
Abstract
Significant advances have been made in deciphering the mechanisms underlying fuel-stimulated insulin secretion by pancreatic beta cells. The contribution of the triggering/ATP-sensitive potassium (KATP)-dependent Ca2+ signalling and KATP-independent amplification pathways, that include anaplerosis and lipid signalling of glucose-stimulated insulin secretion (GSIS), are well established. A proposed model included a key role for a metabolic partitioning ‘switch’, the acetyl-CoA carboxylase (ACC)/malonyl-CoA/carnitine palmitoyltransferase-1 (CPT-1) axis, in beta cell glucose and fatty acid signalling for insulin secretion. This model has gained overwhelming support from a number of studies in recent years and is now refined through its link to the glycerolipid/NEFA cycle that provides lipid signals through its lipolysis arm. Furthermore, acetyl-CoA carboxylase may also control beta cell growth. Here we review the evidence supporting a role for the ACC/malonyl-CoA/CPT-1 axis in the control of GSIS and its particular importance under conditions of elevated fatty acids (e.g. fasting, excess nutrients, hyperlipidaemia and diabetes). We also document how it is linked to a more global lipid signalling system that includes the glycerolipid/NEFA cycle.
Similar content being viewed by others


Introduction
Insulin secretion in response to various fuel stimuli, such as glucose, some amino acids and fatty acids, involves transduction systems that require metabolism of the fuel stimulus in the pancreatic beta cell. Although much progress has been made in recent years, we still have not entirely elucidated the pathways and signalling molecules involved. A glucose-induced rise in the cytosolic ATP/ADP ratio leads to inhibition of ATP-sensitive potassium (KATP) channels and depolarisation of the beta cell, followed by an increase in cytosolic Ca2+, which promotes insulin granule exocytosis [1, 2]. The KATP channel-independent actions of glucose in beta cell signalling, also known as the amplification pathways, involve several metabolic coupling factors (MCFs) that link fuel metabolism to insulin exocytosis [3, 4].
Of the numerous amplification pathways that contribute to glucose-stimulated insulin secretion (GSIS), the following are thought to be important players: anaplerosis/cataplerosis, the ATP citrate lyase (ACL)/acetyl-CoA carboxylase (ACC)/malonyl-CoA/carnitine palmitoyltransferase-1 (CPT-1) axis, the glycerolipid/NEFA cycle [5], post-translational attachment of small ubiquitin-like modifier to target lysine residues (SUMOylation) [6], NADPH [7], reactive oxygen species [8] and the redox control of exocytosis proteins [5, 9] (Figs 1, 2). The idea that anaplerosis/cataplerosis and pyruvate cycling provide some of the essential MCFs is well accepted and this has been reviewed extensively [5, 9,10,11,12,13].

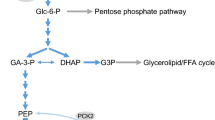
Metabolic signal transduction of the beta cell in response to glucose and NEFA. Glucose metabolism gives rise to various regulatory (glutamate, GTP, citrate) and effectory (ADP, ATP, NADPH, ROS) MCFs. The signalling actions of these key metabolites contribute to the facilitation of insulin granule exocytosis at different steps, including Ca2+ influx following the closure of KATP channels. Malonyl-CoA formed from glucose metabolism in beta cells controls the flux of NEFA through β-oxidation, an ‘off’ pathway of GSIS, by inhibiting the rate-limiting step of β-oxidation (catalysed by CPT-1). The accumulating FA-CoA thus is diverted towards the lipogenesis arm of the glycerolipid/NEFA cycle (see Fig. 2), leading to the formation of the lipolysis-derived MAG, which is an effectory MCF. 1-MAG directly binds and activates Munc13–1, an insulin granule exocytosis-facilitating protein. 1-MAG may be hydrolysed by the 1-MAG hydrolase ABHD6 (see Fig. 2) to generate NEFA for subsequent oxidation, thereby negatively affecting the secretion of insulin. Importantly, the inhibition of NEFA oxidation by malonyl-CoA prevents the catabolism of lipid signalling molecules, such as MAG. Figure 1 summarises the lipid signalling pathways that are shown in more detail in Fig. 2. We previously proposed that two types of MCF can be defined: (1) regulatory MCFs that modulate key metabolic pathways and networks involved in fuel-induced insulin secretion; and (2) effectory MCFs that are directly involved in the triggering and amplification arms of fuel-induced insulin secretion at late steps of their signalling cascade (e.g. exocytosis or membrane ionic events) [9]. ROS are produced in mitochondria, during electron transport, when there is excess supply of electron donors, such as glycerol 3-phosphate. DHAP, dihydroxyacetone phosphate; Glu, glutamate; Gro3P, glycerol 3-phosphate; ROS, reactive oxygen species. This figure is available as part of a downloadable slideset

Lipid signalling and the ACC/malonyl-CoA/CPT-1 metabolic signalling network in beta cell function. Pyruvate produced from glucose in the beta cells enters the mitochondria, where it is metabolised via the Krebs cycle to generate citrate. Under conditions of high glucose availability, a significant amount of citrate exits the mitochondria (cataplerosis) and is used by cytosolic ACL to form acetyl-CoA, which in turn is carboxylated by ACC1 to generate malonyl-CoA. Elevated glucose levels result in increased production of malonyl-CoA, which acts as a metabolic switch by inhibiting CPT-1, the rate-limiting enzyme that controls the entry of fatty acyl groups into mitochondria for β-oxidation. Inhibition of CPT-1 results in diversion of FA-CoA towards the glycerolipid/NEFA cycle, which produces signals that act as MCFs for insulin secretion. The entry of FA-CoA into the lipogenic arm of the glycerolipid/NEFA cycle is dependent on the availability of glycolysis-derived glycerol 3-phosphate, the cytosolic levels of which are controlled by G3PP, which negatively regulates the amplification of GSIS. As it is important to maintain FA-CoA levels, normal beta cells do not express ACOT7, which hydrolyses FA-CoA. The lipolysis arm of the glycerolipid/NEFA cycle generates 1-MAG, which activates Munc13–1 (insulin exocytosis facilitator), and ABHD6 hydrolyses 1-MAG to negatively control GSIS (not illustrated). ABHD6 is a more predominant MAG hydrolase than the classical monoacylglycerol lipase (MAGL) in pancreatic beta cells. NEFA generated via the lipolysis arm of the glycerolipid/NEFA cycle can exit the cell and activate free fatty acid-activated receptor-1 (FFAR-1), resulting in a signalling cascade of 1,2-DAG/ protein kinase D (PKD), stimulating insulin exocytosis. Thus, malonyl-CoA, by controlling fat oxidation and the glycerolipid/NEFA cycle flux, can regulate the lipid amplification of GSIS at several steps. Blue arrows indicate an activating effect on insulin exocytosis. Red arrows indicate pathways that negatively regulate GSIS. Dotted lines indicate inhibitory effect on the corresponding target molecules. Molecules in blue are key regulatory MCF signalling molecules, while those in brick-red colour are effectory MCF molecules. Metabolic enzymes are shown in green ovals. ψ, membrane potential; Ac-CoA, acetyl-CoA; ACSL, acyl-CoA synthetase, long-chain; ATGL, adipose triglyceride lipase; C1, phorbol esters/diacylglycerol binding domain of Munc13–1; C2, calcium-dependent phospholipid binding domain of Munc13–1; [Ca2+]i, intracellular calcium concentration; CGI58, comparative gene identification-58; GL, glycerolipid; Gro3P, glycerol 3-phosphate; HSL, hormone-sensitive lipase; KC, Krebs cycle; Kir6.2, a major subunit of the ATP-sensitive K+ channel, an inward-rectifier potassium ion channel; LPA, lysophosphatidic acid; Mal-CoA, malonyl-CoA; β-Ox, β-oxidation; PA, phosphatidic acid; Pyr, pyruvate; SUR1, sulfonylurea receptor-1; TG, triacylglycerol; VDCC, voltage-dependent calcium channel. This figure is available as part of a downloadable slideset
Lipid signalling is essential for GSIS. Thus, a fatty acid-dependent step is critically important for both GSIS and non-glucose-stimulated insulin secretion in vivo [14] and ex vivo [15] and if islets are deprived of NEFA their response to GSIS is compromised [16]. Lipid signalling of GSIS was proposed to involve three mechanisms. Extracellular lipid signalling is mediated by NEFA activation of free fatty acid-activated receptor-1 (FFAR1, also known as GPR40), leading to generation of intracellular diacylglycerol (DAG) and inositol trisphosphate [17]. The intracellular pathways involve the ACC/malonyl-CoA/CPT-1 network and generation of lipid molecules via the glycerolipid/NEFA cycle [18, 19] (Fig. 2). Our laboratory identified two important enzymes of the glycerolipid/NEFA cycle: glycerol 3-phosphate phosphatase (G3PP) and α/β-hydrolase domain 6 (ABHD6). G3PP hydrolyses glucose-derived glycerol 3-phosphate, the precursor for lipogenesis [20], whereas ABHD6 controls the last step of lipolysis by hydrolysing 1-monoacylglycerol (MAG) [21]. Importantly, 1-MAG is an MCF of GSIS by activating Munc13-1, an exocytosis-facilitating protein [21] (Figs. 1, 2).
This review focuses on intracellular lipid signalling for glucose- and NEFA-induced insulin secretion, highlighting the role of the ACC/malonyl-CoA/CPT-1 network.
What is the ACC/malonyl-CoA/CPT-1 metabolic signalling network?
The hypothesis proposing an intracellular lipid amplification arm for GSIS was originally laid out by us [22,23,24,25]. We initially proposed that glucose-metabolism-derived malonyl-CoA, by inhibiting CPT-1, diverts long-chain fatty acyl-CoA (FA-CoA) from mitochondrial β-oxidation towards the synthesis of complex lipids, such as DAGs, that can act as signals for insulin secretion. The subsequent realisation that lipolysis plays a key role in GSIS led us to refine the model by linking the ACC/malonyl-CoA/CPT-1 network to the glycerolipid/NEFA cycle (Fig. 1). In this revised model malonyl-CoA acts as a ‘metabolic switch’ signal by modulating fuel partitioning (the relative rates of glucose and NEFA oxidation) and is a regulatory MCF in insulin secretion, whereas the lipid signals generated via lipolysis in the glycerolipid/NEFA cycle act as effector signals [9]. Thus, inhibition of CPT-1 and fat oxidation allows continuous operation of the glycerolipid/NEFA cycle.
Here, we review the evidence for and against the ACC/malonyl-CoA/CPT-1 hypothesis and present a consensus view that emerges (Figs. 1, 2).
In vitro evidence for the role of the ACC/malonyl-CoA/CPT-1 metabolic signalling network in metabolic signalling
Biochemical evidence
It was initially noticed using HITβ cells and rat islets, that glucose stimulation causes marked alterations in the acyl-CoA profile, with early change occurring in malonyl-CoA levels [22,23,24,25]. The rise in malonyl-CoA that preceded insulin release and correlated with the dose dependency of GSIS [22,23,24,25] was confirmed in several studies using metabolomics approaches in INS-1(832/13) cells [26, 27]. As predicted by the hypothesis, glucose caused a decrease in NEFA oxidation in association with a rise in citrate and lipogenesis in rodent islets and beta cell lines, and both correlated with the glucose dose dependence of insulin secretion [9, 10, 26]. Finally, glucose decreased ACC phosphorylation and increased its activity in a beta cell line and this was closely related to insulin secretion [28].
Pharmacological evidence
CPT-1 inhibition
In early studies, inhibition of pancreatic islet fatty acid oxidation by 2-bromostearate was shown to restore GSIS in fasted islets [29]. In addition, inhibition of CPT-1 by 2-bromopalmitate in isolated rat islets promoted GSIS while blocking β-oxidation [30]. Similarly, the CPT-1 inhibitor etomoxir reduced β-oxidation in isolated rat islets and this was associated with enhanced GSIS [30]. The reduced GSIS in db/db mouse islets could be restored to near normal levels by incubating the islets with etomoxir [31]. Evidence suggesting a role for CPT-1 in regulating GSIS was also obtained in studies using CPT-1-overexpressing INS1E cells; these cells showed a reduced GSIS response, which could be restored by etomoxir [32]. Acute addition of etomoxir to the INS1E cells also partially reversed the decreased GSIS in cells chronically exposed to NEFA, a condition wherein NEFA oxidation is enhanced [31].
ATP citrate lyase inhibition
Besides its participation in the Krebs cycle, citrate exits mitochondria via the dicarboxylate/tricarboxylate carrier. In the cytoplasm, citrate is cleaved by ACL to oxaloacetate and acetyl-CoA. ACC converts cytosolic acetyl-CoA to malonyl-CoA, which regulates β-oxidation by inhibiting CPT-1, the rate-limiting enzyme involved in the transport of fatty acyl groups into mitochondria (Fig. 2). Further evidence to support the ACC/malonyl-CoA/ CPT-1 hypothesis was garnered in a rat pancreas perfusion study, which showed that inhibition of ACL by hydroxycitrate caused a profound decline in GSIS [30]. The functional importance of cataplerosis (the exit of Krebs cycle intermediates into the cytoplasm) via citrate for GSIS is further supported by the observation that radicicol, another ACL inhibitor, partially blocked GSIS in purified rat beta cells [33] and INS832/13 cells [34].
Pyruvate carboxylase inhibition
The Krebs cycle generates citrate via the condensation of acetyl-CoA and oxaloacetate formed by pyruvate carboxylase. Malonyl-CoA is derived from cytosolic citrate. GSIS in beta cells was found to be reduced by the inhibition of oxaloacetate formation by pyruvate carboxylase using phenylacetate [10, 11].
Inhibition of ACC
It was noticed that ACC1 is the predominant isoform expressed in pancreatic islets and INS-1(832/13) beta cells and that inhibitors of ACC1, CP-640186 and 5-(tetradecyloxy)-2-furoic acid (TOFA), which curtailed lipogenesis, inhibited GSIS [35].
FA-CoA synthase inhibition
Triacsin C, a FA-CoA synthase inhibitor, curtailed GSIS in INS832/13 cells only in the presence of added NEFA, emphasising the importance of incubation conditions in addressing the role of lipid signalling for GSIS [15]. This important point, which is at the root of the controversy in the field, will be further discussed below. In fact, depletion of NEFA by exhaustive washing with bovine serum albumin in MIN6 beta cells was found to lower GSIS, which could be restored by the addition of NEFA [16].
Evidence from altered expression of relevant genes
ACL and mitochondrial citrate carrier
Higher expression levels of ACL in the islets (a non-lipogenic tissue) in comparison with liver, both in rodents and in humans [36], supports a role for this enzyme in GSIS. The expression of ACL was found to be reduced by 60% in islets from individuals with vs without type 2 diabetes, suggesting that a reduction in malonyl-CoA formation in diabetic beta cells could contribute to compromised GSIS [37]. Indeed, RNAi-knockout of Acl (also known as Acly) in INS-1(832/13) cells inhibited GSIS and the KATP-independent pathway of insulin secretion [34]. In a recent study, the relative increase in GSIS response in pancreatic islets from the juvenile stage to adulthood was attributed to elevated pyruvate–citrate cycling, ACL expression and malonyl-CoA production [38]. RNAi-knockdown of the mitochondrial citrate carrier [39] in INS-1(832/13) cells resulted in a decline in GSIS, which is compatible with a role for malonyl-CoA signalling.
The possibility of cytosolic acetyl-CoA being generated from glucose-derived pyruvate by alternative routes was suggested by the observation that inhibition of ACL still allows glucose carbon incorporation into lipids without compromising GSIS, probably via the acetoacetate pathway [40]. Thus, redundant pathways exist that can support malonyl-CoA formation and the generation of lipid MCFs for GSIS.
ACC-1
Employing INS-1 cells stably expressing antisense Acc mRNA, it was noticed that a decline in ACC protein expression was associated with reduced malonyl-CoA formation, elevated β-oxidation and decreased GSIS response [41].
Malonyl-CoA decarboxylase
Overexpression of cytosolically targeted malonyl-CoA decarboxylase (MCD) in INS-1(832/13) cells and rat islets resulted in lowered malonyl-CoA levels in association with elevated fatty acid oxidation and reduced GSIS in the presence of added NEFA, but not in their absence [15]. This underscores the importance of added NEFA in ascertaining the significance of lipid signalling for GSIS, a key point that will be discussed below in the section related to evidence against the hypothesis.
CPT-1
Studies in which wild-type CPT-1 was overexpressed in INS1E cells, showed reduced GSIS, restorable by etomoxir or supply of NEFA [32]. This strongly supports a role for CPT-1 in the negative control of GSIS. In addition, overexpression of malonyl-CoA-insensitive mutant CPT-1 (M593S) in INS-1(832/13) cells and rat islets curtailed GSIS and enhanced fatty acid oxidation [42].
Long-chain acyl-CoA synthase
The significance of FA-CoA in GSIS was demonstrated in a study showing that RNAi-knockdown of long-chain acyl-CoA synthases (either ACSL3 or ACSL4), two enzymes concentrated in the insulin granules and synthesising FA-CoA, led to a decline in GSIS in human islets and INS-1(832/13) cells [43]. In addition, FA-CoA directly promotes exocytosis of insulin granules in beta cells [44].
Acyl-CoA thioesterase-7
About 60 genes expressed ubiquitously are relatively silenced (disallowed) in beta cells. One such gene encodes acyl-CoA thioesterase-7 (ACOT7), which hydrolyses FA-CoA. ACOT7 downregulation is needed if FA-CoA signalling is central to insulin secretion. Acot7-overexpressing INS-1(832/13) cells showed impaired GSIS, supporting a role for FA-CoA or its derivatives in GSIS [45]. Of note, all Acot family members are poorly expressed in mouse islets [45].
Carnitine acylcarnitine translocase
An important step in fatty acid oxidation is the transport of fatty acylcarnitines into mitochondria by carnitine acylcarnitine translocase (CACT) [46]. Deficiency of the CACT-encoding gene (Slc25a20) is associated with hypoketotic hypoglycaemia, though insulin levels were not reported. Downregulation of CACT by miR-132 or miR-212 in beta cells led to elevated fatty acylcarnitines and increased insulin secretion [47]. Although the authors of this study proposed that acylcarnitines may directly modulate exocytosis, the possibility of a build-up of FA-CoA for lipid signalling still remains.
In vivo studies in support of ACC/malonyl-CoA/CPT-1 signalling for insulin secretion
Pharmacological studies
CPT-1 inhibition
Supranormal GSIS response was noticed when 24 h-fasted rats under hyperglycaemic clamp were maintained at high plasma levels of NEFA, and infused with etomoxir to block fatty acid oxidation [48].
Suppression of ACC1 activity
Support for the role of ACC1 in insulin secretion has been reported in studies using the ACC inhibitor ND-630, which lowered malonyl-CoA levels in tissues and reduced GSIS in Sprague Dawley rats fed a high-sucrose high-fat diet, and also in ZDF rats [49].
Inhibition of ACL activity
Inhibition of ACL in vivo by bempedoic acid in high-fat-diet-fed mice curtailed hyperinsulinaemia and improved glucose tolerance [50]. Interestingly, a crossover study involving 3 days of administration of the ACL inhibitor hydroxycitrate revealed a consistent decrease in plasma insulin levels, though the results were not significant possibly due to the low number of participants (n = 10) [51].
Genetic studies
Fatty acid oxidation enzymes
Many genetic defects in fatty acid oxidation, including those of CPT-1, CPT-2, CACT, long-chain acyl-CoA dehydrogenase and short-chain hydroxy acyl-CoA dehydrogenase (HADHSC), are associated with hypoketotic hypoglycaemia, although the precise causes are unknown [52]. At least in the case of the HADHSC defect, it was found to be associated with hyperinsulinism [53, 54]. In many other cases, insulin levels were not reported. Thus, it would be interesting to know whether the hypoglycaemia is partly due to elevated plasma insulin levels, besides other contributions such as reduced gluconeogenesis.
Acot7-overexpressing mice
Transgenic expression of mitochondrial Acot7 specifically in the beta cells of mice led to glucose intolerance and reduced GSIS [45]. The changes in GSIS were related to reduced islet FA-CoA and ATP/ADP. Of interest, islet levels of DAG and MAG, which are thought to be MCFs for insulin secretion, were reduced by 45% and 60%, respectively, although they did not reach significance possibly due to the large SD.
Modulation of transcription factors controlling lipid metabolism genes
An inverse relationship between fatty acid β-oxidation and GSIS was noticed in studies wherein the transcription factors peroxisome proliferator-activated receptor (PPAR)δ [55] or PPARα [56, 57], which control the expression of β-oxidation genes, including Cpt-1 (also known as Cpt1A), were genetically deleted. Pparα (also known as Ppara)-knockout mice develop hyperinsulinaemic hypoglycaemia in the fasting state [56]. Similarly, using a mouse model in which three isoforms of forkhead box protein O (FOXO) were knocked down specifically in beta cells, we observed that there is a preferential utilisation of lipids by the beta cells, associated with elevated β-oxidation and curtailed GSIS [58]. Also, a knockin mouse model expressing deacetylated FOXO1 (activated form) specifically in beta cells displayed reduced mitochondrial β-oxidation and enhanced GSIS response in vivo [59].
Acc1-knockout and -knockin mice
A recent study employing beta cell-specific Acc1 (also known as Acaca)-knockout mice demonstrated that ACC1 is critical for GSIS [60]. Interestingly, ex vivo islet studies showed that the reduced secretion occurs at low and intermediate glucose concentrations and that the inhibitory effect was overridden at high (20 mmol/l) glucose. A simple explanation for why high glucose overrides the inhibition of GSIS in the beta cell Acc1-knockout mice may be that other amplification pathways take over in order to ensure secretion. In our view, this paper provides direct key support for the role of malonyl-CoA in GSIS. The authors mentioned that Acc1-knockout mouse islets did not show changes in fatty acid oxidation or lipid signalling molecules. Unfortunately, they measured these variables at a concentration of glucose where secretion was unchanged (20 mmol/l) and they did not show under their experimental condition that high glucose itself was able to inhibit fat oxidation, as anticipated. Interestingly, this study reported a novel role of ACC1 related to the control of beta cell mass prior to adulthood [60].
Regulation of ACC1 by AMP-activated protein kinase (AMPK) is well established [61]. It was found that mice with serine-to-alanine knockin mutations in both Acc1 (Ser79) and Acc2 (also known as Acacb) (Ser212), which render ACC constitutively active and not susceptible to inhibitory phosphorylation by AMPK, display elevated fasting plasma insulin levels [62]. Thus, it appears that in vivo, when ACC activity is reduced and there is less malonyl-CoA production in the beta cell, insulin secretion is reduced; conversely, when ACC is constitutively active, beta cells secrete more insulin.
Liver kinase B1-knockout mice
Beta cell-specific Lkb1-knockout mice show enhanced insulin secretion by an undefined mechanism [63,64,65]. They display impaired mitochondrial metabolism and lower ATP levels following glucose stimulation, yet compensate for this by upregulating the production of citrate. It was found that at low-glucose Lkb1−/− beta cells failed to inhibit ACC1 and consequently accumulated malonyl-CoA derived fatty acids [65]. Thus, this study also supports a role for ACC/malonyl-CoA signalling in insulin secretion.
Evidence against a role for ACC/malonyl-CoA/CPT1 signalling for insulin secretion
In vitro pharmacological approaches
Despite overwhelming support for the role of malonyl-CoA/CPT-1/fatty acyl-CoA metabolic signalling in the regulation of GSIS, a few studies have raised doubts about this model. These discrepant results can be explained by the methodology that was employed.
Inhibition of fatty acid oxidation by bromopalmitate
In a study using normal mouse islets incubated in the presence of 30 mmol/l KCl plus the KATP opener diazoxide to examine KATP-independent signalling, the fatty acid oxidation inhibitor bromopalmitate failed to modify basal (3 mmol/l) and high-glucose (20 mmol/l)-stimulated insulin secretion, which would be against a role for the malonyl-CoA switch [66]. However, at supramaximal concentrations of glucose, fat oxidation is maximally inhibited such that bromopalmitate cannot further change fat oxidation. Conversely, at 3 mmol/l glucose, cytosolic Ca2+ is low and because a rise in Ca2+ is a prerequisite for the amplification pathways of secretion, even if bromopalmitate reduced fat oxidation, it should not change secretion. In addition, fat oxidation was not measured, and this study examined pathways under interesting but totally unphysiological conditions (very high KCl and cytosolic Ca2+ plus diazoxide) so its relevance to a normal situation is unclear.
Inhibition of ACC, ACL and fatty acyl-CoA synthase
An inhibitor of ACC1, TOFA, was found to have no effect on GSIS in INS-1 cells [67]. Similarly, the ACL inhibitor hydroxycitrate lacked effect on GSIS in INS1(832/13) cells and rat islets [68]. In 13C isotopomer flux measurements, inhibiting the flux of glucose carbons into malonyl-CoA by hydroxycitrate had no effect on GSIS in INS1 cells [40]. Finally, the use of the FA-CoA synthase inhibitor, triascin C, did not reveal a modification of GSIS in INS-1 cells [69] or INS 832/13 cells [70]. However, in all these studies, no fatty acid was present during incubations. The premise for the lipid signalling hypothesis is that substantial endogenous triacylglycerol stores or exogenous NEFA are required because the first step of this process is the synthesis of FA-CoA. These experiments were in fact performed in fatty-acid-depleted cells due to the presence of 0.2% fatty-acid-free BSA, which traps cellular fatty acids. At the time these studies were done, scientists were not aware of the potential caveats related to depleting beta cells of NEFA in examining lipid signalling for GSIS. Thus, the experimental conditions were not appropriate for testing the hypothesis.
In vitro gene expression studies
MCD overexpression
One of the arguments against the role of malonyl-CoA as a regulator of GSIS is that overexpression of MCD in INS-1 cells had no effect on GSIS in one study [69]. However, this study suffers from drawbacks: (1) the MCD construct used was not directed to cytosol and may not have lowered the cytosolic pool of malonyl-CoA and (2) the INS-1 cell line used does not elicit the KATP channel-independent pathway of GSIS, as discovered subsequently [71]. To address this caveat, a later study by the same group employed an MCD construct that lacked both mitochondrial and peroxisomal targeting sequences for overexpression in INS-1(832/13) cells and reported a lack of effect on GSIS at 15 mmol/l glucose, both in the presence and absence of NEFA [70]. If one closely examines the results in cells cultured in the presence of NEFA, they do indicate a substantial (40%) reduction of GSIS in MCD-overexpressing cells, compared with controls, although the difference was deemed not significant [70]. Additionally, 15 mmol/l glucose is a supramaximal concentration for INS832/13 cells; no intermediate glucose concentration was tested and very high glucose concentrations override the inhibition of GSIS, as discussed above. Thus, the inhibition of GSIS in Acc1-knockout mouse islets is observed at basal and intermediate but not maximal glucose concentrations [60]. In a subsequent study, we repeated the MCD overexpression experiments and found similar results, observing that GSIS is markedly reduced in MCD-overexpressing INS832/13 cells but only if the medium is supplemented with exogenous fatty acids to permit intracellular lipid signalling [15].
Suppression of ACL
ACL knockdown was reported to have no effect on GSIS in INS-1(832/13) cells when measured only at maximal 16.7 mmol/l glucose, possibly due to the absence of NEFA in the incubation medium for insulin secretion as well as GSIS not being tested at submaximal glucose [68]. The lack of effect could also be due to the presence of alternate routes that provide cytosolic acetyl-CoA, as mentioned above [40]. We reported that ACL knockdown in the same cell line under similar experimental conditions does result in reduced GSIS but that the inhibitory effect is most prominent in the presence of exogenous NEFA or at intermediate glucose [34].
Where does the balance tip?
Collectively, the above summarises the comprehensive information accumulated over the years and provides overwhelming evidence in favour of an ACC1/malonyl-CoA/CPT-1/fatty acyl-CoA network playing a key role in the regulation of beta cell glucose and fatty acid signalling for insulin secretion. This network appears to be particularly involved in GSIS regulation under conditions where fatty acids are elevated (fasting, cells chronically exposed to NEFA, obesity and diabetes) (see Textbox). In vitro and in vivo studies, as well as data from human islets, addressing various components of this network employing biochemical, pharmacological, molecular and genetic tools directly support this view. In contrast, the evidence against the hypothesis is not strong and is based on questionable experimental approaches. Furthermore, these experiments have been exclusively in vitro without support from in vivo experiments.
Conclusion and perspective
The consensus that emerges places malonyl-CoA as a signal that acts as a ‘metabolic switch’, playing a critical role in regulating insulin secretion promoted by glucose and other fuel stimuli, particularly when islet fat oxidation is elevated as under conditions of fasting, obesity and diabetes, via modulation of β-oxidation flux (see Textbox). Thus, the Textbox shows many situations where the ACC/malonyl-CoA/CPT1 network was shown to be involved in GSIS. The role of this network as a metabolic on/off switch is also in line with the significance of the glycerolipid/NEFA cycle and its lipolysis arm in GSIS (Fig. 1). Nevertheless, unlike MAG, which acts as an ‘effectory’ MCF [9] at the late event of insulin exocytosis by activating Munc13-1, malonyl-CoA is a ‘regulatory’ MCF, controlling insulin secretion at earlier steps in the amplification pathway. Besides functioning as a switch for redirecting fatty acid flux, malonyl-CoA has also been ascribed a central fuel-sensing role in the hypothalamus and as an anorectic mediator in the central control of feeding [72, 73]. Interestingly, these actions may in part be independent of fatty acid oxidation regulation. Thus, future studies should be directed towards delineating the underlying mechanisms that encompass the various regulatory roles of malonyl-CoA in beta cell function, some independent of CPT-1, possibly involving protein malonylation [74]. Studies of these processes may also be relevant to fuel signalling in other tissues.
Abbreviations
- ABHD6:
-
α/β-Hydrolase domain 6
- ACC:
-
Acetyl-CoA carboxylase
- ACL:
-
ATP citrate lyase
- ACOT7:
-
Acyl-CoA thioesterase-7
- AMPK:
-
AMP-activated protein kinase
- CACT:
-
Carnitine acylcarnitine translocase
- CPT-1:
-
Carnitine palmitoyltransferase-1
- DAG:
-
Diacylglycerol
- FA-CoA:
-
Fatty acyl-CoA
- FOXO:
-
Forkhead box protein O
- G3PP:
-
Glycerol 3-phosphate phosphatase
- GSIS:
-
Glucose-stimulated insulin secretion
- HADHSC:
-
Short-chain hydroxy acyl-CoA dehydrogenase
- KATP :
-
ATP-sensitive potassium
- MAG:
-
Monoacylglycerol
- MCD:
-
Malonyl-CoA decarboxylase
- MCF:
-
Metabolic coupling factor
- NEFA:
-
Non-esterified fatty acid
- PPAR:
-
Peroxisome proliferator-activated receptor
- TOFA:
-
5-(Tetradecyloxy)-2-furoic acid
References
Prentki M (1996) New insights into pancreatic β-cell metabolic signaling in insulin secretion. Eur J Endocrinol 134(3):272–286. https://doi.org/10.1530/eje.0.1340272
Fridlyand LE, Ma L, Philipson LH (2005) Adenine nucleotide regulation in pancreatic β-cells: modeling of ATP/ADP-Ca2+ interactions. Am J Physiol Endocrinol Metab 289(5):E839–E848. https://doi.org/10.1152/ajpendo.00595.2004
Aizawa T, Sato Y, Ishihara F et al (1994) ATP-sensitive K+ channel-independent glucose action in rat pancreatic β-cell. Am J Phys 266(3 Pt 1):C622–C627. https://doi.org/10.1152/ajpcell.1994.266.3.C622
Gembal M, Gilon P, Henquin JC (1992) Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse β cells. J Clin Invest 89(4):1288–1295. https://doi.org/10.1172/JCI115714
Prentki M, Madiraju SR (2012) Glycerolipid/free fatty acid cycle and islet beta-cell function in health, obesity and diabetes. Mol Cell Endocrinol 353(1–2):88–100. https://doi.org/10.1016/j.mce.2011.11.004
Ferdaoussi M, Fu J, Dai X et al (2017) SUMOylation and calcium control syntaxin-1A and secretagogin sequestration by tomosyn to regulate insulin exocytosis in human ss cells. Sci Rep 7(1):248. https://doi.org/10.1038/s41598-017-00344-z
Ivarsson R, Quintens R, Dejonghe S et al (2005) Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 54(7):2132–2142. https://doi.org/10.2337/diabetes.54.7.2132
Pi J, Bai Y, Zhang Q et al (2007) Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56(7):1783–1791. https://doi.org/10.2337/db06-1601
Prentki M, Matschinsky FM, Madiraju SR (2013) Metabolic signaling in fuel-induced insulin secretion. Cell Metab 18(2):162–185. https://doi.org/10.1016/j.cmet.2013.05.018
Farfari S, Schulz V, Corkey B, Prentki M (2000) Glucose-regulated anaplerosis and cataplerosis in pancreatic beta-cells: possible implication of a pyruvate/citrate shuttle in insulin secretion. Diabetes 49(5):718–726. https://doi.org/10.2337/diabetes.49.5.718
Fransson U, Rosengren AH, Schuit FC, Renstrom E, Mulder H (2006) Anaplerosis via pyruvate carboxylase is required for the fuel-induced rise in the ATP:ADP ratio in rat pancreatic islets. Diabetologia 49(7):1578–1586. https://doi.org/10.1007/s00125-006-0263-y
Schuit F, De Vos A, Farfari S et al (1997) Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J Biol Chem 272(30):18572–18579. https://doi.org/10.1074/jbc.272.30.18572
Jensen MV, Gooding JR, Ferdaoussi M et al (2017) Metabolomics applied to islet nutrient sensing mechanisms. Diabetes Obes Metab 19(Suppl 1):90–94. https://doi.org/10.1111/dom.13010
Dobbins RL, Chester MW, Stevenson BE, Daniels MB, Stein DT, McGarry JD (1998) A fatty acid- dependent step is critically important for both glucose- and non-glucose-stimulated insulin secretion. J Clin Invest 101(11):2370–2376. https://doi.org/10.1172/JCI1813
Roduit R, Nolan C, Alarcon C et al (2004) A role for the malonyl-CoA/long-chain acyl-CoA pathway of lipid signaling in the regulation of insulin secretion in response to both fuel and nonfuel stimuli. Diabetes 53(4):1007–1019. https://doi.org/10.2337/diabetes.53.4.1007
Hauke S, Keutler K, Phapale P, Yushchenko DA, Schultz C (2018) Endogenous fatty acids are essential signaling factors of pancreatic β-cells and insulin secretion. Diabetes 67(10):1986–1998. https://doi.org/10.2337/db17-1215
Ghislain J, Poitout V (2017) The role and future of FFA1 as a therapeutic target. Handb Exp Pharmacol 236:159–180. https://doi.org/10.1007/164_2016_51
Nolan CJ, Madiraju MS, Delghingaro-Augusto V, Peyot ML, Prentki M (2006) Fatty acid signaling in the beta-cell and insulin secretion. Diabetes 55(Suppl 2):S16–S23. https://doi.org/10.2337/db06-S003
Nolan CJ, Leahy JL, Delghingaro-Augusto V et al (2006) Beta cell compensation for insulin resistance in Zucker fatty rats: increased lipolysis and fatty acid signalling. Diabetologia 49(9):2120–2130. https://doi.org/10.1007/s00125-006-0305-5
Mugabo Y, Zhao S, Seifried A et al (2016) Identification of a mammalian glycerol-3-phosphate phosphatase: role in metabolism and signaling in pancreatic beta-cells and hepatocytes. Proc Natl Acad Sci U S A 113(4):E430–E439. https://doi.org/10.1073/pnas.1514375113
Zhao S, Mugabo Y, Iglesias J et al (2014) α/β-Hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion. Cell Metab 19(6):993–1007. https://doi.org/10.1016/j.cmet.2014.04.003
Corkey BE, Glennon MC, Chen KS, Deeney JT, Matschinsky FM, Prentki M (1989) A role for malonyl-CoA in glucose-stimulated insulin secretion from clonal pancreatic beta-cells. J Biol Chem 264(36):21608–21612
Prentki M, Corkey BE (1996) Are the β-cell signaling molecules malonyl-CoA and cytosolic long-chain acyl-CoA implicated in multiple tissue defects of obesity and NIDDM? Diabetes 45(3):273–283. https://doi.org/10.2337/diab.45.3.273
Liang Y, Matschinsky FM (1991) Content of CoA-esters in perifused rat islets stimulated by glucose and other fuels. Diabetes 40(3):327–333. https://doi.org/10.2337/diab.40.3.327
Prentki M, Vischer S, Glennon MC, Regazzi R, Deeney JT, Corkey BE (1992) Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J Biol Chem 267(9):5802–5810
Lorenz MA, El Azzouny MA, Kennedy RT, Burant CF (2013) Metabolome response to glucose in the beta-cell line INS-1 832/13. J Biol Chem 288(15):10923–10935. https://doi.org/10.1074/jbc.M112.414961
Mugabo Y, Zhao S, Lamontagne J et al (2017) Metabolic fate of glucose and candidate signaling and excess-fuel detoxification pathways in pancreatic beta-cells. J Biol Chem 292(18):7407–7422. https://doi.org/10.1074/jbc.M116.763060
Zhang S, Kim KH (1995) Glucose activation of acetyl-CoA carboxylase in association with insulin secretion in a pancreatic beta-cell line. J Endocrinol 147(1):33–41. https://doi.org/10.1677/joe.0.1470033
Tamarit-Rodriguez J, Vara E, Tamarit J (1984) Starvation-induced secretory changes of insulin, somatostatin, and glucagon and their modification by 2-bromostearate. Horm Metab Res 16(3):115–119. https://doi.org/10.1055/s-2007-1014715
Chen S, Ogawa A, Ohneda M, Unger RH, Foster DW, McGarry JD (1994) More direct evidence for a malonyl-CoA-carnitine palmitoyltransferase I interaction as a key event in pancreatic β-cell signaling. Diabetes 43(7):878–883. https://doi.org/10.2337/diab.43.7.878
Zhou YP, Berggren PO, Grill V (1996) A fatty acid-induced decrease in pyruvate dehydrogenase activity is an important determinant of β-cell dysfunction in the obese diabetic db/db mouse. Diabetes 45(5):580–586. https://doi.org/10.2337/diab.45.5.580
Rubi B, Antinozzi PA, Herrero L et al (2002) Adenovirus-mediated overexpression of liver carnitine palmitoyltransferase I in INS1E cells: effects on cell metabolism and insulin secretion. Biochem J 364(Pt 1):219–226. https://doi.org/10.1042/bj3640219
Flamez D, Berger V, Kruhoffer M, Orntoft T, Pipeleers D, Schuit FC (2002) Critical role for cataplerosis via citrate in glucose-regulated insulin release. Diabetes 51(7):2018–2024. https://doi.org/10.2337/diabetes.51.7.2018
Guay C, Madiraju SR, Aumais A, Joly E, Prentki M (2007) A role for ATP-citrate lyase, malic enzyme, and pyruvate/citrate cycling in glucose-induced insulin secretion. J Biol Chem 282(49):35657–35665. https://doi.org/10.1074/jbc.M707294200
MacDonald MJ, Dobrzyn A, Ntambi J, Stoker SW (2008) The role of rapid lipogenesis in insulin secretion: insulin secretagogues acutely alter lipid composition of INS-1 832/13 cells. Arch Biochem Biophys 470(2):153–162. https://doi.org/10.1016/j.abb.2007.11.017
MacDonald MJ, Longacre MJ, Warner TF, Thonpho A (2013) High level of ATP citrate lyase expression in human and rat pancreatic islets. Horm Metab Res 45(5):391–393. https://doi.org/10.1055/s-0032-1329987
MacDonald MJ, Longacre MJ, Langberg EC et al (2009) Decreased levels of metabolic enzymes in pancreatic islets of patients with type 2 diabetes. Diabetologia 52(6):1087–1091. https://doi.org/10.1007/s00125-009-1319-6
Wortham M, Benthuysen JR, Wallace M et al (2018) Integrated in vivo quantitative proteomics and nutrient tracing reveals age-related metabolic rewiring of pancreatic beta cell function. Cell Rep 25(10):2904–2918 e2908. https://doi.org/10.1016/j.celrep.2018.11.031
Joseph JW, Jensen MV, Ilkayeva O et al (2006) The mitochondrial citrate/isocitrate carrier plays a regulatory role in glucose-stimulated insulin secretion. J Biol Chem 281(47):35624–35632. https://doi.org/10.1074/jbc.M602606200
El Azzouny M, Longacre MJ, Ansari IH, Kennedy RT, Burant CF, MacDonald MJ (2016) Knockdown of ATP citrate lyase in pancreatic beta cells does not inhibit insulin secretion or glucose flux and implicates the acetoacetate pathway in insulin secretion. Mol Metab 5(10):980–987. https://doi.org/10.1016/j.molmet.2016.07.011
Zhang S, Kim KH (1998) Essential role of acetyl-CoA carboxylase in the glucose-induced insulin secretion in a pancreatic beta-cell line. Cell Signal 10(1):35–42. https://doi.org/10.1016/S0898-6568(97)00070-3
Herrero L, Rubi B, Sebastian D et al (2005) Alteration of the malonyl-CoA/carnitine palmitoyltransferase I interaction in the beta-cell impairs glucose-induced insulin secretion. Diabetes 54(2):462–471. https://doi.org/10.2337/diabetes.54.2.462
Ansari IH, Longacre MJ, Stoker SW et al (2017) Characterization of Acyl-CoA synthetase isoforms in pancreatic beta cells: gene silencing shows participation of ACSL3 and ACSL4 in insulin secretion. Arch Biochem Biophys 618(March 15):32–43. https://doi.org/10.1016/j.abb.2017.02.001
Deeney JT, Gromada J, Hoy M et al (2000) Acute stimulation with long chain acyl-CoA enhances exocytosis in insulin-secreting cells (HIT T-15 and NMRI beta-cells). J Biol Chem 275(13):9363–9368. https://doi.org/10.1074/jbc.275.13.9363
Martinez-Sanchez A, Pullen TJ, Chabosseau P et al (2016) Disallowance of Acot7 in β-cells is required for normal glucose tolerance and insulin secretion. Diabetes 65(5):1268–1282. https://doi.org/10.2337/db15-1240
Pande SV, Murthy MS (1994) Carnitine-acylcarnitine translocase deficiency: implications in human pathology. Biochim Biophys Acta 1226(3):269–276. https://doi.org/10.1016/0925-4439(94)90037-X
Soni MS, Rabaglia ME, Bhatnagar S et al (2014) Downregulation of carnitine acyl-carnitine translocase by miRNAs 132 and 212 amplifies glucose-stimulated insulin secretion. Diabetes 63(11):3805–3814. https://doi.org/10.2337/db13-1677
Stein DT, Esser V, Stevenson BE et al (1996) Essentiality of circulating fatty acids for glucose-stimulated insulin secretion in the fasted rat. J Clin Invest 97(12):2728–2735. https://doi.org/10.1172/JCI118727
Harriman G, Greenwood J, Bhat S et al (2016) Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc Natl Acad Sci U S A 113(13):E1796–E1805. https://doi.org/10.1073/pnas.1520686113
Samsoondar JP, Burke AC, Sutherland BG et al (2017) Prevention of diet-induced metabolic dysregulation, inflammation, and atherosclerosis in Ldlr−/− mice by treatment with the ATP-citrate lyase inhibitor bempedoic acid. Arterioscler Thromb Vasc Biol 37(4):647–656. https://doi.org/10.1161/ATVBAHA.116.308963
Kriketos AD, Thompson HR, Greene H, Hill JO (1999) (−)-Hydroxycitric acid does not affect energy expenditure and substrate oxidation in adult males in a post-absorptive state. Int J Obes Relat Metab Disord 23(8):867–873. https://doi.org/10.1038/sj.ijo.0800965
Houten SM, Violante S, Ventura FV, Wanders RJ (2016) The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu Rev Physiol 78(1):23–44. https://doi.org/10.1146/annurev-physiol-021115-105045
Clayton PT, Eaton S, Aynsley-Green A et al (2001) Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. J Clin Invest 108(3):457–465. https://doi.org/10.1172/JCI200111294
Pepin E, Guay C, Delghingaro-Augusto V, Joly E, Madiraju SR, Prentki M (2010) Short-chain 3-hydroxyacyl-CoA dehydrogenase is a negative regulator of insulin secretion in response to fuel and non-fuel stimuli in INS832/13 beta-cells. J Diabetes 2(3):157–167. https://doi.org/10.1111/j.1753-0407.2010.00076.x
Iglesias J, Barg S, Vallois D et al (2012) PPARβ/δ affects pancreatic β cell mass and insulin secretion in mice. J Clin Invest 122(11):4105–4117. https://doi.org/10.1172/JCI42127
Gremlich S, Nolan C, Roduit R et al (2005) Pancreatic islet adaptation to fasting is dependent on peroxisome proliferator-activated receptor alpha transcriptional up-regulation of fatty acid oxidation. Endocrinology 146(1):375–382. https://doi.org/10.1210/en.2004-0667
Ramakrishnan SK, Khuder SS, Al-Share QY et al (2016) PPARα (peroxisome proliferator-activated receptor α) activation reduces hepatic CEACAM1 protein expression to regulate fatty acid oxidation during fasting-refeeding transition. J Biol Chem 291(15):8121–8129. https://doi.org/10.1074/jbc.M116.714014
Kim-Muller JY, Zhao S, Srivastava S et al (2014) Metabolic inflexibility impairs insulin secretion and results in MODY-like diabetes in triple FoxO-deficient mice. Cell Metab 20(4):593–602. https://doi.org/10.1016/j.cmet.2014.08.012
Kim-Muller JY, Kim YJ, Fan J et al (2016) FoxO1 deacetylation decreases fatty acid oxidation in β-cells and sustains insulin secretion in diabetes. J Biol Chem 291(19):10162–10172. https://doi.org/10.1074/jbc.M115.705608
Cantley J, Davenport A, Vetterli L et al (2018) Disruption of beta cell acetyl-CoA carboxylase-1 in mice impairs insulin secretion and beta cell mass. Diabetologia 62(1):99–111. https://doi.org/10.1007/s00125-018-4743-7
Rourke JL, Hu Q, Screaton RA (2018) AMPK and friends: central regulators of β cell biology. Trends Endocrinol Metab 29(2):111–122. https://doi.org/10.1016/j.tem.2017.11.007
Fullerton MD, Galic S, Marcinko K et al (2013) Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med 19(12):1649–1654. https://doi.org/10.1038/nm.3372
Kone M, Pullen TJ, Sun G et al (2014) LKB1 and AMPK differentially regulate pancreatic beta-cell identity. FASEB J 28(11):4972–4985. https://doi.org/10.1096/fj.14-257667
Sun G, Tarasov AI, McGinty J et al (2010) Ablation of AMP-activated protein kinase alpha1 and alpha2 from mouse pancreatic beta cells and RIP2.Cre neurons suppresses insulin release in vivo. Diabetologia 53(5):924–936. https://doi.org/10.1007/s00125-010-1692-1
Sun G, Tarasov AI, McGinty JA et al (2010) LKB1 deletion with the RIP2.Cre transgene modifies pancreatic beta-cell morphology and enhances insulin secretion in vivo. Am J Physiol Endocrinol Metab 298(6):E1261–E1273. https://doi.org/10.1152/ajpendo.00100.2010
Sato Y, Henquin JC (1998) The K+-ATP channel-independent pathway of regulation of insulin secretion by glucose: in search of the underlying mechanism. Diabetes 47(11):1713–1721. https://doi.org/10.2337/diabetes.47.11.1713
Ronnebaum SM, Joseph JW, Ilkayeva O et al (2008) Chronic suppression of acetyl-CoA carboxylase 1 in beta-cells impairs insulin secretion via inhibition of glucose rather than lipid metabolism. J Biol Chem 283(21):14248–14256. https://doi.org/10.1074/jbc.M800119200
Joseph JW, Odegaard ML, Ronnebaum SM et al (2007) Normal flux through ATP-citrate lyase or fatty acid synthase is not required for glucose-stimulated insulin secretion. J Biol Chem 282(43):31592–31600. https://doi.org/10.1074/jbc.M706080200
Antinozzi PA, Segall L, Prentki M, McGarry JD, Newgard CB (1998) Molecular or pharmacologic perturbation of the link between glucose and lipid metabolism is without effect on glucose-stimulated insulin secretion. A re-evaluation of the long-chain acyl-CoA hypothesis. J Biol Chem 273(26):16146–16154. https://doi.org/10.1074/jbc.273.26.16146
Mulder H, Lu D, Finley J et al (2001) Overexpression of a modified human malonyl-CoA decarboxylase blocks the glucose-induced increase in malonyl-CoA level but has no impact on insulin secretion in INS-1-derived (832/13) beta-cells. J Biol Chem 276(9):6479–6484. https://doi.org/10.1074/jbc.M010364200
Straub SG, Sharp GW (2002) Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes Metab Res Rev 18(6):451–463. https://doi.org/10.1002/dmrr.329
Wolfgang MJ, Cha SH, Millington DS et al (2008) Brain-specific carnitine palmitoyl-transferase-1c: role in CNS fatty acid metabolism, food intake, and body weight. J Neurochem 105(4):1550–1559. https://doi.org/10.1111/j.1471-4159.2008.05255.x
Gao S, Moran TH, Lopaschuk GD, Butler AA (2013) Hypothalamic malonyl-CoA and the control of food intake. Physiol Behav 122:17–24. https://doi.org/10.1016/j.physbeh.2013.07.014
Hirschey MD, Zhao Y (2015) Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol Cell Proteomics 14(9):2308–2315. https://doi.org/10.1074/mcp.R114.046664
Acknowledgements
We thank C. Nolan (Department of Endocrinology, the Canberra Hospital, Australian National University Medical, Australia), O. Shirihai (David Geffen School of Medicine at University of California Los Angeles) and C. Guay (University of Lausanne, Switzerland) for helpful discussions and critical review of the manuscript.
Funding
Work in the authors’ laboratories is supported by funds from Canadian Institutes of Health Research (to MP and SRMM). MP holds the Canada Research Chair in Diabetes and Metabolism.
Author information
Authors and Affiliations
Contributions
All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Corresponding author
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Slideset of figures
(PPTX 444 kb)
Rights and permissions
About this article

Cite this article
Prentki, M., Corkey, B.E. & Madiraju, S.R.M. Lipid-associated metabolic signalling networks in pancreatic beta cell function. Diabetologia 63, 10–20 (2020). https://doi.org/10.1007/s00125-019-04976-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-019-04976-w