
Abstract
Aims/hypothesis
Targeting regulators of adipose tissue lipoprotein lipase could enhance adipose lipid clearance, prevent ectopic lipid accumulation and consequently ameliorate insulin resistance and type 2 diabetes. Angiopoietin-like 8 (ANGPTL8) is an insulin-regulated lipoprotein lipase inhibitor strongly expressed in murine adipose tissue. However, Angptl8 knockout mice do not have improved insulin resistance. We hypothesised that pharmacological inhibition, using a second-generation antisense oligonucleotide (ASO) against Angptl8 in adult high-fat-fed rodents, would prevent ectopic lipid accumulation and insulin resistance by promoting adipose lipid uptake.
Methods
ANGPTL8 expression was assessed by quantitative PCR in omental adipose tissue of bariatric surgery patients. High-fat-fed Sprague Dawley rats and C57BL/6 mice were treated with ASO against Angptl8 and insulin sensitivity was assessed by hyperinsulinaemic–euglycaemic clamps in rats and glucose tolerance tests in mice. Factors mediating lipid-induced hepatic insulin resistance were assessed, including lipid content, protein kinase Cε (PKCε) activation and insulin-stimulated Akt phosphorylation. Rat adipose lipid uptake was assessed by mixed meal tolerance tests. Murine energy balance was assessed by indirect calorimetry.
Results
Omental fat ANGPTL8 mRNA expression is higher in obese individuals with fatty liver and insulin resistance compared with BMI-matched insulin-sensitive individuals. Angptl8 ASO prevented hepatic steatosis, PKCε activation and hepatic insulin resistance in high-fat-fed rats. Postprandial triacylglycerol uptake in white adipose tissue was increased in Angptl8 ASO-treated rats. Angptl8 ASO protected high-fat-fed mice from glucose intolerance. Although there was no change in net energy balance, Angptl8 ASO increased fat mass in high-fat-fed mice.
Conclusions/interpretation
Disinhibition of adipose tissue lipoprotein lipase is a novel therapeutic modality to enhance adipose lipid uptake and treat non-alcoholic fatty liver disease and insulin resistance. In line with this, adipose ANGPTL8 is a candidate therapeutic target for these conditions.
Similar content being viewed by others



Introduction
Ectopic accumulation of specific lipid species impairs liver and muscle insulin signalling, causes insulin resistance and accounts for the link between obesity and type 2 diabetes. However, not all obese individuals are insulin resistant. This phenotypic heterogeneity may lie in differences in adipose biology [1]. Notably, some genes linked to forms of lipodystrophy are also dysregulated in individuals with more common forms of insulin resistance [2], lending genetic support for a key role for adipose dysfunction in individuals who develop insulin resistance [3]. Thiazolidinediones (TZDs) are the only medications currently available that improve adipose function. These peroxisome proliferator activated receptor γ agonists increase adipogenesis and adipose lipid storage and decrease adipose lipolysis, effectively redistributing lipid from ectopic storage sites (liver, muscle etc.) to eutopic sites (subcutaneous adipose tissue) [4,5,6]. This dramatically improves insulin resistance and glycaemic control with the added benefit of reducing the risk for cerebrovascular and acute coronary events [7, 8]. TZDs are also beneficial in individuals with type 2 diabetes and non-alcoholic fatty liver disease (NAFLD) or non-alcoholic steatohepatitis [9,10,11]. Unfortunately, numerous side effects (e.g. weight gain, volume retention and osteoporotic fractures) limit the clinical utility of TZDs [12]. Thus, newer agents are needed.
The transfer of lipids from circulating lipoproteins to tissues is largely catalysed by lipoprotein lipase (LPL) and is tightly regulated. The combined effect of a cohort of endogenous activators and inhibitors determines the quantity, timing and location of tissue lipid uptake during feeding and fasting [13, 14]. Changes in endogenous regulators of LPL can influence the propensity for insulin resistance. For example, alterations in the LPL inhibitor APOC3 have been seen in individuals with an increased susceptibility to insulin resistance [15, 16]. Thus, pharmacological modulation of endogenous LPL regulatory factors to promote the storage of lipid in eutopic sites may be a novel paradigm to prevent or treat insulin resistance and NAFLD.
The angiopoietin-like (ANGPTL) proteins ANGPTL3, ANGPTL4 and ANGPTL8 are a family of differentially-regulated LPL inhibitors [14]. ANGPTL8 was once thought to have a trophic effect on pancreatic beta cells, although this is no longer believed to be the case [17,18,19]. Circulating ANGPTL8 is lower in humans with dyslipidaemia (with the caveat that circulating ANGPTL8 in humans has never been measured with a validated assay), particularly those with low HDL-cholesterol or high triacylglycerols [20]. However, ANGPTL8’s primary functions may be better ascribed to local, tissue-specific actions. ANGPTL8 is an LPL inhibitor that is expressed strongly in the white adipose tissue (WAT) [17, 21]; it complexes with circulating ANGPTL3 to potently inhibit LPL [22]. Notably, ANGPTL8 expression is increased by insulin stimulation [23], so in the immediate postprandial period, a high level of circulating insulin increases ANGPTL8 expression and decreases adipose tissue LPL action and lipid uptake, potentially balancing insulin’s primary actions at adipose tissue, suppression of lipolysis and promotion of lipid storage.
Surprisingly, Angptl8 knockout mice do not manifest a dramatic metabolic phenotype [24], exhibiting no change in glucose tolerance or insulin tolerance and no meaningful effect on ectopic tissue triacylglycerol content. Nonetheless, pharmacological knockdown of ANGPTL8 expression may provide new insights into the relationship between endogenous regulation of LPL activity and the deposition of ectopic lipid and lipotoxic insulin resistance. In this study, we used a second-generation 2′-O-methoxyethyl antisense oligonucleotide (ASO) against ANGPTL8 in rats and mice, evaluating the potential for adipose-specific disinhibition of LPL to prevent ectopic lipid deposition and lipid-induced insulin resistance.
Methods
Animals
Male Sprague Dawley (SD) rats (250–275 g) and C57BL/6 mice (8–11 weeks old) were obtained from Charles River Laboratories (Waltham, MA, USA) and Jackson Laboratory (Bar Harbor, ME, USA), respectively. Animals were housed under controlled temperature (23°C) and lighting (12 h light/dark cycle, lights on at 07:00) with free access to water and food. For the hyperinsulinaemic–euglycaemic clamp study, jugular venous and carotid artery catheters were placed; for the mixed-meal tolerance tests (MMTTs), jugular venous and gastric catheters were placed. Rodents were maintained on standard regular chow (RC) (Envigo 2108S, Envigo, Madison WI, USA); or rats were placed on a high-fat diet (HFD; D12451, Research Diets, New Brunswick, NJ, USA); or mice were placed on an HFD (Research Diets D12492). Rodents were treated with 2′-O-methoxyethyl chimeric ASO against Angptl8 or with a control ASO that does not target any known rat, mouse or human gene; ASO was delivered intraperitoneally (25 mg kg−1 week−1). Basal tissues were taken after a 6 h fast and infusions were performed after a 12–16 h fast.
All procedures were approved by the Institutional Animal Care and Use Committee of the Yale University School of Medicine. Surgeries were performed under isoflurane anaesthesia and carprofen analgesia was provided in the postoperative period. Animals were killed either with intravenous pentobarbital or under isoflurane anaesthesia. Care was taken throughout the study to minimise suffering.
Tissue lipid isolation and measurements
All lipids were extracted from tissues in 2:1 chloroform:methanol. For hepatic diacylglycerol measurements, lipids were first extracted from cytosolic/lipid droplet and membrane-associated fractions by a modification of a previously published protocol [25]. Homogenates were separated by ultracentrifugation. The pellet contains plasma membrane lipids while the supernatant contains cytosolic lipids, including lipid droplets. The chloroform:methanol extraction was then performed from each fraction. Lipids from the organic layer were resuspended in hexane:methylene chloride:ether (89:10:1). Diacylglycerols were separated from triacylglycerols using a diol-bonded solid-phase extraction column (Waters, Milford, MA, USA) [26]. Triacylglycerols were measured using the Triglyceride-SL kit (Sekisui, Charlottetown, PEI, Canada). Diacylglycerols and ceramides were measured by LC-MS/MS [26].
Plasma biochemical analysis
Glucose concentrations were determined using a YSI 2700 select (Yellow Springs Instruments, Yellow Springs, OH, USA) or a standard kit (Sekisui). Kits were also used to measure NEFAs (Wako, Mountain View, CA, USA) and triacylglycerols (Sekisui). Insulin was measured by radioimmunoassay (EMD Millipore, Burlington, MA, USA). Leptin was measured by ELISA (Abcam, Cambridge, UK).
Hyperinsulinaemic–euglycaemic clamp studies
Clamps were performed as previously described [27, 28] with primed-continuous infusion of [6,6-2H2]glucose tracer with a 2 h basal period and a 2 h period in which the rats received insulin (4 mU kg−1 min−1). During the clamp period, the 20% dextrose solution used to maintain euglycaemia was enriched with tracer to approximately 2.5%.
Lipoprotein lipase activity assay
Plasma LPL activity was assessed by a fluorometric assay (Cell Biolabs, San Diego, CA, USA). Adipose tissue LPL activity was determined by incubation of 80–120 mg adipose tissue at 37°C for 1 h in PBS (Sigma, St. Louis, MO, USA) with 5 U/ml heparin (Sagent, Schaumburg, IL, USA) and 2 mg/ml bovine serum albumin (Sigma); samples were centrifuged (900 g, 4°C, 15 min) and supernatant LPL activity assayed in the presence of heat-inactivated rat serum with a fluorometric kit (Cell Biolabs).
MMTTs
A mixed meal (1.16 ml/kg vegetable oil labelled with 1.8 MBq/ml 3H triolein [Perkin Elmer, Waltham, MA, USA] or 0.21 MBq/ml 3H retinyl palmitate [American Radiolabeled Chemicals, St. Louis, MO, USA] and 2.84 ml/kg Ensure Plus [Abbott Nutrition, Columbus, OH, USA]) was delivered through gastric catheters, while blood was collected from venous catheters.
Tissue solubilisation for scintillation counting
Tissues were solubilised (in 400 mM sodium hydroxide, 1% NP-40, 1% Triton X-100). Liver samples were decolourised in 1.5% hydrogen peroxide at 50°C. Samples were mixed with scintillation cocktail (Ultima Gold, Perkin Elmer) and beta counting was performed.
IPGTT
IPGTTs were conducted as previously described [29]. Mice were injected intraperitoneally with glucose (1 mg/kg body weight). Blood samples were taken by tail bleed.
Body composition and metabolic cage studies
Murine body composition was measured by 1H magnetic resonance spectroscopy (Bruker, Billerica, MA, USA). Energy expenditure and caloric intake were measured in a Comprehensive Laboratory Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH, USA).
Western blotting
Proteins from tissue lysate were resolved by SDS-PAGE using a 4–12% gradient gel and electroblotted onto polyvinylidene difluoride membranes (EMD Millipore). Secondary antibodies were horseradish peroxidase-conjugated (Cell Signaling Technology, Danvers, MA, USA) and detection was performed with enhanced chemiluminescence. For protein kinase Cε (PKCε) translocation, cytoplasm and plasma membrane fractions were separated by ultracentrifugation as previously described [30, 31]. Proteins assessed are listed in Table 1 and electronic supplementary material (ESM) Table 1.
Quantitative PCR
RNA was extracted with a standard kit (RNeasy, Qiagen, Germantown, MD, USA). cDNA was generated with the QuantiTect Reverse Transcription Kit (Qiagen). Abundance of transcripts was assessed by real-time PCR with a SYBR Green detection system (Bio-Rad, Hercules, CA, USA). Expression for the genes of interest and an invariant control were determined using amplification efficiencies [32]. Genes assessed are listed in Table 1 and ESM Table 2.
PKCε activity assay
The PKCε activity assay is modified from previously described assays [33, 34] with reagents from a commercially available PKC assay kit (EMD Millipore). Liver tissue was homogenised and protein was extracted; 500 μg of hepatic protein was diluted in immunoprecipitation (IP) buffer and precleared with protein A/G agarose beads. IP was performed with anti-PKCε antibody (Cell Signaling). IP beads were washed and resuspended in PKC reaction buffer. The PKC activity kit was used to assess PKC activity, with 0.37 MBq [γ-32P] ATP per reaction for detection.
Human adipose and liver tissue
Visceral WAT and liver biopsies were obtained intra-operatively from individuals with extreme obesity undergoing bariatric surgery at the Geisinger Clinic (Danville, PA, USA). Liver tissue was preserved in formalin for histological analysis [35]. Adipose tissue was preserved in RNALater (Qiagen) and stored at −80°C [36]. Individuals were enrolled in a standardised clinical programme at the Center for Nutrition and Weight Management during which phenotypic data were collected as previously described [37]. All research participants provided written informed consent using protocols adherent to the Code of Ethics of the World Medical Association (Declaration of Helsinki). The Institutional Review Boards of Geisinger Health System and the Temple University School of Medicine approved the research.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA). Groups were compared by the Student’s unpaired t test or a one-way ANOVA analysis followed by Tukey’s Multiple Comparison test. One-way ANCOVA analysis was performed using a web-based calculator provided by Vassar College (vassarstats.net; accessed 14 Dec 2017). All data are expressed as mean ± SEM. p values less than 0.05 were considered significant.
Results
Adipose ANGPTL8 expression is increased in insulin-resistant adults
Visceral adipose tissue biopsies were obtained from a cohort of BMI-matched obese individuals categorised as ‘insulin sensitive’ (HOMA-IR < 3) or ‘insulin resistant’ (HOMA-IR > 7) (Table 2). Diabetic individuals taking a TZD were excluded. As expected, individuals with insulin resistance had lower plasma HDL-cholesterol and higher plasma triacylglycerol concentrations. Insulin-resistant individuals also had more advanced NAFLD, as reflected by higher NAFLD activity scores on liver biopsy (Table 2). ANGPTL8 gene expression normalised to TATA-binding protein gene expression was 7.6-fold higher (p < 0.05) in insulin-resistant individuals as compared with the insulin-sensitive individuals (Table 2).
Increased adipose ANGPTL8 expression could reduce postprandial adipose LPL activity and predispose to NAFLD. We performed a series of rodent experiments to determine if inhibiting ANGPTL8 with a specific ASO prevented ectopic lipid accumulation and insulin resistance.
Angptl8 ASO treatment reduces fasting plasma triacylglycerols and insulin in fat-fed rats
Three weeks of Angptl8 ASO treatment in HFD-fed male SD rats decreased fasting Angptl8 expression by 73% in the epididymal fat pad and 82% in the liver. The expression of other hepatic negative regulators of LPL such as Angptl3, Apoc3, and Apoe was increased (ESM Fig. 1a,b). ASO treatment did not cause transaminitis or changes in plasma blood urea nitrogen (BUN). Body weight was not different, though it did tend to be lower (ESM Fig. 1c, Table 3). As expected, Angptl8 ASO decreased fasting plasma triacylglycerol (59% reduction; Table 3). Plasma glucose after a 6 h fast was unchanged, while plasma insulin was reduced by 54% (Table 3), consistent with improved insulin sensitivity. There was no difference in leptin and adiponectin levels after an overnight fast (ESM Table 3). Macrophage-associated gene Cd68 expression was modestly increased in overnight fasted adipose tissue (ESM Table 3).
Angptl8 ASO treatment prevents lipid-induced hepatic insulin resistance
HFD-fed male SD rats were treated with ASO for 3 weeks; whole-body and tissue-specific insulin action was assessed by the hyperinsulinaemic–euglycaemic clamp technique in awake, unrestrained animals. Angptl8 ASO-treated rats had a slightly lower body weight prior to clamp (control ASO: 424 g ± 13; Angptl8 ASO: 382 g ± 7; p < 0.01), potentially due to slower recovery from surgery. The glucose infusion rate required to maintain euglycaemia was similar between the two groups (Fig. 1a, b) and insulin-stimulated whole-body glucose disposal was also unchanged (Fig. 1c). Basal rates of endogenous glucose production, reflecting hepatic glucose production, were unchanged between control and Angptl8 ASO-treated groups (Fig. 1d). Angptl8 ASO had no impact on insulin-stimulated whole-body glucose disposal (Fig. 1c) but improved the suppression of endogenous glucose production (Fig. 1e). Surprisingly, plasma insulin concentration was approximately 17% lower during the clamped portion of the experiment (p < 0.05). Insulin-mediated suppression of circulating NEFA concentration was not changed by Angptl8 ASO treatment (Fig. 1f). Thus, Angptl8 ASO specifically improved hepatic insulin sensitivity.

Hyperinsulinaemic–euglycaemic clamps. Control ASO- and Angptl8 ASO-treated rats underwent hyperinsulinaemic–euglycaemic clamps. (a) Plasma glucose and (b) Glucose infusion rate (GINF) time course throughout the clamp procedure (white squares: control ASO; black squares: Angptl8 ASO). (c) Whole-body insulin-stimulated glucose disposal (Rd). (d) Endogenous glucose production (EGP) measured in both the basal and clamped condition. (e) Insulin-mediated suppression of endogenous glucose production. (f) Plasma NEFA concentrations in basal and clamped conditions. In all figure parts, data are the mean ± SEM of n=10 (control ASO) or n=13 (Angptl8 ASO). **p<0.01 vs control ASO; ††p<0.01 Angptl8 ASO clamp vs control clamp
Angptl8 ASO attenuates HFD-induced hepatic steatosis
Angptl8 ASO reduced hepatic triacylglycerol content by 66% in comparison with control ASO-treated rats (Fig. 2a). Total diacylglycerol content was reduced in Angptl8 ASO-treated rats (Fig. 2b). Diacylglycerol (DAG) was reduced both in a plasma membrane fraction and in a lipid-droplet-containing cytoplasmic fraction (ESM Fig. 2). The primary DAG species in both control and Angptl8 ASO-treated groups contained C18:1, C18:2 and C16:0 fatty acyl moieties, largely reflecting the lipid content of the lard-based diet. Total hepatic ceramide content was unchanged (Fig. 2c). Two ceramide species implicated in resistance, C16 ceramides [38, 39] and C18 ceramides [40], were actually increased. C24:1 ceramides were elevated, while C20 ceramides were decreased (ESM Fig. 3).

Hepatic lipid content and hepatic insulin signalling. (a) Hepatic triacylglycerol (TAG) content after a 6 h fast. (b) Hepatic DAG content after a 6 h fast. (c) Hepatic ceramide content after a 6 h fast. (d) Hepatic cell lysate PKCε activity and (e) PKCε translocation after a 6 h fast. (f) Hepatic Akt phosphorylation, comparing basal (B) 6 h fasted livers with clamped (C) insulin-stimulated livers. Membrane:cytoplasm ratio quantified with respect to housekeeping proteins sodium–potassium ATPase (Na+/K+-ATPase) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Graph in (e) represents membrane:cytoplasm PKCε abundance ratio. (f) Hepatic Akt phosphorylation, comparing basal 6 h fasted livers with clamped insulin-stimulated livers. Graph represents the ratio of Akt phosphorylation in the insulin-stimulated state to Akt phosphorylation in the basal state. For hepatic lipids and PKCε activity, data are the mean ± SEM of n=8; for PKCε translocation, data are the mean ± SEM of n=6 (control ASO) and mean ± SEM of n=8 (Angptl8 ASO); for hepatic Akt phosphorylation, data are the mean ± SEM of n=6. *p< 0.05; **p< 0.01; ***p<0.001 vs control
DAGs impair hepatic insulin signalling by activating PKCε, which phosphorylates the insulin receptor at an inhibitory threonine, thereby reducing insulin receptor signalling through Akt [41, 42]. Hepatic PKCε enzymatic activity was decreased by 43% in Angptl8 ASO-treated rats (Fig. 2d). Similarly, PKCε translocation to the plasma membrane, a readout of novel PKC activation, was decreased by 51% in Angptl8 ASO-treated rats (Fig. 2e). Improved hepatocellular insulin action was reflected in improved insulin stimulation of hepatic Akt phosphorylation (Fig. 2f).
Angptl8 ASO treatment increases adipose tissue chylomicron triacylglycerol uptake
We hypothesised that Angptl8 ASO prevents ectopic lipid accumulation by enhancing adipose lipid uptake. Angptl8 ASO did not affect plasma or epididymal adipose LPL activity (Fig. 3a,b). However, subcutaneous adipose LPL activity was increased by approximately threefold (Fig. 3c). We quantified adipose tissue mealtime triacylglycerol clearance with labelled MMTTs. Mixed meals were given in combination either with 3H-retinyl palmitate or, in a separate cohort, 3H-triolein. Retinyl palmitate is primarily taken up by the liver and is used to quantify hepatic chylomicron remnant clearance. Triolein is used to quantify fatty acid uptake into all lipid-storing tissues. To avoid possible effects of altered body weight on triacylglycerol clearance, these studies were performed after one week of treatment with ASO when Angptl8 gene expression was decreased by half (ESM Fig. 4a) without body weight divergence; ASO was given during the last week of the 4 week fat-feeding period.

LPL activity and tissue triacylglycerol uptake. LPL activity was determined in plasma and tissues of rats euthanised in the fed state. MMTTs were performed in HFD-fed rats after an overnight fast. Tissue lipid uptake was assessed 4 h after delivery of mixed meal. (a) Plasma LPL activity. (b) Epididymal adipose LPL activity, expressed relative to control ASO treatment. (c) Subcutaneous adipose LPL activity, expressed relative to control ASO treatment. (d) Plasma triacylglycerol during the MMTT (white squares: control ASO; black squares: Angptl8 ASO). (e) Hepatic chylomicron remnant uptake assessed by 3H retinyl palmitate incorporation. (f) Epididymal adipose tissue triacylglycerol uptake (counts per mg tissue) normalised to hepatic triacylglycerol uptake (counts per mg tissue). (g) Subcutaneous adipose tissue triacylglycerol uptake normalised to hepatic triacylglycerol uptake. For LPL activity measurements, data are the mean ± SEM of n=5–6. For chylomicron remnant uptake, data are the mean ± SEM of n=5 (control ASO) or mean ± SEM of n=7 (Angptl8 ASO). For plasma triacylglycerol, data are the mean ± SEM of n=10–14 rats per group; for adipose triacylglycerol uptake, data are the mean ± SEM of n=10 (control ASO) or mean ± SEM of n=12 (Angptl8 ASO). **p<0.01 vs control
Hepatic retinyl palmitate uptake 4 h after delivery of the mixed meal was similar between control ASO and Angptl8 ASO-treated rats (Fig. 3e), suggesting that chylomicron remnant clearance was unchanged by Angptl8 ASO. MMTTs with triolein label were performed in both insulin-sensitive RC-fed rats and in insulin-resistant 4 week HFD-fed rats (Fig. 3 and ESM Fig. 5). The ratio of adipose triolein uptake per gram of adipose to hepatic triolein uptake per gram of liver was measured. Under both RC- and HFD-fed conditions, plasma triacylglycerol concentrations were reduced during the MMTT in Angptl8 ASO-treated rats (Fig. 3d and ESM Fig. 5a). Subcutaneous adipose triacylglycerol uptake was increased in the RC-fed Angptl8 ASO-treated rats (ESM Fig. 5b,c). In HFD-fed rats, incorporation of labelled triolein was increased by Angptl8 ASO in both subcutaneous and epididymal adipose tissue (Fig. 3f,g). Together, these data demonstrate that reducing Angptl8 expression enhances adipose lipid uptake.
Angptl8 ASO protects fat-fed mice from hepatic steatosis and insulin resistance
We assessed the robustness of the effect of Angptl8 ASO in a second rodent model, the HFD-fed C57BL/6 mouse, a widely studied preclinical model of diet-induced insulin resistance that also enabled us to perform indirect calorimetry to assess energy balance.
Three weeks of Angptl8 ASO treatment in fat-fed mice reduced fasting adipose ANGPTL8 expression by 43% (ESM Fig. 4b) and hepatic triacylglycerol by 37% compared with control ASO-treated mice (Fig. 4a). Furthermore, IPGTTs were performed. Plasma glucose after intraperitoneal glucose injection was unchanged by Angptl8 ASO but the insulin level required to maintain these glucose levels was significantly reduced (Fig. 4b,c).

Murine protection from hepatic steatosis and glucose intolerance. (a) Hepatic triacylglycerol (TAG) content. (b, c) IPGTT (control ASO: white triangles; Angptl8 ASO: black triangles), measurements of plasma glucose (b) and plasma insulin (c) presented vs time, together with plasma insulin AUC. For hepatic triacylglycerol, data are the mean ± SEM of n=5 (control ASO) or mean ± SEM of n=6 (Angptl8 ASO). For IPGTT, data are the mean ± SEM of n=8 (control ASO) or mean ± SEM of n=7 (Angptl8 ASO). *p<0.05; **p<0.01; ***p<0.001 vs control
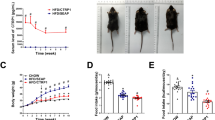
Angptl8 ASO alters body composition but not net energy metabolism in mice
We assessed the effect of ANGPTL8 knockdown on calorie intake and energy expenditure. Metabolic cage experiments were performed with both RC-fed and HFD-fed cohorts of C57BL/6 mice. Angptl8 ASO did not alter the growth curve of mice on either diet (Fig. 5a,b). Angptl8 ASO treatment decreased body-fat percentage in RC-fed mice (Fig. 5c) but increased body-fat percentage in HFD-fed mice (Fig. 5d). Angptl8 ASO had no measurable effect on food intake in RC-fed mice, but reduced food intake in HFD-fed mice (Fig. 5e,f). As assessed by ANCOVA, energy expenditure correlated with muscle mass in both RC-fed mice (R2 = 0.51) and HFD-fed mice (R2 = 0.62); adjusted for muscle mass, Angptl8 ASO had no effect on energy expenditure in RC-fed mice (p = 1.0) while it reduced muscle-mass-adjusted energy expenditure in HFD-fed mice (p < 0.001) (Fig. 5g,h and ESM Fig. 6).

Murine growth curves and energy homeostasis. Whole-body adipose content assessed by 1H magnetic resonance spectroscopy; energy homeostasis assessed by metabolic cage analysis: Growth curve in (a) RC-fed mice and (b) HFD-fed mice. Fat mass accretion in (c) RC-fed mice and (d) HFD-fed mice (control ASO: white triangles; Angptl8 ASO: black triangles). Food intake assessed in (e) RC-fed mice and (f) HFD-fed mice. Energy expenditure assessed in (g) RC-fed mice and (h) HFD-fed mice. RQ assessed in (i) RC-fed mice and (j) HFD-fed mice. Metabolic cage variables were normalised to lean body mass. For RC-fed growth curves and fat mass assessments, energy expenditure and RQ, data are the mean ± SEM of n=8 in both groups. For HFD-fed growth curves and fat mass assessments, data are the mean ± SEM of n=7 (control ASO) or mean ± SEM of n=8 (Angptl8 ASO). For RC-fed food intake, HFD-fed energy expenditure and RQ, data are the mean ± SEM of n=7 in both groups. For HFD-fed food intake, data are the mean ± SEM of n=5 (control ASO) or mean ± SEM of n = 7 (Angptl8 ASO). *p<0.05; **p<0.01; ***p<0.001 vs control
Discussion
Adipose dysfunction leads to ectopic lipid deposition and, in turn, insulin resistance, type 2 diabetes, NAFLD and cardiovascular disease. Improving adipocyte function with TZDs is a well-established treatment for all these conditions [4, 7,8,9, 11]. However, weight gain, fluid retention and increased fracture risk limit their clinical utility. Adipose lipid storage could be improved by enhancing adipose LPL activity, potentially through targeting ANGPTL8. This paradigm would leverage a specific biological effect to achieve the same clinical benefits as TZDs, while minimising off-target adverse effects. However, Angptl8 knockout mice did not demonstrate significant improvements in ectopic lipid or glucose metabolism [24]. In contrast, the data in this study suggest that dysregulation of ANGPTL8 is associated with NAFLD and insulin resistance, and pharmacological inhibition using a second-generation ASO against Angptl8 improves adipose function, increasing postprandial triacylglycerol uptake and preventing ectopic lipid accumulation and lipid-induced insulin resistance.
We observed an association between adipose ANGPTL8 expression and metabolic disease. Prior studies of ANGPTL8 in human blood reported increases in circulating ANGPTL8 in individuals with the metabolic syndrome [43] and type 2 diabetes [44, 45]; furthermore, serum ANGPTL8 levels correlate with BMI [45] and hepatic steatosis [46]. However, circulating ANGPTL8 may not accurately reflect adipose tissue ANGPTL8 content and has not been measured using validated assays. We observed that in BMI-matched adult human participants with extreme obesity, increased visceral adipose ANGPTL8 expression is associated with insulin resistance and NAFLD, independent of the documented association with BMI. Increased adipose ANGPTL8 may inhibit adipose LPL activity, decreasing eutopic adipose lipid storage and promoting ectopic lipid accumulation. Adipose ANGPTL8 expression may be induced by the resulting hyperinsulinaemia, contributing to a pathogenic cycle that further sustains ectopic lipid accumulation and insulin resistance.
Our data support the hypothesis that inhibition of ANGPTL8 expression reduces ectopic lipid accumulation by improving adipose lipid uptake. First, Angptl8 ASO increased subcutaneous adipose LPL activity and improved post-prandial adipose triacylglycerol uptake in both RC-fed and HFD-fed rats. Angptl8 ASO also decreased hepatic Angptl8 expression. Since the liver is a major site for ANGPTL8 expression, this could have impacted circulating ANGPTL8 and possibly plasma LPL activity [47]. However, we did not detect any change in plasma LPL activity, suggesting the physiological effect of the ASO is specifically due to its impact on adipose Angptl8. The lack of effect on global circulating LPL activity may in part be explained by the dramatic upregulation of other hepatically produced LPL inhibitory factors seen in the Angptl8 ASO-treated animals.
Second, in HFD-fed rats, improved adipose lipid clearance reduced hepatic steatosis and improved hepatic insulin action. The reduction in hepatic diacylglycerol content decreased PKCε activation and improved insulin signalling. Angptl8 ASO-treated C57BL/6 mice were also protected from HFD-induced hepatic steatosis and had improved glucose tolerance. Notably, liver ceramide content did not correlate with changes in tissue insulin action. Taken together, these findings suggest that diversion of lipid from the liver to the adipose tissue protects rodents from hepatic DAG–PKCε mediated insulin resistance.
Some subtleties of the methodology warrant discussion. Tissue-specific triacylglycerol clearance is usually assessed by oral challenge with a labelled lipid mixture (e.g. vegetable oil with radiolabelled triolein) or intravenous challenge with radiolabelled lipoproteins. Here, we labelled the triacylglycerol component of an MMTT. This allowed us to assess the disposition of mealtime triacylglycerol from physiologically produced chylomicrons in the context of dietary carbohydrate and prandial insulin secretion. This is relevant to studies of the function of ANGPTL8, as Angptl8 gene expression is stimulated by insulin [23]. This test may help to explain the difference between this study and the previous study of knockout mice, in which no change in adipose tissue lipid uptake was observed. Of course, there is also a difference in biology between the models, reflecting the difference between knockdown and complete knockout of ANGPTL8 (which may impact development and/or lead to compensatory changes).
The protective effects of Angptl8 ASO are independent of changes in net energy balance or weight gain. Angptl8 ASO did not change energy expenditure or food intake under RC-fed conditions. In HFD-fed mice, there were subtle but matched decreases in both energy expenditure and food intake. These results contrast with a recent report that a monoclonal anti-ANGPTL8 antibody increased energy expenditure and weight loss [48], a difference which may be due to the different treatment modalities. The effects of Angptl8 ASO on body composition are intriguing. Angptl8 ASO treatment decreased fat mass in RC-fed mice but increased fat mass accretion in HFD-fed mice. This divergence reflects the different pathways by which adipose fat is delivered with high-carbohydrate diets and HFDs. In the HFD-fed rodents, dietary fat is the main source of adipose lipid, and Angptl8 ASO increased lipid uptake from chylomicrons into adipose tissue in HFD-fed rats. Subcutaneous adipose lipid uptake was also increased in RC-fed rats, but RC-fed mice had a decrease in overall adiposity. In RC-fed rodents, most tissue lipid is produced via hepatic de novo lipogenesis, which would be delivered to the adipose tissue in triacylglycerol-rich VLDL particles. VLDL secretion is highest in the post-absorptive state, when Angptl8 expression is normally low, hence when Angptl8 ASO is unlikely to increase lipid uptake. This is consistent with the prior report demonstrating decreased WAT triacylglycerol uptake of intravenously infused VLDL in Angptl8 knockout mice [24].
These studies have some limitations. In the clamp study, the Angptl8 ASO-treated rats were 10% smaller than control ASO-treated rats. The reduced body weight may favour improved insulin sensitivity. However, there were still improvements in glucose tolerance in the weight-matched mice and improvements in insulin level in 6 h fasted rats, suggesting that the improvements in insulin action are independent of changes in body weight. Moreover, the plasma insulin concentrations achieved during the hyperinsulinaemic–euglycaemic clamps in Angptl8 ASO-treated rats were lower than the concentrations in the control ASO-treated rats, possibly due to increased insulin clearance. This unintended difference would reduce the observed effect of the Angptl8 ASO on insulin resistance; under matched insulin concentrations, an even greater suppression of hepatic glucose production in the Angptl8 ASO-treated animals is expected.
In contrast to prior studies with adipose-specific Angptl8 knockout mice [24], the present set of studies suggest that targeting adipose ANGPTL8 can prevent lipid-induced insulin resistance. Reduction in Angptl8 improved adipose lipid uptake and reduced the accumulation of ectopic lipid, addressing a root cause of insulin resistance. These results are intriguing and support further study of this system, its role in modulating LPL in fed and fasted conditions, and in modulating fat mass and possibly energy metabolism. Moreover, additional studies are needed to determine whether the increases in adipose ANGPTL8 in humans are a cause or consequence of insulin resistance. More broadly, campaigns focused on compounds that specifically activate or disinhibit adipose LPL may lead to exciting new therapeutic opportunities.
Data availability
Data are available on request from the authors.
Abbreviations
- ANGPTL3/4/8:
-
Angiopoietin-like 3/4/8
- ASO:
-
Antisense oligonucleotide
- BUN:
-
Blood urea nitrogen
- DAG:
-
Diacylglycerol
- GINF:
-
Glucose infusion rate
- HFD:
-
High-fat diet
- IP:
-
Immunoprecipitation
- LPL:
-
Lipoprotein lipase
- MMTT:
-
Mixed-meal tolerance test
- NAFLD:
-
Non-alcoholic fatty liver disease
- PKCε:
-
Protein kinase Cε
- qPCR:
-
Quantitative PCR
- RC:
-
Regular chow
- SD:
-
Sprague Dawley
- TZD:
-
Thiazolidinedione
- WAT:
-
White adipose tissue
References
Cuthbertson DJ, Steele T, Wilding JP et al (2017) What have human experimental overfeeding studies taught us about adipose tissue expansion and susceptibility to obesity and metabolic complications? Int J Obes 41:853–865
Lotta LA, Gulati P, Day FR et al (2017) Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet 49:17–26
Shulman GI (2000) Cellular mechanisms of insulin resistance. J Clin Invest 106:171–176
Soccio RE, Chen ER, Lazar MA (2014) Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 20:573–591
Mayerson AB, Hundal RS, Dufour S et al (2002) The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 51:797–802
Kim JK, Fillmore JJ, Gavrilova O et al (2003) Differential effects of rosiglitazone on skeletal muscle and liver insulin resistance in A-ZIP/F-1 fatless mice. Diabetes 52:1311–1318
Young LH, Viscoli CM, Curtis JP et al (2017) Cardiac outcomes after ischemic stroke or TIA: effects of pioglitazone in patients with insulin resistance without diabetes. Circulation 20:1882–1893
Kernan WN, Viscoli CM, Furie KL et al (2016) Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med 374:1321–1331
Belfort R, Harrison SA, Brown K et al (2006) A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 355:2297–2307
Gastaldelli A, Harrison SA, Belfort-Aguilar R et al (2009) Importance of changes in adipose tissue insulin resistance to histological response during thiazolidinedione treatment of patients with nonalcoholic steatohepatitis. Hepatology 50:1087–1093
Cusi K, Orsak B, Bril F et al (2016) Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med 165: 305–315
Rizos CV, Kei A, Elisaf MS (2016) The current role of thiazolidinediones in diabetes management. Arch Toxicol 90:1861–1881
Kersten S (2014) Physiological regulation of lipoprotein lipase. Biochim Biophys Acta 1841:919–933
Zhang R (2016) The ANGPTL3-4-8 model, a molecular mechanism for triglyceride trafficking. Open Biol 6:150272
Petersen KF, Dufour S, Hariri A et al (2010) Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N Engl J Med 362:1082–1089
Pollex RL, Ban MR, Young TK et al (2007) Association between the -455T>C promoter polymorphism of the APOC3 gene and the metabolic syndrome in a multi-ethnic sample. BMC Med Genet 8:80
Quagliarini F, Wang Y, Kozlitina J et al (2012) Atypical angiopoietin-like protein that regulates ANGPTL3. Proc Natl Acad Sci U S A 109:19751–19756
Yi P, Park JS, Melton DA (2013) Betatrophin: a hormone that controls pancreatic beta cell proliferation. Cell 153:747–758
Cox AR, Barrandon O, Cai EP et al (2016) Resolving discrepant findings on ANGPTL8 in beta-cell proliferation: a collaborative approach to resolving the betatrophin controversy. PLoS One 11:e0159276
Gomez-Ambrosi J, Pascual-Corrales E, Catalan V et al (2016) Altered concentrations in dyslipidemia evidence a role for ANGPTL8/betatrophin in lipid metabolism in humans. J Clin Endocrinol Metab 101:3803–3811
Ren G, Kim JY, Smas CM (2012) Identification of RIFL, a novel adipocyte-enriched insulin target gene with a role in lipid metabolism. Am J Phys Endocrinol Metab 303:E334–E351
Haller JF, Mintah IJ, Shihanian LM et al (2017) ANGPTL8 requires ANGPTL3 to inhibit lipoprotein lipase and plasma triglyceride clearance. J Lipid Res 58:1166–1173
Nidhina Haridas PA, Soronen J, Sadevirta S et al (2015) Regulation of angiopoietin-like proteins (ANGPTLs) 3 and 8 by insulin. J Clin Endocrinol Metab 100:E1299–E1307
Wang Y, Quagliarini F, Gusarova V et al (2013) Mice lacking ANGPTL8 (Betatrophin) manifest disrupted triglyceride metabolism without impaired glucose homeostasis. Proc Natl Acad Sci U S A 110:16109–16114
Bogan JS, McKee AE, Lodish HF (2001) Insulin-responsive compartments containing GLUT4 in 3T3-L1 and CHO cells: regulation by amino acid concentrations. Mol Cell Biol 21:4785–4806
Yu C, Chen Y, Cline GW et al (2002) Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 277:50230–50236
Samuel VT, Liu ZX, Wang A et al (2007) Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest 117:739–745
Vatner DF, Weismann D, Beddow SA et al (2013) Thyroid hormone receptor-beta agonists prevent hepatic steatosis in fat-fed rats but impair insulin sensitivity via discrete pathways. Am J Phys Endocrinol Metab 305:E89–E100
Lee HY, Birkenfeld AL, Jornayvaz FR et al (2011) Apolipoprotein CIII overexpressing mice are predisposed to diet-induced hepatic steatosis and hepatic insulin resistance. Hepatology 54:1650–1660
Kumashiro N, Erion DM, Zhang D et al (2011) Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 108:16381–16385
Qu X, Seale JP, Donnelly R (1999) Tissue and isoform-selective activation of protein kinase C in insulin-resistant obese Zucker rats - effects of feeding. J Endocrinol 162:207–214
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Sando JJ, Beals JK (2003) Enzyme assays for protein kinase C activity. Methods Mol Biol 233:63–76
Ping P, Zhang J, Huang S et al (1999) PKC-dependent activation of p46/p54 JNKs during ischemic preconditioning in conscious rabbits. Am J Phys 277:H1771–H1785
Petrick A, Benotti P, Wood GC et al (2015) Utility of ultrasound, transaminases, and visual inspection to assess nonalcoholic fatty liver disease in bariatric surgery patients. Obes Surg 25:2368–2375
Gerhard GS, Styer AM, Strodel WE et al (2014) Gene expression profiling in subcutaneous, visceral and epigastric adipose tissues of patients with extreme obesity. Int J Obes 38:371–378
Wood GC, Chu X, Manney C et al (2012) An electronic health record-enabled obesity database. BMC Med Inform Decis Mak 12:45
Turpin SM, Nicholls HT, Willmes DM et al (2014) Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab 20:678–686
Raichur S, Wang ST, Chan PW et al (2014) CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab 20:687–695
Blachnio-Zabielska AU, Chacinska M, Vendelbo MH, Zabielski P (2016) The crucial role of C18-Cer in fat-induced skeletal muscle insulin resistance. Cell Physiol Biochem 40:1207–1220
Petersen MC, Madiraju AK, Gassaway BM et al (2016) Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest 126:4361–4371
Samuel VT, Shulman GI (2016) The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 126:12–22
Liu D, Li S, He H et al (2017) Increased circulating full-length betatrophin levels in drug-naive metabolic syndrome. Oncotarget 8:17510–17517
Ebert T, Kralisch S, Hoffmann A et al (2014) Circulating angiopoietin-like protein 8 is independently associated with fasting plasma glucose and type 2 diabetes mellitus. J Clin Endocrinol Metab 99:E2510–E2517
Fu Z, Berhane F, Fite A, Seyoum B, Abou-Samra AB, Zhang R (2014) Elevated circulating lipasin/betatrophin in human type 2 diabetes and obesity. Sci Rep 4:5013
von Loeffelholz C, Pfeiffer AF, Lock JF et al (2017) ANGPTL8 (betatrophin) is expressed in visceral adipose tissue and relates to human hepatic steatosis in two independent clinical collectives. Horm Metab Res 49:343–349
Zhang R (2012) Lipasin, a novel nutritionally-regulated liver-enriched factor that regulates serum triglyceride levels. Biochem Biophys Res Commun 424:786–792
Gusarova V, Banfi S, Alexa-Braun CA et al (2017) ANGPTL8 blockade with a monoclonal antibody promotes triglyceride clearance, energy expenditure and weight loss in mice. Endocrinology 158:1252–1259
Acknowledgements
We would like to thank M. Kahn for LC-MS measurements; and J. Dong, G. Butrico, C. Todeasa, M. Batsu and the Yale Diabetes Research Core facility for excellent technical support. All are at Yale University School of Medicine, New Haven, CT, USA.
Contribution statement
DFV conceived the study, developed methodology, performed experiments, supervised the study and wrote the manuscript; LG designed and performed experiments and assisted with writing the manuscript; JGC, KL, ARN and DZ performed experiments, analysed and interpreted data; SB and SFM conceived, designed and performed experiments in the generation of ASOs; CDS and GSG conceived, designed, and performed experiments, provided human tissues for analysis and assisted with writing the manuscript; GIS and VTS conceived the study, supervised the study and assisted with writing the manuscript. All authors revised the manuscript critically for important intellectual content, reviewed the manuscript and approved the final version. DFV and VTS are responsible for the integrity of the work as a whole.
Funding
We would like to thank our funding sources: the National Institutes of Health (K23 DK-10287, R01 DK-40936, U24 DK-059635, P30 DK-045735) and the Veterans Health Administration (Merit Review Award I01 BX000901).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
SB and SFM report being employees and shareholders of Ionis Pharmaceuticals. The other authors declare that there is no duality of interest associated with their contribution to this manuscript.
Electronic supplementary material
ESM
(PDF 501 kb)
Rights and permissions
About this article

Cite this article
Vatner, D.F., Goedeke, L., Camporez, JP.G. et al. Angptl8 antisense oligonucleotide improves adipose lipid metabolism and prevents diet-induced NAFLD and hepatic insulin resistance in rodents. Diabetologia 61, 1435–1446 (2018). https://doi.org/10.1007/s00125-018-4579-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-018-4579-1