundley WG, Massing MW, Bonds DE, Burke GL, Goff DC Jr (2004) Heart failure prevalence, incidence, and mortality in the elderly with diabetes. Diabetes Care 27(3):699–703. https://doi.org/10.2337/diacare.27.3.699
Boudina S, Abel ED (2007) Diabetic cardiomyopathy revisited. Circulation 115(25):3213–3223. https://doi.org/10.1161/CIRCULATIONAHA.106.679597
Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83(3):731–801. https://doi.org/10.1152/physrev.00029.2002
Fujitani K, Kambayashi J, Sakon M et al (1997) Identification of mu-, m-calpains and calpastatin and capture of mu-calpain activation in endothelial cells. J Cell Biochem 66(2):197–209. https://doi.org/10.1002/(SICI)1097-4644(19970801)66:2<197::AID-JCB7>3.0.CO;2-L
Tan Y, Dourdin N, Wu C, De Veyra T, Elce JS, Greer PA (2006) Conditional disruption of ubiquitous calpains in the mouse. Genesis 44(6):297–303. https://doi.org/10.1002/dvg.20216
Li Y, Ma J, Zhu H et al (2011) Targeted inhibition of calpain reduces myocardial hypertrophy and fibrosis in mouse models of type 1 diabetes. Diabetes 60(11):2985–2994. https://doi.org/10.2337/db10-1333
Li S, Zhang L, Ni R et al (2016) Disruption of calpain reduces lipotoxicity-induced cardiac injury by preventing endoplasmic reticulum stress. Biochim Biophys Acta 1862(11):2023–2033. https://doi.org/10.1016/j.bbadis.2016.08.005
Ni R, Zheng D, Xiong S et al (2016) Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type 1 diabetic hearts: a novel mechanism contributing to diabetic cardiomyopathy. Diabetes 65:255–268
Aronson D, Edelman ER (2014) Coronary artery disease and diabetes mellitus. Cardiol Clin 32(3):439–455. https://doi.org/10.1016/j.ccl.2014.04.001
Beckman JA, Paneni F, Cosentino F, Creager MA (2013) Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part II. Eur Heart J 34(31):2444–2452. https://doi.org/10.1093/eurheartj/eht142
Hinkel R, Hoewe A, Renner S et al (2017) Diabetes mellitus-induced microvascular destabilization in the myocardium. J Am Coll Cardiol 69(2):131–143. https://doi.org/10.1016/j.jacc.2016.10.058
Muris DM, Houben AJ, Schram MT, Stehouwer CD (2012) Microvascular dysfunction is associated with a higher incidence of type 2 diabetes mellitus: a systematic review and meta-analysis. Arterioscler Thromb Vasc Biol 32(12):3082–3094. https://doi.org/10.1161/ATVBAHA.112.300291
Sabanayagam C, Lye WK, Klein R et al (2015) Retinal microvascular calibre and risk of diabetes mellitus: a systematic review and participant-level meta-analysis. Diabetologia 58(11):2476–2485. https://doi.org/10.1007/s00125-015-3717-2
Stalker TJ, Gong Y, Scalia R (2005) The calcium-dependent protease calpain causes endothelial dysfunction in type 2 diabetes. Diabetes 54(4):1132–1140. https://doi.org/10.2337/diabetes.54.4.1132
Scalia R, Gong Y, Berzins B, Zhao LJ, Sharma K (2007) Hyperglycemia is a major determinant of albumin permeability in diabetic microcirculation: the role of mu-calpain. Diabetes 56(7):1842–1849. https://doi.org/10.2337/db06-1198
Smolock AR, Mishra G, Eguchi K, Eguchi S, Scalia R (2011) Protein kinase C upregulates intercellular adhesion molecule-1 and leukocyte-endothelium interactions in hyperglycemia via activation of endothelial expressed calpain. Arterioscler Thromb Vasc Biol 31(2):289–296. https://doi.org/10.1161/ATVBAHA.110.217901
Chen B, Zhao Q, Ni R et al (2014) Inhibition of calpain reduces oxidative stress and attenuates endothelial dysfunction in diabetes. Cardiovasc Diabetol 13(1):88. https://doi.org/10.1186/1475-2840-13-88
Randriamboavonjy V, Kyselova A, Elgheznawy A, Zukunft S, Wittig I, Fleming I (2017) Calpain 1 cleaves and inactivates prostacyclin synthase in mesenteric arteries from diabetic mice. Basic Res Cardiol 112(1):10. https://doi.org/10.1007/s00395-016-0596-8
Hinder LM, O’Brien PD, Hayes JM et al (2017) Dietary reversal of neuropathy in a murine model of prediabetes and metabolic syndrome. Dis Model Mech 10(6):717–725. https://doi.org/10.1242/dmm.028530
Kusakabe T, Tanioka H, Ebihara K et al (2009) Beneficial effects of leptin on glycaemic and lipid control in a mouse model of type 2 diabetes with increased adiposity induced by streptozotocin and a high-fat diet. Diabetologia 52(4):675–683. https://doi.org/10.1007/s00125-009-1258-2
Li Y, Li Y, Feng Q, Arnold M, Peng T (2009) Calpain activation contributes to hyperglycaemia-induced apoptosis in cardiomyocytes. Cardiovasc Res 84(1):100–110. https://doi.org/10.1093/cvr/cvp189
Baker M, Robinson SD, Lechertier T et al (2011) Use of the mouse aortic ring assay to study angiogenesis. Nat Protoc 7(1):89–104. https://doi.org/10.1038/nprot.2011.435
Dror E, Dalmas E, Meier DT et al (2017) Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol 18(3):283–292. https://doi.org/10.1038/ni.3659
Zheng D, Ma J, Yu Y et al (2015) Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia 58(8):1949–1958. https://doi.org/10.1007/s00125-015-3622-8
Liang CC, Park AY, Guan JL (2007) In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2(2):329–333. https://doi.org/10.1038/nprot.2007.30
Graesser D, Solowiej A, Bruckner M et al (2002) Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest 109(3):383–392. https://doi.org/10.1172/JCI0213595
Oskarsson G (2004) Coronary flow and flow reserve in children. Acta Paediatr Suppl 93:20–25
Nakagami H, Kaneda Y, Ogihara T, Morishita R (2005) Endothelial dysfunction in hyperglycemia as a trigger of atherosclerosis. Curr Diabetes Rev 1(1):59–63. https://doi.org/10.2174/1573399052952550
Cohen G, Riahi Y, Alpert E, Gruzman A, Sasson S (2007) The roles of hyperglycaemia and oxidative stress in the rise and collapse of the natural protective mechanism against vascular endothelial cell dysfunction in diabetes. Arch Physiol Biochem 113(4-5):259–267. https://doi.org/10.1080/13813450701783513
Heather LC, Clarke K (2011) Metabolism, hypoxia and the diabetic heart. J Mol Cell Cardiol 50(4):598–605. https://doi.org/10.1016/j.yjmcc.2011.01.007
Costa PZ, Soares R (2013) Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sci 92(22):1037–1045. https://doi.org/10.1016/j.lfs.2013.04.001
Benetti R, Copetti T, Dell’Orso S et al (2005) The calpain system is involved in the constitutive regulation of beta-catenin signaling functions. J Biol Chem 280(23):22070–22080. https://doi.org/10.1074/jbc.M501810200
Shen Y, Zuo S, Wang Y et al (2016) Thromboxane governs the differentiation of adipose-derived stromal cells toward endothelial cells in vitro and in vivo. Circ Res 118(8):1194–1207. https://doi.org/10.1161/CIRCRESAHA.115.307853
Galderisi M (2006) Diastolic dysfunction and diabetic cardiomyopathy: evaluation by Doppler echocardiography. J Am Coll Cardiol 48(8):1548–1551. https://doi.org/10.1016/j.jacc.2006.07.033
Ganote C, Armstrong S (1993) Ischaemia and the myocyte cytoskeleton: review and speculation. Cardiovasc Res 27(8):1387–1403. https://doi.org/10.1093/cvr/27.8.1387
Moe GW, Marin-Garcia J (2016) Role of cell death in the progression of heart failure. Heart Fail Rev 21(2):157–167. https://doi.org/10.1007/s10741-016-9532-0
Crea F, Camici PG, Bairey Merz CN (2014) Coronary microvascular dysfunction: an update. Eur Heart J 35(17):1101–1111. https://doi.org/10.1093/eurheartj/eht513
Stalker TJ, Skvarka CB, Scalia R (2003) A novel role for calpains in the endothelial dysfunction of hyperglycemia. FASEB J 17(11):1511–1513. https://doi.org/10.1096/fj.02-1213fje
Bodnar RJ, Rodgers ME, Chen WC, Wells A (2013) Pericyte regulation of vascular remodeling through the CXC receptor 3. Arterioscler Thromb Vasc Biol 33(12):2818–2829. https://doi.org/10.1161/ATVBAHA.113.302012
Yildirim C, Favre J, Weijers EM et al (2015) IFN-β affects the angiogenic potential of circulating angiogenic cells by activating calpain 1. Am J Physiol Heart Circ Physiol 309(10):H1667–H1678. https://doi.org/10.1152/ajpheart.00810.2014
Su Y, Cui Z, Li Z, Block ER (2006) Calpain-2 regulation of VEGF-mediated angiogenesis. FASEB J 20(9):1443–1451. https://doi.org/10.1096/fj.05-5354com
Miyazaki T, Honda K, Ohata H (2010) m-Calpain antagonizes RhoA overactivation and endothelial barrier dysfunction under disturbed shear conditions. Cardiovasc Res 85(3):530–541. https://doi.org/10.1093/cvr/cvp311
Zhang Y, Li Q, Youn JY, Cai H (2017) Protein phosphotyrosine phosphatase 1B (PTP1B) in calpain-dependent feedback regulation of vascular endothelial growth factor receptor (VEGFR2) in endothelial cells: implications in VEGF-dependent angiogenesis and diabetic wound healing. J Biol Chem 292(2):407–416. https://doi.org/10.1074/jbc.M116.766832
Li G, Iyengar R (2002) Calpain as an effector of the Gq signaling pathway for inhibition of Wnt/beta -catenin-regulated cell proliferation. Proc Natl Acad Sci U S A 99(20):13254–13259. https://doi.org/10.1073/pnas.202355799
Rios-Doria J, Kuefer R, Ethier SP, Day ML (2004) Cleavage of beta-catenin by calpain in prostate and mammary tumor cells. Cancer Res 64(20):7237–7240. https://doi.org/10.1158/0008-5472.CAN-04-1048
Xi XH, Wang Y, Li J, Wang FW, Tian GH, Yin MS, Mu YL, Chong ZZ (2015) Activation of Wnt/beta-catenin/GSK3beta signaling during the development of diabetic cardiomyopathy. Cardiovasc Pathol 24(3):179–186. https://doi.org/10.1016/j.carpath.2014.12.002
Ying Y, Zhu H, Liang Z, Ma X, Li S (2015) GLP1 protects cardiomyocytes from palmitate-induced apoptosis via Akt/GSK3b/b-catenin pathway. J Mol Endocrinol 55(3):245–262. https://doi.org/10.1530/JME-15-0155
Huang CJ, Gurlo T, Haataja L et al (2010) Calcium-activated calpain-2 is a mediator of beta cell dysfunction and apoptosis in type 2 diabetes. J Biol Chem 285(1):339–348. https://doi.org/10.1074/jbc.M109.024190
Shanab AY, Nakazawa T, Ryu M et al (2012) Metabolic stress response implicated in diabetic retinopathy: the role of calpain, and the therapeutic impact of calpain inhibitor. Neurobiol Dis 48(3):556–567. https://doi.org/10.1016/j.nbd.2012.07.025
Funding
This work was supported by operating grants from the National Natural Science Foundation of China [81470499] and the Canadian Institutes of Health Research [MOP-133657], and by the Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT, IRT1075).
Author information
Authors and Affiliations
Contributions
XT designed the experiments, researched and analysed data, drafted the manuscript and approved the submitted version. CJ researched and interpreted data, drafted the manuscript and approved the submitted version. HZ researched and collected data, drafted the manuscript and approved the submitted version. DZ designed the experiments, researched data, drafted the manuscript and approved the submitted version. RN researched data, drafted the manuscript and approved the submitted version. DJH contributed to the experimental design and discussion, reviewed/edited the manuscript and approved the submitted version. SX contributed to the discussion and the experimental design, reviewed/edited the manuscript, and approved the submitted version. G-CF contributed to the design and discussion, reviewed/revised the manuscript and approved the submitted version. PAG contributed to the generation of endothelial cell Capns1 knockout mice, the design and discussion, reviewed/revised the manuscript and approved the submitted version. ZS contributed to the early conception and design and the discussion, reviewed/revised the manuscript and approved the submitted version. TP designed the study, analysed data, wrote/revised the manuscript and approved/submitted the final version. TP is the guarantor of this work.
Corresponding author
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM
(PDF 740 kb)
Rights and permissions
About this article

Cite this article
Teng, X., Ji, C., Zhong, H. et al. Selective deletion of endothelial cell calpain in mice reduces diabetic cardiomyopathy by improving angiogenesis. Diabetologia 62, 860–872 (2019). https://doi.org/10.1007/s00125-019-4828-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-019-4828-y