
Abstract
Aims/hypothesis
Individuals exposed to maternal diabetes in utero are more likely to develop metabolic and cardiovascular diseases later in life. This may be partially attributable to epigenetic regulation of gene expression. We performed an epigenome-wide association study to examine whether differential DNA methylation, a major source of epigenetic regulation, can be observed in offspring of mothers with type 2 diabetes during the pregnancy (OMD) compared with offspring of mothers with no diabetes during the pregnancy (OMND).
Methods
DNA methylation was measured in peripheral blood using the Illumina HumanMethylation450K BeadChip. A total of 423,311 CpG sites were analysed in 388 Pima Indian individuals, mean age at examination was 13.0 years, 187 of whom were OMD and 201 were OMND. Differences in methylation between OMD and OMND were assessed.
Results
Forty-eight differentially methylated CpG sites (with an empirical false discovery rate ≤0.05), mapping to 29 genes and ten intergenic regions, were identified. The gene with the strongest evidence was LHX3, in which six CpG sites were hypermethylated in OMD compared with OMND (p ≤ 1.1 × 10−5). Similarly, a CpG near PRDM16 was hypermethylated in OMD (1.1% higher, p = 5.6 × 10−7), where hypermethylation also predicted future diabetes risk (HR 2.12 per SD methylation increase, p = 9.7 × 10−5). Hypermethylation near AK3 and hypomethylation at PCDHGA4 and STC1 were associated with exposure to diabetes in utero (AK3: 2.5% higher, p = 7.8 × 10−6; PCDHGA4: 2.8% lower, p = 3.0 × 10−5; STC1: 2.9% lower, p = 1.6 × 10−5) and decreased insulin secretory function among offspring with normal glucose tolerance (AK3: 0.088 SD lower per SD of methylation increase, p = 0.02; PCDHGA4: 0.08 lower SD per SD of methylation decrease, p = 0.03; STC1: 0.072 SD lower per SD of methylation decrease, p = 0.05). Seventeen CpG sites were also associated with BMI (p ≤ 0.05). Pathway analysis of the genes with at least one differentially methylated CpG (p < 0.005) showed enrichment for three relevant biological pathways.
Conclusions/interpretation
Intrauterine exposure to diabetes can affect methylation at multiple genomic sites. Methylation status at some of these sites can impair insulin secretion, increase body weight and increase risk of type 2 diabetes.
Similar content being viewed by others
Introduction
Unbalanced nutrition in utero, such as hyperglycaemia, has been associated with the development of metabolic diseases in later life [1]. In particular, intrauterine exposure to maternal diabetes increases the risk that the child will subsequently develop obesity and diabetes [2]. Among Pima Indian siblings discordant for intrauterine exposure to diabetes, the risk was higher in those born after their mother developed diabetes than in those born before their mother developed diabetes, providing evidence that the increased risk is due to an effect of the diabetic intrauterine environment in addition to any effect conferred by inheritance of diabetes susceptibility alleles [3]. Studies in Pima Indians with normal glucose tolerance (NGT) have also shown that insulin secretion is lower in young adults whose mothers were diabetic during pregnancy compared with those whose mothers developed diabetes at an early age but after the birth of the child, suggesting that exposure to a diabetic intrauterine environment increases the risk of diabetes, in part because of decreased insulin secretion [4]. This reduction in insulin secretion may be mediated by epigenetic modifications during key developmental periods of the fetus [5].
One source of epigenetic modification is DNA methylation at CpG sites. In some cases, methylation at a specific site is stable; thus, assessment after birth can provide information on methylation patterns established in utero [6]. Two recent studies have reported epigenome-wide analyses that identified differentially methylated regions as a consequence of intrauterine exposure to maternal diabetes [7, 8]. One of these studies was our prior examination of promoter regions in DNA from 28 Pima Indians which did not identify any specific differentially methylated promoter that met statistical significance [7]. A third study which examined methylation of GNAS and IGF2 in 168 Chinese newborns found an association between maternal diabetes and methylation of GNAS, in which methylation levels were also associated with obesity [9]. However, these three studies had limited statistical power due to their small sample sizes. In the current study, we sought to understand the effect of maternal diabetes on DNA methylation in 388 Pima Indian offspring, which to our knowledge is the largest epigenome-wide report to date. These individuals were, on average, 13 years old when examined, so the current study sought to determine methylation changes due to exposure to diabetes in utero that were stable over many years. The Pima Indian population is at high risk of type 2 diabetes, but type 1 diabetes does not occur [10].
Methods
Study participants
Participants in the present study were a subset of individuals from a longitudinal study among residents of an American Indian community in Arizona who are predominately Pima Indian [2]. Biennial health examinations, beginning at age ≥5 years, included a 75 g OGTT; in a substudy of pregnant women, a 75 g OGTT was also offered in the third trimester. In the current study, data for the OGTT during pregnancy were obtained either at a biennial examination or at a third trimester examination. Diabetes was defined by World Health Organization criteria for non-pregnant individuals, regardless of whether a woman was pregnant, i.e. 2 h post-load plasma glucose ≥11.1 mmol/l, fasting plasma glucose ≥7.0 mmol/l or a diagnosis observed during clinical care [11]. DNA analysed in the current study was isolated from peripheral blood collected at the offspring’s first examination at which they were not diabetic. All available offspring of mothers with diabetes during pregnancy (OMD), defined as having a mother diagnosed with type 2 diabetes before the child’s birth, were selected for methylation studies (N = 187). Of these, 169 (90%) were offspring of a mother diagnosed more than 9 months before the child’s birth. Of the remaining 18 (10%) whose mother’s diabetes diagnosis was less than 9 months before the child’s birth, six were born to a mother with no previous examination, four were born to a mother with a non-diabetic examination <1 year before the child’s birth and eight were born to a mother whose last non-diabetic examination was ≥1 year before the child’s birth. Thus, the current study largely examines the effect on the intrauterine environment of type 2 diabetes that predates the pregnancy. For comparison, DNA samples (N = 201) were randomly selected from offspring of mothers with no diabetes during the pregnancy (OMND), where the mother did not have diabetes diagnosed before the child’s birth and the mother had a non-diabetic OGTT (2 h post-load plasma glucose <11.1 mmol/l) in the longitudinal study ≥12 months after the child’s birth. OMND were frequency-matched to OMD by sex, age (5 year strata) and self-reported Pima heritage (full vs partial), and all were required to have at least 50% American Indian heritage by self-report. All studies were approved by the institutional review board of the National Institute of Diabetes and Digestive and Kidney Diseases, and all participants provided written informed consent.
Characteristics of the 388 offspring analysed in the study are provided in Table 1. The mean age of the offspring when their DNA was obtained was 13.0 (range 5.2–41.6) years. The OMD group, on average, had a higher maternal age and BMI, and was more likely to have a sibling included in the analyses, compared with the OMND group (Table 1).
Statistical analysis to identify differential methylation
DNA methylation at CpG sites was measured using the Infinium HumanMethylation450K BeadChip (Illumina, San Diego, CA, USA; methods detailed in electronic supplementary material [ESM] Methods). To stabilise the variance for purposes of analyses, Beta-values (the ratio of methylated probe intensity and overall intensity [the sum of methylated and unmethylated probe intensities]) were converted to M-values (the log2 ratio of methylated probe intensity vs unmethylated probe intensity) and standardised to a mean of 0 and an SD of 1 [12]. The standardised M-value was adjusted by linear regression for age, sex, a genetic estimate of American Indian ancestry in the offspring, mother’s age when the child was born, array batch number, and the first four principal components (PCs) to account for cellular heterogeneity or other unmeasured confounders [13]. In these analyses, American Indian ancestry was estimated according to the method of Hanis et al [14] from 45 ancestry-informative markers [15]. The PCs were calculated using the R software package pcaMethods (R Foundation for Statistical Computing, Vienna, Austria), and the methylation Beta-values from 47,340 probes, with one probe selected from each gene and CpG island. The residuals of the standardised M-values obtained from this regression were tested for association with intrauterine exposure to diabetes using Procedure GLM in SAS, version 9.3 (SAS Institute, Cary, NC, USA), with M-value as the dependent variable. To estimate the effect size in biologically meaningful terms, the same model was analysed with the original Beta-value as the dependent variable. In Table 2, effect sizes are shown for the model with Beta-value as the dependent variable, while p values are reported for the model with M-value as the dependent variable (which generally conforms better to the assumptions of linear regression).
The empirical false discovery rate (FDR) was estimated using a nested permutation procedure to generate the null distribution. In brief, the methylation data (M-values, PCs) were permuted independently from the phenotypic data (exposure to maternal diabetes status, age, sex, maternal age and ancestry estimate). To account for familial dependence, all phenotypic data for a sibship were permuted together; thus, permutations were conducted across sibships of the same size. With 1000 permutations, the empirical FDR p values were calculated using the method proposed by Millstein and Volfson [16].
Differentially methylated regions were analysed to determine whether there was a general tendency for these CpGs to be hypo- or hypermethylated in OMD relative to OMND. The number of hypermethylated regions (effect >0) and hypomethylated regions (effect <0) were counted and subjected to a binomial test for the methylation proportion using the R function binom.test (R Foundation for Statistical Computing), in comparison with the expected percentage of hypermethylated regions of 50%.
Assessing differentially methylated sites for prediction of impaired secretory function, obesity and diabetes risk
Secretory function could be assessed in 230 of the 388 offspring who had NGT (2 h post-load plasma glucose <7.8 mmol/l) at baseline. The 2 h corrected insulin response (CIR), a surrogate measure of insulin secretion, was derived from OGTT measurements at the time of the DNA sample collection [17]. Fourteen of these NGT offspring had further been assessed for their acute insulin response (AIR) to a 25 g intravenous glucose challenge at a later time in their life [4]. The association between the insulin secretory function (defined as standardised logarithm of CIR or AIR) and standardised M-value residuals for individual CpG sites was calculated with adjustments as detailed in the Table 3 legend.
BMI could be assessed in 376 of the 388 offspring who had both height and weight measured at baseline, while risk of type 2 diabetes could be assessed in 303 offspring who had data from at least one outpatient follow-up examination (58 out of 303 had documented diabetes at follow-up). The association between the logarithm of BMI and standardised M-value, as well as the association between diabetes risk and standardised M-value, was calculated with adjustments as detailed in the Table 3 legend. A Cox proportional hazards model (Procedure PHREG in SAS, version 9.3; SAS Institute) was used for the analysis of diabetes risk. To assess the extent to which observed methylation differences may account for the increased diabetes risk in OMD, a formal mediation analysis was conducted as detailed in ESM Text [18, 19].
Pathway enrichment analysis
Genes with differentially methylated CpG sites (p ≤ 0.005) were analysed for pathway enrichment (KEGG pathways [20]; release date 21 March 2011; access date 4 November 2015) compared with Homo sapiens genetic background using the WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) [21]. The enrichment p values were adjusted for multiple tests by the FDR method. Pathways with FDR p ≤ 0.001 and more than three differentially methylated genes were considered enriched.
Results
Thirty-nine unique differentially methylated regions are associated with intrauterine diabetes exposure
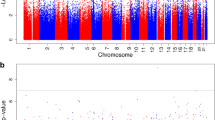
Methylation data from 423,311 CpG sites passed our quality control metrics. When analysed in aggregate, baseline DNA methylation mean Beta-values across all 423,311 sites did not differ (p = 0.70) between the OMD group (N = 187; Beta-value 0.513 [SD 0.006]) and the OMND group (N = 201; Beta-value 0.513 [SD 0.012]). Analysis of each individual CpG site in OMD compared with OMND is shown by chromosome in a Manhattan plot (Fig. 1a), and the corresponding quantile–quantile plot is also shown (Fig. 1b). Forty-eight CpGs representing 39 unique regions were significantly associated with intrauterine diabetes exposure (empirical FDR ≤0.05; Table 2). At these sites, the adjusted median absolute value of the difference in DNA methylation between OMD and OMND was 2.2% (range 0.6–4.6%). Among these 48 CpGs (17 in gene bodies or 3′ untranslated regions [UTRs], 20 in regions close to transcription start sites [TSSs] or 5′ UTRs, 11 in intergenic regions [with two CpGs being found in the same intergenic region]), 29 were hypermethylated and 19 were hypomethylated; the proportion of hypermethylated CpGs was not significantly greater than 0.5 (binominal p = 0.097). Since there was a large difference in pre-pregnancy maternal BMI between the OMD and OMND groups (Table 1), we further adjusted for pre-pregnancy BMI in the 296 individuals (154 OMD and 142 OMND) for whom maternal BMI data were available. The effect sizes were generally unchanged with the additional adjustment for maternal pre-pregnancy BMI (ESM Table 1).

The genome-wide differential methylation signals. (a) Manhattan plot of differentially methylated CpGs in OMND vs OMD. Genome-wide significant differences are highlighted in green. (b) Quantile–quantile plot of the differential signals. The red line is the identity line
Methylation at some CpG sites which associate with intrauterine exposure also associate with impaired insulin secretory function, higher BMI or future risk of type 2 diabetes
We further assessed whether any of the 48 CpG sites that associated with exposure to intrauterine diabetes also associated with reduction in insulin secretory function, higher BMI or future type 2 diabetes. Three of the 48 sites were modestly associated with CIR (Table 3). The CpG site (cg24049468) located in the gene body of AK3 was hypermethylated in the OMD group, and hypermethylation at this site was associated with a reduced CIR in the 230 NGT participants. Similarly, CpG sites located in the gene body of PCDHGA4 (cg25949304) and close to the TSS of STC1 (cg04645534) were hypomethylated in the OMD group, and hypomethylation at these sites was associated with a reduced CIR in the 230 NGT participants. Methylation at these three CpG sites was further analysed in 14 individuals who remained NGT and had participated in a subsequent inpatient examination (median follow-up was 1.8 + 5.8 years) in our clinical research centre to characterise insulin secretory function via a 25 g IVGTT. In this very small sample, those who were hypomethylated at STC1 similarly tended to have a reduced AIR (effect 3.7% per SD; p = 0.058).
Among the 48 CpGs differentially methylated between OMD and OMND, 17 were associated with baseline BMI of the offspring (p < 0.05). However, only CpG sites in SBK1 (cg04413090, p = 4.1 × 10−4) and PRDM16 (cg12140144, p = 1.2 × 10−3) and two intergenic CpGs (cg10772621, p = 2.3 × 10−4; cg07464358, p = 2.3 × 10−4) remained significantly associated after correction for multiple tests (assuming 39 independent tests). Three of the CpG sites, cg04413090 (SBK1), cg12140144 (PRDM16) and cg07464358, were hypermethylated in OMD, and hypermethylation was associated with higher BMI (5.3%, 6.0% and 9.2% higher per SD of standardised M-value, respectively). Similarly, the CpG site cg10772621 was hypomethylated in OMD, and hypomethylation was associated with higher BMI (by 4.9% per SD of standardised M-value).
Among the 303 offspring with data from a follow-up examination, 43 OMD and 15 OMND developed diabetes later in life. The adjusted HR for developing diabetes for OMD compared with OMND was 5.48 (95% CI 2.85, 10.52; p = 3.2 × 10−7). Among these 303 individuals, methylation at cg12140144 in the TSS of PRDM16 was associated with a higher risk of diabetes (Table 3; HR 2.12 [95% CI 1.74, 2.50] per SD of M-value; p = 9.7 × 10−5). Another differentially methylated CpG site, cg08911291, which is located in an intergenic region, also carried diabetes risk with marginal significance (Table 3; HR 1.47 [95% CI 1.09, 1.85] per SD of M-value; p = 0.05). Among the 303 individuals with data on development of diabetes, the adjusted standardised M-value at the cg12140144 site in PRDM16 was 0.51 SD higher in OMD compared with OMND. After adjustment for methylation at this site, the HR for developing diabetes in OMD compared with OMND was 3.89 (95% CI 1.98, 7.65; p = 8.0 × 10−5). In mediation analyses, this corresponded to a percentage mediation of 20% (two-sided p = 0.026). The corresponding value for cg08911291 was 7% (p = 0.27).
Differentially methylated genes clustered in known pathways
The pathway analysis included 641 genes with a differentially methylated CpG site (p ≤ 0.005). The differentially methylated genes were enriched for: (1) metabolic pathways (37 genes, adjusted p = 0.0002); (2) Wnt signalling pathways (11 genes, adjusted p = 0.0004); and (3) protein digestion and absorption (eight genes, adjusted p = 0.0005) (ESM Table 2).
Database searches (www.ncbi.nlm.nih.gov/gene, accessed 1 September 2016) revealed that 11 of the 29 genes have known roles in embryonic development (ESM Table 3), and several genes have a physiological function consistent with a role in beta cell function/type 2 diabetes.
Discussion
We investigated genome-wide DNA methylation in peripheral blood in relation to intrauterine diabetes exposure. Compared with published studies of intrauterine exposure to maternal diabetes, our study featured a larger sample size. The Infinium (Illumina) methylation assay used in the current study covers 96% of CpG islands and 99% of reference sequence genes with an average of 17 CpG sites per gene. Forty-eight CpG sites were identified, with a statistically significant (FDR ≤0.05) difference in methylation among people exposed to maternal diabetes in utero compared with those not exposed. For differentially methylated sites, the median adjusted absolute difference in methylation between OMD and OMND was 2.2% (range 0.6–4.6%). Exposure to diabetes in utero is strongly associated with higher maternal BMI, and genetically raised maternal BMI is associated with birthweight, which in turn is associated with long-term developmental outcomes and adult diseases [22, 23]. However, the additional adjustment for maternal pre-pregnancy BMI did not materially change the effect sizes of our 48 top signals.
The strongest result in our study, in terms of statistical significance, is the differential methylation of the transcriptional activator LHX3. Six of the 23 CpG sites assayed on the array from this region were hypermethylated in OMD (p ≤ 1.1 × 10−5). Mutations in LHX3 have been associated with combined pituitary hormone deficiency (CPHD), which is often characterised by reduced growth and short stature [24]. Lhx3 expression is regulated by DNA methylation in mice, where decreased methylation has been shown to activate expression in pituitary cells that do not normally express Lhx3 [25]. In humans, LHX3 is broadly expressed in many tissues, including the pancreas. Although this particular transcription factor has not been previously reported to affect insulin secretory pathways, it does interact with several other transcription factors including insulin gene enhancer protein ISL1 and LIM homeobox transcription factor 1-alpha (LMX1A), which are known regulators of insulin gene expression [26].
Several of the genes identified as differentially methylated between OMD and OMND have established roles in beta cell function, including a CpG site in STC1 that was hypomethylated in the OMD group, where hypomethylation of the same CpG was also associated with both reduced CIR and AIR, as well as increased BMI at baseline. STC1 encodes the polypeptide hormone stanniocalcin-1 (STC-1), which co-localises with insulin in mouse pancreatic islets, and it has been proposed that STC-1 regulates beta cell mitochondria membrane potential, which is a crucial component in regulation of insulin release [27]. Another gene, CACNA1C, which was hypomethylated in the OMD group, has also been shown to be important for beta cell function [28]. However, hypomethylation at CACNA1C was not associated with reduction in insulin secretory response in the present data.
Two CpG sites that were significantly differentially methylated between OMD and OMND on an epigenome-wide basis also had nominally significant associations with diabetes risk in later life; however, only the CpG site in PRMD16 (cg12140144) remained associated after correction for multiple testing (assuming 39 independent differentially methylated regions). This CpG was hypermethylated in OMD by 1.1% on average, and a 1 SD increase in methylation was associated with a doubled diabetes risk. Mediation analysis suggested that differential methylation at this site could potentially explain 20% of the excess risk of diabetes for OMD compared with OMND, and this mediation effect was nominally statistically significant. This site maps within a CpG island (chr1:2983925−2987962) that spans both 5′ UTRs of LINC00982 and the promoter of PRDM16. In human placental tissue, methylation at two CpG sites in PRDM16 was associated with maternal fasting glucose in the second and third gestational trimesters [29]. CpGs in PRDM16 were also identified in another study which investigated differential DNA methylation in pancreatic islets of human donors with type 2 diabetes [30]. PRDM16 has been predominately studied in relation to brown adipose tissue (BAT), where it regulates differentiation from myoblastic precursors to BAT [31]. A SNP (rs12409277) in its 5′ flanking region was associated with lean body mass in a Japanese population, and the same SNP was demonstrated to affect the transcriptional activity of PRDM16 [32]. An essential role for PRDM16 in pancreatic islet development from a single fetal progenitor cell has recently been described [33], suggesting that this gene also has an important role in pancreatic development. In mice, the function of Prdm16 is regulated by epigenetic histone modification [34]. Taken together, multiple evidence shows that PRDM16 could be the regulatory target of hyperglycaemia and responsible in part for increased diabetes risk in OMD. The differentially methylated CpG site in PRDM16 identified in peripheral blood might be transferable to other tissues, making it a promising biomarker for diabetes risk screening.
While our previous in vivo clinical studies in Pima Indians have shown that exposure to diabetes in utero increases the risk of type 2 diabetes primarily via impairment of beta cell function, it is possible that reduced insulin sensitivity either as a primary effect or secondary to increased adiposity may also contribute to the higher risk of type 2 diabetes. For example, PPP1R3B, which was differentially methylated in our study, codes for the GL regulatory subunit of protein phosphatase 1 (PP1), which targets PP1 to activate glycogen synthase, thereby increasing insulin sensitivity of liver and skeletal muscle [35, 36]. A decreased rate of glycogen synthesis in the skeletal muscle in response to insulin is a characteristic feature of individuals with type 2 diabetes [37], and polymorphisms in another regulatory subunit (PPP1R3A), which also targets PP1 activation of glycogen synthase in the skeletal muscle, were associated with insulin resistance and type 2 diabetes in several different populations [38–42]. Differential methylation of a second gene, PM20D1, could also affect insulin sensitivity via its effect on body weight. In humans, methylation of PM20D1 in cord blood is associated with birthweight centile [43]. Methylation of PM20D1 has also been associated with several adverse health events during early life, indicating that PM20D1 may be a target gene for in utero epigenetic programming in multiple organs [44, 45]. Differential methylation of PM20D1 in obese and non-obese individuals at older ages suggests that in utero programming of PM20D1 has sustainable effects much later in life [46].
Among the 29 genes identified in the current study which are differentially methylated in OMD but not in OMND, only TBL1X has been previously implicated in exposure to maternal diabetes. In our study, a CpG in the 5′ UTR of TBL1X was hypermethylated in the OMD group, and others have reported a 27.22-fold decrease in expression following exposure to maternal diabetes [47]. TBL1X encodes a mediator of β-catenin proteolysis through ubiquitination in the canonical Wnt pathway, which was highlighted by our pathway analysis (ESM Table 2).
The strength of this study is its sample size and detailed clinical measures of a well-phenotyped, longitudinal cohort. There are, however, several weaknesses. First, since blood samples were not obtained at birth in our longitudinal study, the DNA analysed for methylation was from individuals with a mean age of 13 years. Thus, our study can only detect in utero differences in methylation that are stable throughout many years, and our results could also be influenced by differences in methylation that arise postnatally. Prior studies of in utero methylation have shown evidence of both transient and stable methylation sites. For example, a longitudinal analysis showed that sites that were differentially methylated at birth and associated with birthweight and gestational age were no longer different when the child reached 7 years of age [48]. By contrast, DNA methylation differences associated with in utero exposure to maternal smoking have been reported to be long-lasting [49, 50]. A second weakness of our study is that we analyse DNA from peripheral blood, although we believe that the pancreas and corresponding beta cell function are the key tissues responsible for the development of early-onset type 2 diabetes as a consequence of in utero exposure to diabetes. Therefore, our study will only detect differences in methylation that affect pancreatic development/function that are similarly differentially methylated in the blood. Fine mapping of CpG sites from DNA isolated from specific target tissues is required to further understand the detailed molecular mechanisms.
In conclusion, our study identified differentially methylated CpGs in 39 genomic regions that achieved epigenome-wide significance in their association with exposure to a diabetic intrauterine environment. Methylation at three sites also nominally associated with insulin secretion, while a fourth site associated with future risk of type 2 diabetes. However, our studies of insulin secretion and future diabetes risk were conducted in relatively small numbers of individuals and thus may be subject to considerable stochastic variation; therefore, replication in other cohorts will be very informative. In addition, further investigations on quantitative gene expression as a consequence of DNA methylation at these CpG sites, as well as causal CpG mapping, will lead to a better understanding of the mechanism of diabetes and its complications.
Abbreviations
- AIR:
-
Acute insulin response
- BAT:
-
Brown adipose tissue
- Beta-value:
-
Ratio of methylated probe intensity and overall intensity
- CIR:
-
Corrected insulin response
- FDR:
-
False discovery rate
- M-value:
-
Log2 ratio of methylated probe intensity vs unmethylated probe intensity
- NGT:
-
Normal glucose tolerance
- OMD:
-
Offspring of mothers with type 2 diabetes during pregnancy
- OMND:
-
Offspring of mothers with no type 2 diabetes during pregnancy
- PC:
-
Principle component
- PP1:
-
Protein phosphatase 1
- TSS:
-
Transcription start site
- UTR:
-
Untranslated region
References
Lehnen H, Zechner U, Haaf T (2013) Epigenetics of gestational diabetes mellitus and offspring health: the time for action is in early stages of life. Mol Hum Reprod 19:415–422
Pettitt DJ, Aleck KA, Baird HR, Carraher MJ, Bennett PH, Knowler WC (1988) Congenital susceptibility to NIDDM. Role of intrauterine environment. Diabetes 37:622–628
Dabelea D, Hanson RL, Lindsay RS et al (2000) Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes 49:2208–2211
Gautier JF, Wilson C, Weyer C et al (2001) Low acute insulin secretory responses in adult offspring of people with early onset type 2 diabetes. Diabetes 50:1828–1833
Ruchat SM, Hivert MF, Bouchard L (2013) Epigenetic programming of obesity and diabetes by in utero exposure to gestational diabetes mellitus. Nutr Rev 71(Suppl 1):S88–S94
Yan J, Yang H (2014) Gestational diabetes mellitus, programing and epigenetics. J Mater Fetal Neonatal Med 27:1266–1269
del Rosario MC, Ossowski V, Knowler WC, Bogardus C, Baier LJ, Hanson RL (2014) Potential epigenetic dysregulation of genes associated with MODY and type 2 diabetes in humans exposed to a diabetic intrauterine environment: an analysis of genome-wide DNA methylation. Metab Clin Exp 63:654–660
West NA, Kechris K, Dabelea D (2013) Exposure to maternal diabetes in utero and DNA methylation patterns in the offspring. Immunometabolism 1:1–9
Chen D, Zhang A, Fang M et al (2014) Increased methylation at differentially methylated region of GNAS in infants born to gestational diabetes. BMC Med Genet 15:108
Dabelea D, Palmer JP, Bennett PH, Pettitt DJ, Knowler WC (1999) Absence of glutamic acid decarboxylase antibodies in Pima Indian children with diabetes mellitus. Diabetologia 42:1265–1266
Alberti KG, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 15:539–553
Du P, Zhang X, Huang CC et al (2010) Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinforma 11:587
Jaffe AE, Irizarry RA (2014) Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 15:R31
Hanis CL, Chakraborty R, Ferrell RE, Schull WJ (1986) Individual admixture estimates: disease associations and individual risk of diabetes and gallbladder disease among Mexican-Americans in Starr County, Texas. Am J Phys Anthropol 70:433–441
Tian C, Hinds DA, Shigeta R et al (2007) A genomewide single-nucleotide-polymorphism panel for Mexican American admixture mapping. Am J Hum Genet 80:1014–1023
Millstein J, Volfson D (2013) Computationally efficient permutation-based confidence interval estimation for tail-area FDR. Front Genet 4:179
Hanson RL, Pratley RE, Bogardus C et al (2000) Evaluation of simple indices of insulin sensitivity and insulin secretion for use in epidemiologic studies. Am J Epidemiol 151:190–198
Baron RM, Kenny DA (1986) The moderator–mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J Pers Soc Psychol 51:1173–1182
Preacher KJ, Kelley K (2011) Effect size measures for mediation models: quantitative strategies for communicating indirect effects. Psychol Methods 16:93–115
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Zhang B, Kirov S, Snoddy J (2005) WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33:W741–W748
Barker DJ, Godfrey KM, Fall C, Osmond C, Winter PD, Shaheen SO (1991) Relation of birth weight and childhood respiratory infection to adult lung function and death from chronic obstructive airways disease. BMJ 303:671–675
Tyrrell J, Richmond RC, Palmer TM et al (2016) Genetic evidence for causal relationships between maternal obesity-related traits and birth weight. JAMA 315:1129–1140
Netchine I, Sobrier ML, Krude H et al (2000) Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet 25:182–186
Malik RE, Rhodes SJ (2014) The role of DNA methylation in regulation of the murine Lhx3 gene. Gene 534:272–281
Gadd MS, Jacques DA, Nisevic I et al (2013) A structural basis for the regulation of the LIM-homeodomain protein islet 1 (Isl1) by intra- and intermolecular interactions. J Biol Chem 288:21924–21935
Zaidi D, Turner JK, Durst MA, Wagner GF (2012) Stanniocalcin-1 co-localizes with insulin in the pancreatic islets. ISRN Endocrinol 2012:834359
Nitert MD, Nagorny CL, Wendt A, Eliasson L, Mulder H (2008) CaV1.2 rather than CaV1.3 is coupled to glucose-stimulated insulin secretion in INS-1 832/13 cells. J Mol Endocrinol 41:1–11
Cote S, Gagne-Ouellet V, Guay SP et al (2016) PPARGC1α gene DNA methylation variations in human placenta mediate the link between maternal hyperglycemia and leptin levels in newborns. Clin Epigenetics 8:72
Dayeh T, Volkov P, Salo S et al (2014) Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet 10:e1004160
Kajimura S, Seale P, Kubota K et al (2009) Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature 460:1154–1158
Urano T, Shiraki M, Sasaki N, Ouchi Y, Inoue S (2014) Large-scale analysis reveals a functional single-nucleotide polymorphism in the 5′-flanking region of PRDM16 gene associated with lean body mass. Aging Cell 13:739–743
Sugiyama T, Benitez CM, Ghodasara A et al (2013) Reconstituting pancreas development from purified progenitor cells reveals genes essential for islet differentiation. Proc Natl Acad Sci U S A 110:12691–12696
Ohno H, Shinoda K, Ohyama K, Sharp LZ, Kajimura S (2013) EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature 504:163–167
Doherty MJ, Moorhead G, Morrice N, Cohen P, Cohen PT (1995) Amino acid sequence and expression of the hepatic glycogen-binding (GL)-subunit of protein phosphatase-1. FEBS Lett 375:294–298
Munro S, Cuthbertson DJ, Cunningham J, Sales M, Cohen PT (2002) Human skeletal muscle expresses a glycogen-targeting subunit of PP1 that is identical to the insulin-sensitive glycogen-targeting subunit GL of liver. Diabetes 51:591–598
Bogardus C, Lillioja S, Stone K, Mott D (1984) Correlation between muscle glycogen synthase activity and in vivo insulin action in man. J Clin Invest 73:1185–1190
Hansen L, Hansen T, Vestergaard H et al (1995) A widespread amino acid polymorphism at codon 905 of the glycogen-associated regulatory subunit of protein phosphatase-1 is associated with insulin resistance and hypersecretion of insulin. Hum Mol Genet 4:1313–1320
Xia J, Scherer SW, Cohen PT et al (1998) A common variant in PPP1R3 associated with insulin resistance and type 2 diabetes. Diabetes 47:1519–1524
Maegawa H, Shi K, Hidaka H et al (1999) The 3′-untranslated region polymorphism of the gene for skeletal muscle-specific glycogen-targeting subunit of protein phosphatase 1 in the type 2 diabetic Japanese population. Diabetes 48:1469–1472
Wang G, Qian R, Li Q, Niu T, Chen C, Xu X (2001) The association between PPP1R3 gene polymorphisms and type 2 diabetes mellitus. Chin Med J 114:1258–1262
Doney AS, Fischer B, Cecil JE et al (2003) Male preponderance in early diagnosed type 2 diabetes is associated with the ARE insertion/deletion polymorphism in the PPP1R3A locus. BMC Genet 4:11
Haworth KE, Farrell WE, Emes RD et al (2014) Methylation of the FGFR2 gene is associated with high birth weight centile in humans. Epigenomics 6:477–491
Gunawardhana LP, Baines KJ, Mattes J, Murphy VE, Simpson JL, Gibson PG (2014) Differential DNA methylation profiles of infants exposed to maternal asthma during pregnancy. Pediatr Pulmonol 49:852–862
Suderman M, Borghol N, Pappas JJ et al (2014) Childhood abuse is associated with methylation of multiple loci in adult DNA. BMC Med Genet 7:13
Feinberg AP, Irizarry RA, Fradin D et al (2010) Personalized epigenomic signatures that are stable over time and covary with body mass index. Sci Transl Med 2:49ra67
Pavlinkova G, Salbaum JM, Kappen C (2009) Maternal diabetes alters transcriptional programs in the developing embryo. BMC Genomics 10:274
Simpkin AJ, Suderman M, Gaunt TR et al (2015) Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum Mol Genet 24:3752–3763
Terry MB, Ferris JS, Pilsner R et al (2008) Genomic DNA methylation among women in a multiethnic New York City birth cohort. Cancer Epidemiol Biomarkers Prev 17:2306–2310
Richmond RC, Simpkin AJ, Woodward G et al (2015) Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet 24:2201–2217
Acknowledgements
This study used the computational resources of the Biowulf system at the National Institutes of Health, Bethesda, MD, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Data availability
These data are not publicly available for reasons of privacy. For the present study, the National Institutes of Health (NIH) has granted an exception from the expectation of broad data sharing outlined in the NIH Genomic Data Sharing Policy.
Funding
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript
Contribution statement
CB, WCK, LJB and RLH conceived and designed the study and were responsible for data acquisition. PC, PP, MT, LJB and RLH analysed and interpreted the data. All authors drafted or revised the article and approved the final version of the manuscript. LJB and RLH are responsible for the integrity of the work as a whole.
Additional information
Leslie J. Baier and Robert L. Hanson contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(PDF 126 kb)
Rights and permissions
About this article

Cite this article
Chen, P., Piaggi, P., Traurig, M. et al. Differential methylation of genes in individuals exposed to maternal diabetes in utero. Diabetologia 60, 645–655 (2017). https://doi.org/10.1007/s00125-016-4203-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4203-1