
Abstract
Aims/hypothesis
Beta cell inflammation and demise is a feature of type 1 diabetes. The insulin-sensitising molecule ‘adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1’ (APPL1), which contains an NH2-terminal Bin/Amphiphysin/Rvs domain, a central pleckstrin homology domain and a COOH-terminal phosphotyrosine-binding domain, has been shown to modulate inflammatory response in various cell types but its role in regulating beta cell mass and inflammation in type 1 diabetes remains unknown. Thus, we investigated whether APPL1 prevents beta cell apoptosis and inflammation in diabetes.
Methods
Appl1-knockout mice and their wild-type littermates, as well as C57BL/6N mice injected with adeno-associated virus encoding APPL1 or green fluorescent protein, were treated with multiple-low-dose streptozotocin (MLDS) to induce experimental type 1 diabetes. Their glucose metabolism and beta cell function were assessed. The effect of APPL1 deficiency on beta cell function upon exposure to a diabetogenic cytokine cocktail (CKS; consisting of TNF-α, IL-1β and IFN-γ) was assessed ex vivo.
Results
Expression of APPL1 was significantly reduced in pancreatic islets from mouse models of type 1 diabetes or islets treated with CKS. Hyperglycaemia, beta cell loss and insulitis induced by MLDS were exacerbated by genetic deletion of Appl1 but were alleviated by beta cell-specific overexpression of APPL1. APPL1 preserved beta cell mass by reducing beta cell apoptosis upon treatment with MLDS. Mechanistically, APPL1 deficiency potentiate CKS-induced phosphorylation of NFκB inhibitor, α (IκBα) and subsequent phosphorylation and transcriptional activation of p65, leading to a dramatic induction of NFκB-regulated apoptotic and proinflammatory programs in beta cells. Pharmacological inhibition of NFκB or inducible NO synthase (iNOS) largely abrogate the detrimental effects of APPL1 deficiency on beta cell functions.
Conclusions/interpretation
APPL1 negatively regulates inflammation and apoptosis in pancreatic beta cells by dampening the NFκB–iNOS–NO axis, representing a promising target for treating type 1 diabetes.
Similar content being viewed by others


Introduction
Pancreatic beta cell destruction and dysfunction underlie the pathogenesis of type 1 and 2 diabetes, both of which feature aberrant immunity. Recent studies indicate that local production of diabetogenic cytokines (such as IFN-γ, IL-1β and TNF-α) by infiltrated immune cells in pancreatic islets triggers inflammatory and chemotaxis pathways in beta cells, eventually causing defective glucose-stimulated insulin secretion (GSIS) and beta cell apoptosis in diabetes [1, 2]. Inhibition of islet inflammation or IL-1β alleviates hyperglycaemia through improving beta cell function and/or mass in rodent models of diabetes and humans with diabetes [2–5]. The diabetogenic cytokines bind to their corresponding receptors, which in turn recruit distinct scaffold proteins and elicit activation of a cascade of kinases, leading to activation of NFκB and subsequent induction of an apoptotic and inflammatory program dependent on inducible NO synthase (iNOS) [1, 2, 6].
Adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1 (APPL1), which contains an NH2-terminal Bin/Amphiphysin/Rvs domain, a central pleckstrin homology domain and a COOH-terminal phosphotyrosine-binding domain, is an endosomal protein involved in multiple cellular processes, including cell proliferation, survival, apoptosis, metabolism and inflammation [7, 8]. APPL1 is crucial for glucose and cardiovascular homeostasis by mediating both adiponectin and insulin signalling pathways [9–12]. Genetic ablation of Appl1 not only causes insulin and adiponectin resistance in peripheral tissues but also impairs GSIS in beta cells [13–16]. APPL1 is abundantly expressed in beta cells but its expression is decreased in mouse models of dietary-induced obesity and genetically inherited type 2 diabetes [15]. APPL1 facilitates insulin granule exocytosis by enhancing soluble N-ethylmaleimide-sensitive factor activating protein receptor protein expression via an Akt-dependent pathway in beta cells [15]. In humans, expression of islet APPL1 is positively correlated with GSIS [17]. APPL1 is also crucial for mitochondrial metabolism in beta cells [18]. Whole-exome sequencing identified two large families with a high prevalence of diabetes carrying loss-function mutations in APPL1, which appear to impair insulin-mediated Akt activation in hepatocytes [17]. However, it remains unclear whether APPL1 plays any role in beta cell apoptosis and inflammation in type 1 diabetes.
Methods
Reagents and materials
See electronic supplementary materials (ESM) Methods for further details.
Generation, production and titration of adeno-associated virus
Human APPL1 gene with N-terminal FLAG epitope and green fluorescent protein (GFP) were cloned into adeno-associated virus (AAV) vector consisting of modified mouse insulin promoter, namely AAV-mIP2-APPL1 and AAV-mIP2-GFP. The AAV vector, pRep2Cap8 vector and the helper vector were co-transfected into human embryonic kidney (HEK) 293 T cells, followed by purification using polyethylene glycol/aqueous two-phase partitioning and titration by real-time quantitative PCR analysis using primers specifically targeted for genes encoding APPL1 or GFP (ESM Table 1) [19]. AAV-mIP2-APPL1 and AAV-mIP2-GFP plasmids were used as standard curve.
Animal study
Ten- to twelve-week-old male Appl1-KO mice and their wild-type (WT) littermates with C57BL/6N background [9], male C57BL/6N mice (Laboratory Animal Unit, The University of Hong Kong) and 20-week-old female NOD mice (Jackson laboratory, Bar Harbor, MA, USA) were used. The animals were housed in a room with temperature (23 ± 1°C) and light (12 h light-dark cycle) control and had free access to water and diet (unless otherwise noted). The investigators were not blinded to the experimental groups, unless otherwise noted. Hyperglycaemia or diabetes was defined as a random non-fasted blood glucose level >15 mmol/l. All animal experimental protocols were approved by the Animal Ethics Committee of The University of Hong Kong. For the methodologies of multiple-low-dose streptozotocin (MLDS) treatment, AAV injection and GTT, please refer to ESM Methods.
Immunohistochemical and morphological analysis
Pancreases from the different mouse models (including Appl1-KO mice and their WT littermates, female NOD mice and their WT controls, C57BL/6N mice injected with AAV-APPL1 or AAV-GFP) were fixed with 4% wt/vol. paraformaldehyde, embedded in paraffin and cut into sections (5 μm thickness) as described in our previous study [15]. Detailed procedures for immunohistochemical, TUNEL staining and morphological analyses are described in ESM Methods.
Cell culture, cytokine treatment and transfection
Rat INS-1E cells (a kind gift from C. B. Wollheim, University of Geneva, Geneva, Switzerland) and HEK293T cells (ATCC, Manassas, VA, USA) (free of mycoplasma contamination) were cultured in RPMI1640 or DMEM supplemented with 10% vol./vol. FBS, penicillin (100 U/ml) and streptomycin (100 μg/ml), respectively. INS-1E cells were transfected with small interfering RNA (siRNA) or plasmid DNA using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). INS-1E cells or pancreatic islets were treated with cytokine cocktail (CKS), consisting of TNF-α (50 ng/ml), IL-1β (100 ng/ml) and IFN-γ (100 ng/ml), or PBS containing 0.1% wt/vol. fatty-acid-free BSA as vehicle. For details of islet isolation, see ESM Methods.
Pancreatic insulin content
Pancreatic tissues from Appl1-KO mice and WT controls (Fig. 3) and C57BL/6N mice injected with AAV-APPL1 or AAV-GFP (Fig. 8) were homogenised in acid–ethanol (1.5% vol./vol. HCl in 70% vol./vol. ethanol) and the insulin content of the supernatant fraction was measured using the insulin ELISA kit (Antibody and Immunoassay Services, The University of Hong Kong). See ESM Methods for further details.
NO production and monocyte chemotactic protein 1 secretion in the conditioned medium and intracellular caspase-3 activity
Isolated pancreatic islets (50 islets per well) or INS-1E cells were cultured in a 24-well plate and then treated with CKS for 20 h. NO in the conditioned culture medium was measured using a Nitrate/Nitrite Colorimetric Assay Kit (Cayman, Michigan, MO, USA) and monocyte chemotactic protein 1 (MCP1) was measured using Mouse CCL2/JE/MCP1 DuoSet ELISA kit (R&D Systems) following the manufacturers’ instructions. Caspase-3 activity was determined by Caspase-3 Fluorometric Assay Kit (BioVision, Mountain View, CA, USA), following the manufacturer’s instructions. Data was normalised with total protein concentration and expressed as fold change over vehicle control.
DNA binding ability of p65
Nuclear extracts were isolated from INS-1E cells and p65 DNA binding activity was measured using NFκB p50/p65 Transcription Factor Assay Kit (Abcam, Cambridge, UK), following the manufacturer’s instruction. p65 DNA binding activity was normalised with nuclear protein concentration and expressed as fold change over scramble-vehicle controls.
RNA extraction, reverse transcription reaction and real-time quantitative PCR
Total RNA was extracted from pancreatic islets or INS-1E cells using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) and cDNA was synthesised using ImProm-II reverse transcription kit (Promega, Madison, WI, USA). Real-time quantitative PCR was performed using SYBR Green QPCR system (Qiagen, Valencia, CA, USA) on Applied Biosystems StepOnePlus Real-time PCR System (Foster City, CA, USA). Gene-specific primers used for quantitative PCR analysis are listed in ESM Table 1. The expression level of target gene was normalised with Gapdh.
NFκB luciferase assay
INS-1E cells were co-transfected with NFκB firefly luciferase reporter (BD Bioscience, San Jose, CA, USA), pRL-TK renilla reporter (Promega) and siRNA against Appl1 or scramble control using Lipofectamine 3000 for 48 h, followed by treatment with CKS for 20 h. The luciferase activity in the cell lysate was measured using a dual luciferase reporter assay kit (Promega).
Statistical analysis and inclusion/exclusion criteria
All experiments were performed at least three times and results are presented as means ± SEM. Representative images are shown from at least three independent experiments or biological samples. Statistical significance was determined by one-way ANOVA or unpaired Student’s t test; p < 0.05 indicated statistical significance. No inclusion or exclusion criteria were used.
Results
Expression of APPL1 is reduced in pancreatic islets in a mouse model of type 1 diabetes
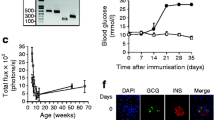
Despite the significantly reduced levels of APPL1 seen in rodent models of type 2 diabetes [15], whether APPL1 expression in beta cells is altered in type 1 diabetes is unknown. Therefore, we examined APPL1 protein expression in isolated pancreatic islets and pancreatic sections from C57BL/6N mice after injection with MLDS for 5 days and from female NOD mice with or without diabetes. Both MLDS-treated mice and diabetic NOD mice exhibited hyperglycaemia when compared with their corresponding controls (Fig. 1a,b). Immunofluorescence staining revealed that protein expression of APPL1 and insulin was downregulated in pancreatic islets from MLDS-treated mice and diabetic NOD mice when compared with islets from vehicle-treated mice and non-diabetic NOD mice, respectively (Fig. 1c). In contrast, glucagon-positive alpha cells were increased in islets of mice treated with MLDS (ESM Fig. 1b). Immunoblotting analysis confirmed the significant reduction of APPL1 in islets isolated from diabetic NOD mice and MLDS-treated mice (Fig. 1d, e).

Expression of APPL1 is decreased in pancreatic islets of type 1 diabetic mouse models. Ten-week-old C57BL/6N mice after treatment with MLDS or vehicle and 20-week-old female NOD mice with or without diabetes were used. (a, b) Fed glucose levels (n = 4 or 5). (c) Immunofluorescence staining for insulin (green) and APPL1 (red) in the pancreatic sections (n = 4). (d, e) Immunoblotting analysis of APPL1 in the isolated islets. For the MLDS experiment, results were obtained on day 10. The relative abundance of APPL1 was measured by densitometric analysis and normalised with GAPDH (n = 4). *p < 0.05, **p < 0.01 and ***p < 0.001 for MLDS vs Vehicle or for Diabetic vs Non-diabetic. Scale bars, 100 μm
Genetic ablation of Appl1 exacerbates the development of MLDS-induced diabetes
To test whether a reduction in APPL1 contributes to the pathogenesis of type 1 diabetes, we injected Appl1-knockout (KO) mice and their WT littermates with MLDS. MLDS-treated Appl1-KO mice displayed a higher fed glucose level when compared with their WT controls on days 10–16 (Fig. 2a). Genetic deletion of Appl1 had no obvious effect on body weight or insulin sensitivity in mice treated with vehicle or MLDS (ESM Fig. 2). Although there was no significant difference in glucose tolerance between vehicle-treated Appl1-KO mice and WT littermates, APPL1 deficiency exacerbated MLDS-induced glucose intolerance (Fig. 2b). The glucose intolerance of the MLDS-treated Appl1-KO mice was, at least in part, due to impaired GSIS (Fig. 2c). In addition, MLDS-treated Appl1-KO mice displayed hypoinsulinaemia (Fig. 2c). Taken together, these findings suggest that Appl1-KO mice are more vulnerable to MLDS-induced beta cell dysfunction and diabetes.

Genetic deletion of Appl1 exacerbates the development of MLDS-induced diabetes. Appl1-KO mice and their WT littermates were injected with MLDS or vehicle. (a) Fed glucose levels (n = 10). (b, c) Serum levels of glucose (b) and insulin (c) during the GTT on day 9 (n = 10). White circles, WT + vehicle; black circles, KO + vehicle; white squares, WT + MLDS; black squares, KO + MLDS.*p < 0.05, KO-MLDS vs WT-MLDS
Appl1-KO mice are more susceptible to MLDS-induced beta cell loss
To investigate the underlying cause of the severe hyperglycaemia in MLDS-treated Appl1-KO mice, we measured pancreatic insulin content and beta cell area. Immunohistochemical staining and ELISA analysis revealed that MLDS treatment significantly reduced pancreatic insulin content and beta cell area in WT controls and that APPL1 deficiency further exacerbated these detrimental effects (Fig. 3a,b). Conversely, alpha cell area was increased in MLDS-treated Appl1-KO mice when compared with WT controls (Fig. 3c). Of note, such a difference was not observed in vehicle-treated mice (Fig. 3c). In situ detection of apoptosis revealed that the percentage of TUNEL-positive beta cells was dramatically increased in Appl1-KO mice treated with MLDS when compared with their WT littermates (Fig. 3d). Noticeably, TUNEL-positive beta cells were virtually undetectable in the vehicle-treated mice (Fig. 3d).

APPL1 deficiency exacerbates MLDS-induced beta cell loss. Pancreases were collected from Appl1-KO mice and WT controls on day 16 after treatment with MLDS or vehicle. (a) Pancreatic insulin content (n = 8). (b) Immunohistochemical staining of insulin in the pancreatic sections and quantification of beta cell area (n = 8). (c) Immunofluorescence staining of glucagon (red) and insulin (green) in the pancreatic sections and quantification of alpha cell area in pancreatic islets (n = 8). (d) Immunofluorescence staining of insulin (red) and TUNEL (green) in the pancreatic sections and quantification of TUNEL-positive beta cells (n = 8). *p < 0.05 and **p < 0.01, MLDS-treated KO vs MLDS-treated WT. Scale bars, 100 μm
To investigate whether the increased susceptibility of Appl1-KO mice to MLDS-induced beta cell loss and diabetes is due to aggravated insulitis, we examined the histology and immunohistology of pancreatic islets from Appl1-KO mice and WT controls. More immune cells, such as macrophages (F4/80 as surface marker of macrophage), infiltrated into the pancreatic islet of Appl1-KO mice, when compared with WT controls, after treatment with MLDS (day 9) (ESM Fig. 3a,b). In addition, the expression of inflammatory factors, such as iNOS, Il-1β and F4/80, was significantly increased in islets isolated from Appl1-KO mice after MLDS treatment, when compared with the expression in islets isolated from WT controls (ESM Fig. 3c,d), indicating that APPL1 deficiency exacerbates MLDS-induced intra-islet inflammation.
APPL1 deficiency potentiates MLDS- and CKS-induced inflammation, apoptosis and NFκB activation in pancreatic islets
MLDS induces beta cell loss via direct DNA alkylation and induction of local production of diabetogenic cytokines by infiltrated immunocytes in the intra-islet environment [20, 21]. Consistent with the findings in the mouse model of type 1 diabetes, APPL1 expression was significantly diminished in islets after stimulation with diabetogenic CKS for 20 h (Fig. 4a). Next, we investigated whether APPL1 deficiency sensitises pancreatic islets to CKS-induced apoptosis in an ex vivo system. Treatment with CKS significantly elicited activation of caspase-3 (a surrogate marker of apoptosis) in islets of WT controls and APPL1 deficiency further augmented the apoptotic effects of CKS (Fig. 4b). Since Appl1-KO mice treated with MLDS exhibited increased insulitis, we measured mRNA levels of chemokines, including Cxcl10 and Mcp1 [also known as Ccl2] (which contribute to the recruitment and activation of immunocytes), in islets isolated from Appl1-KO mice and WT controls exposed to MLDS in an in vivo setting or to CKS in an ex vivo setting. Expression of Cxcl10 and Mcp1 mRNA was undetectable in islets from WT controls and Appl1-KO mice under basal condition (data not shown and Fig. 4c) but APPL1 deficiency significantly augmented the expression of these chemokines upon treatment with MLDS (ESM Fig. 3d) or CKS (Fig. 4c,d). Expression of iNOS and Fas, well-known NFκB targets, was significantly higher in Appl1-KO islets exposed to CKS when compared with islets isolated from WT controls (iNos [also known as Nos2] and Fas mRNA expression is shown in Fig. 4c and iNOS protein expression in Fig. 4e). Exposure to CKS also caused a significant increase in levels of NO (the end-product of iNOS) in Appl1-KO islets, when compared with islets isolated from WT controls (Fig. 4f). APPL1 deficiency also potentiated MLDS-induced Fas expression in pancreatic islets (ESM Fig. 3d). Likewise, siRNA-mediated knockdown of APPL1 by ∼80% potentiated CKS-induced apoptosis (as demonstrated by TUNEL-positive cells, caspase-3 activity and Fas expression), chemokine (Mcp1 and Cxcl10) expression and iNos expression as well as NO formation (ESM Fig. 4).

APPL1 deficiency potentiates CKS-induced expression of chemokines, NO production and apoptosis in islets. (a) Pancreatic islets from 10-week-old male C57BL/6N mice were treated with CKS or vehicle for 20 h. Immunoblotting analysis of APPL1 and GAPDH and the relative abundance of APPL1, determined by densitometric analysis, was normalised with GAPDH (n = 6). (b–f) Pancreatic islets from Appl1-KO mice and their WT littermates were treated with CKS or vehicle for 20 h. Intracellular caspase-3 activity was measured (n = 4) (b). mRNA levels of iNos, Mcp1, Cxcl10 and Fas were quantified by real-time PCR analysis (n = 4) (c). MCP1 secretion in the conditioned medium was measured (n = 5) (d). ND, not detectable. Immunoblotting analysis of iNOS and GAPDH in the islets was performed and the relative abundance of iNOS, normalised with GAPDH is shown (e). Levels of NO in the conditioned medium were measured (n = 4) (f). *p < 0.05, **p < 0.01 and ***p < 0.001 for the indicated comparisons or vs WT
We next investigated how APPL1 inhibits CKS-induced NFκB activation. Knockdown of APPL1 enhanced the activity of NFκB luciferase reporter and enhanced the binding of NFκB subunit p65 to the DNA sequence containing the NFκB-responsive element, upon CKS stimulation (Fig. 5a, b). As expected, CKS induced a marked increase in the phosphorylation of NFkB inhibitor, α (IκBα) at serine 32/36 and p65 at serine 536 in INS-1E cells transfected with scramble control and this induction was further enhanced in beta cells with siRNA-mediated downregulation of Appl1 (Fig. 5c–e). On the other hand, knockdown of APPL1 had no obvious effect on the phosphorylation of c-Jun N-terminal kinase (JNK) and extracellular-signal-regulated kinase (ERK) 1/2 induced by CKS, excluding the possibility that the actions of APPL1 are mediated by these pathways (ESM Fig. 5).

Knockdown of APPL1 enhances CKS-induced NFκB activation in INS-1E cells. (a) INS-1E cells were co-transfected with dual luciferase reporters (NFκB and TK) and siRNA against Appl1 or scramble control for 48 h, followed by CKS treatments for 20 h. Luciferase activity was expressed as fold change over scramble-vehicle (n = 4). (b) DNA binding activity of p65 in siRNA-transfected INS-1E cells treated with CKS or vehicle for 20 h (n = 4). (c) siRNA-transfected INS-1E cells were treated with CKS for indicated time points, followed by immunoblotting using an antibody against APPL1, phospho (p)-IκBα (Ser32/36), IκBα, p-p65 (Ser536), p65 or GAPDH. Representative immunoblot images are shown. (d, e) Densitometric analysis of blot shown in (c) for the relative abundance of p-IκBα and p-p65 (normalised with GAPDH and p65, respectively). White circles, siRNA against scramble; black squares, siRNA against Appl1. *p < 0.05 and **p < 0.01 for indicated comparisons or vs scramble
Inactivation of NFκB or iNOS partially rescues the APPL1-null phenotypes
To test whether the inhibition of NFκB could reverse the increased apoptosis and inflammation in APPL1-deficient beta cells, islets isolated from Appl1-KO mice were pre-treated with the NFκB inhibitor BAY11-7082 for 30 min before exposure to CKS [22, 23]. Real-time PCR analysis revealed that the augmented mRNA expression of chemokines (Mcp1 and Cxcl10), iNos and Fas in islets of Appl1-KO mice was largely reversed by pre-treatment with BAY11-7082 (Fig. 6a, b). These effects of BAY11-7082 were accompanied by reduced NO formation and caspase-3 activity (Fig. 6c, d).

Inhibition of NFκB partially reverses the pro-apoptotic and proinflammatory effects of APPL1 deficiency on pancreatic islets. Pancreatic islets isolated from Appl1-KO mice and their WT controls were pre-treated with BAY11-7082 (BAY) or DMSO as vehicle control for 30 min, followed by treatment with CKS for 20 h. (a) Real-time PCR analysis of Mcp1, iNos, Cxcl10 and Fas mRNA levels in the islets (n = 4). (b, c) MCP1 secretion (b) and NO production (c) in the conditioned medium (n = 4). (d) Activity of caspase-3 in the treated islets (n = 4–6). ND, not detectable. *p < 0.05, **p < 0.01 and ***p < 0.001 for indicated comparisons
Since the deteriorative effect of NFκB on beta cells is mainly mediated via the iNOS–NO pathway [24], we next examined whether pharmacological inhibition of iNOS could reverse the augmented apoptosis and inflammation in APPL1-deficient beta cells. Pre-treatment with the iNOS inhibitor L-NIL largely abolished CKS-induced NO formation in islets isolated from Appl1-KO mice and WT controls (ESM Fig. 6a). The elevation of caspase-3 activity in APPL1-null islets and control islets in response to CKS treatment was attenuated by treatment with L-NIL (ESM Fig. 6b).
Recombinant AAV-mediated beta cell-specific expression of APPL1 attenuates MLDS-induced beta cell loss and diabetes
To test whether beta cell-specific overexpression of APPL1 relieves MLDS-induced diabetes, we gave C57BL/6N mice an intraperitoneal injection of AAV encoding human APPL1 or GFP under the control of modified mouse insulin promoter 2. Exogenous FLAG-tagged APPL1 was detected in islets but not in exocrine cells of mice injected with AAV-APPL1 (Fig. 7a). Immunofluorescent staining revealed that the reduction of APPL1 in beta cells induced by treatment with MLDS was partially rectified by AAV-mediated expression of APPL1 (Fig. 7b). Random non-fasted blood glucose and glucose tolerance were similar in AAV-APPL1 and AAV-GFP mice treated with vehicle, whereas MLDS-induced hyperglycaemia and glucose intolerance were markedly alleviated by beta cell-specific overexpression of APPL1 (Fig. 7c,d). The improvement in glucose homeostasis in AAV-APPL1 mice treated with MLDS was associated with increased pancreatic insulin content and beta cell area (Fig. 8a,b). The alpha cell areas in islets of vehicle-treated AAV-APPL1 and AAV-GFP mice were comparable (Fig. 8c). The increase in alpha cell area induced by MLDS was partially abolished by overexpression of APPL1 (Fig. 8c). Furthermore, beta cell apoptosis induced by MLDS was decreased in AAV-APPL1 mice when compared with AAV-GFP controls (Fig. 8d).

AAV-mediated beta cell-specific overexpression of APPL1 relieves MLDS-induced diabetes. C57BL/6N mice infected with AAV encoding APPL1 or GFP were injected intraperitoneally with MLDS or vehicle. (a) Immunohisto-chemical staining with an anti-FLAG antibody in pancreatic sections of the AAV-infected mice treated with vehicle. (b) Immunofluorescence staining of insulin (red) and APPL1 (green) in the pancreatic sections (day 16). (c) Fed glucose level (MLDS group n = 10, vehicle group n = 5). (d) Glucose levels during the GTT on day 9 (n = 5). White circles, GFP–vehicle; black circles, APPL1–vehicle; white squares, GFP–MLDS; black squares, APPL1–MLDS. *p < 0.05 for AAV-GFP–MLDS vs AAV-APPL1–MLDS. Scale bars, 100 μm

Beta cell-specific overexpression of APPL1 reduces MLDS-induced beta cell loss. Pancreases isolated from AAV-injected mice were treated with MLDS or vehicle on day 16. (a) Pancreatic insulin content (n = 10). (b) Immunohisto-chemical staining of insulin in the pancreatic sections and quantification of beta cell area (n = 8). (c) Immunofluorescence staining for glucagon (red) and insulin (green), and quantification of alpha cell area (n = 8). (d) TUNEL staining (green) and immunofluorescence staining of insulin (red), and quantitative analysis of TUNEL-positive beta cells in the MLDS-treated mice (n = 8). *p < 0.05 for indicated comparisons. Scale bars, 100 μm
Discussion
Pancreatic beta cell loss is a major contributor to the pathogenesis of both type 1 and type 2 diabetes. Our study shows that APPL1 protects beta cells from diabetogenic agents at least in part by dampening the NFκB–iNOS–NO pathway. The deleterious effects of diabetogenic agents on beta cell functions are exacerbated by APPL1 deficiency and are alleviated by beta cell overexpression of APPL1.
In pancreatic beta cells, activation of NFκB induces a cluster of genes related to inflammation, chemotaxis and apoptosis, leading to diabetes [6]. Inactivation of NFκB by overexpression of a degradation-resistant IκBα in beta cells markedly attenuates MLDS-induced diabetes in vivo and CKS-induced apoptosis in vitro [25]. In contrast, constitutive activation of NFκB leads to insulitis and immune-mediated diabetes [25, 26]. The pro-apoptotic effect of NFκB in beta cells is partially mediated by the iNOS–NO pathway [27]. Indeed, genetic ablation of iNOS abrogates MLDS-induced diabetes [28] and, vice versa, transgenic expression of iNOS in pancreatic beta cells results in beta cell loss, hypoinsulinaemia and diabetes and these phenotypes can be reversed by treatment with an iNOS inhibitor [29]. APPL1 has been reported to control NFκB activity in a cell type- and stimulus-specific manner [30–32]. In HEK293T cells, APPL1 appears to enhance basal NFκB activity by promoting nuclear localisation of p65 via stabilisation of the non-canonical NFκB-inducing kinase (NIK) [30]. However, APPL1 mediates the anti-inflammatory actions of adiponectin on endothelial cells and macrophages by attenuating NFκB activation via an unknown mechanism [31, 33]. In the present study, we showed that APPL1 negatively regulates the canonical NFκB pathway in beta cells. The upregulation of NFκB-responsive genes in APPL1-deficient beta cells upon exposure to CKS is due to increased phosphorylation of IκBα at serine 32/36 and p65 at serine 536. Phosphorylation of IκBα at serine 32/36 leads to proteasomal degradation of IκBα, which in turn allows translocation of NFκB from cytoplasm to the nucleus where NFκB initiates the transcription of its target genes [34]. On the other hand, phosphorylation of p65 at serine 536 induces conformational changes that can promote the transcriptional activity of NFκB and/or reduce the nuclear export of NFκB by inhibiting the interaction between IκBα and NFκB [34]. Although pharmacological inhibition of NFκB or iNOS largely abolishes the detrimental effects of APPL1 deficiency on beta cells, we cannot exclude the involvement of other signalling pathways, such as signal transducer and activator of transcription 1 (STAT1, the major downstream target of IFN-γ [35]), in the anti-inflammatory and anti-apoptotic actions of APPL1. Since APPL1 is a key downstream mediator of adiponectin signalling and adiponectin protects beta cells from apoptosis and inflammation, it is possible that the protective effects of APPL1 against MLDS-induced diabetes may be related to its potentiating effects on adiponectin activity [36, 37]. Taken together, our results support the notion that APPL1 protects beta cells from apoptosis and inflammation by attenuating, in part, CKS-induced NFκB activation in beta cells.
Streptozotocin (STZ) enters beta cells via GLUT2 and destroys the cells via DNA alkylation. Our previous study indicated that expression of Glut2 in islets from Appl1-KO mice and WT is similar [15]. This excludes the possibility that the increased susceptibility of Appl1-KO mice to MLDS-induced beta cell damage is due to a change in GLUT2 expression. Apart from its direct cytotoxic effect, MLDS triggers local islet inflammation and the release of proinflammatory cytokines (such as TNF-α, IL-1β and IFN-γ), in part through the recruitment of immunocytes such as macrophages and T cells [20, 21]. Indeed, depletion of immune cells or inhibition of cytokines has been shown to alleviate MLDS-induced beta cell loss and diabetes [38, 39]. In our study, MLDS-induced diabetes and insulitis were exacerbated in Appl1-KO mice. Such change is associated with augmented expression of NFκB-responsive genes, resulting in induction of local inflammation and apoptosis in pancreatic islets. This notion is further supported by the in vitro experiments showing that islets lacking APPL1 or INS-1E cells with decreased expression of APPL1 secreted more MCP1 and NO and exhibited increased apoptosis upon stimulation with CKS.
APPL1 prevents stress-induced apoptosis in neuronal cells, cardiomyocytes and endothelial cells [31, 40, 41]. APPL1 deficiency induces apoptosis and developmental defects in xenopus pancreas and zebrafishes as a result of diminished Akt activity in the early endosome [42, 43]. However, these defects are not observed in Appl1-KO mice fed with a standard chow or a high-fat diet [15, 44]. In the present study, beta cell area was strikingly reduced in Appl1-KO mice treated with MLDS, whereas beta cell-specific overexpression of APPL1 had the opposite effect. Noticeably, transgenic expression of constitutive active form of Akt in beta cells prevents STZ-induced diabetes in mice [45]. However, whether Akt is involved in the anti-apoptotic effects of APPL1 on beta cells in diabetic conditions warrants further investigation.
APPL1 expression is reduced in some inflammatory conditions. For instance, treatment with lipopolysaccharide, resistin and TNF-α leads to reduction of APPL1 in macrophages, hypothalamic cells and myotubes, respectively [46–48]. In macrophages, lipopolysaccharide-dependent proteasomal degradation of APPL1 is mediated via the mitogen-activated protein kinase 1/2–ERK1/2 pathway [46]. We showed here that beta cell APPL1 is reduced in islets treated with CKS and islets from mouse models of chemical-induced and genetically inherited type 1 diabetes (both feature intra-islet inflammation), suggesting that reduced expression of APPL1 may contribute to the pathogenesis of this inflammatory disease.
Our results showed that AAV-mediated overexpression of APPL1 in beta cells partially ameliorates MLDS-induced diabetes. The modest effect of APPL1 overexpression may be due to moderated increase of exogenous APPL1 in the beta cells and/or incomplete transduction of AAV to beta cells [49]. Although intra-islet inflammation also contributes to the development of diabetes in the MLDS model, the autoimmune feature and pathogenesis are distinct from human type 1 diabetes [50]. Therefore, further investigation to determine whether transgenic overexpression of APPL1 in beta cells of diabetic NOD mice (which exhibit aberrant immune phenotypes as in human type 1 diabetes) prevents beta cell apoptosis and inflammation is warranted.
In summary, our study reveals that APPL1 protects beta cells from diabetogenic agent (STZ and CKS)-induced inflammation and apoptosis by diminishing NFκB activation. In mouse models of type 1 diabetes, a reduction in APPL1 expression increases the susceptibility of beta cells to local islet inflammation and subsequent beta cell death (ESM Fig. 7). Thus, therapeutic strategies aimed at restoring beta cell APPL1 expression may represent a promising approach to preserve beta cell mass and function in diabetes.
Abbreviations
- AAV:
-
Adeno-associated virus
- APPL1:
-
Adaptor protein, phosphotyrosine interacting with PH domain and leucine zipper 1
- CKS:
-
Cytokine cocktail
- ERK:
-
Extracellular-signal-regulated kinases
- GFP:
-
Green fluorescent protein
- GSIS:
-
Glucose-stimulated insulin secretion
- IκBα:
-
NFκB inhibitor, α
- iNOS:
-
Inducible NO synthase
- KO:
-
Knockout
- MCP1:
-
Monocyte chemotactic protein 1
- MLDS:
-
Multiple-low-dose streptozotocin
- siRNA:
-
Small interfering RNA
- STZ:
-
Streptozotocin
- WT:
-
Wild-type
References
Cnop M, Welsh N, Jonas J-C, Jörns A, Lenzen S, Eizirik DL (2005) Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54:S97–S107
Imai Y, Dobrian AD, Morris MA, Nadler JL (2013) Islet inflammation: a unifying target for diabetes treatment? Trends Endocrinol Metab 24:351–360
Mandrup-Poulsen T, Pickersgill L, Donath MY (2010) Blockade of interleukin 1 in type 1 diabetes mellitus. Nat Rev Endocrinol 6:158–166
Moran A, Bundy B, Becker DJ et al (2013) Interleukin-1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double-blind, placebo-controlled trials. Lancet 381:1905–1915
Larsen CM, Faulenbach M, Vaag A et al (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356:1517–1526
Eizirik DL, Colli ML, Ortis F (2009) The role of inflammation in insulitis and β-cell loss in type 1 diabetes. Nat Rev Endocrinol 5:219–226
Cheng KK, Lam KS, Wang B, Xu A (2014) Signaling mechanisms underlying the insulin-sensitizing effects of adiponectin. Best Pract Res Clin Endocrinol Metab 28:3–13
Miaczynska M, Christoforidis S, Giner A et al (2004) APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell 116:445–456
Wang Y, Cheng KK, Lam KS et al (2011) APPL1 counteracts obesity-induced vascular insulin resistance and endothelial dysfunction by modulating the endothelial production of nitric oxide and endothelin-1 in mice. Diabetes 60:3044–3054
Cheng KK, Lam KS, Wang Y et al (2007) Adiponectin-induced endothelial nitric oxide synthase activation and nitric oxide production are mediated by APPL1 in endothelial cells. Diabetes 56:1387–1394
Park M, Wu D, Park T et al (2013) APPL1 transgenic mice are protected from high-fat diet-induced cardiac dysfunction. Am J Physiol Endocrinol Metab 305:E795–E804
Mao X, Kikani CK, Riojas RA et al (2006) APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol 8:516–523
Cheng KK, Iglesias MA, Lam KS et al (2009) APPL1 potentiates insulin-mediated inhibition of hepatic glucose production and alleviates diabetes via Akt activation in mice. Cell Metab 9:417–427
Cleasby ME, Lau Q, Polkinghorne E et al (2011) The adaptor protein APPL1 increases glycogen accumulation in rat skeletal muscle through activation of the PI3-kinase signalling pathway. J Endocrinol 210:81–92
Cheng KK, Lam KS, Wu D et al (2012) APPL1 potentiates insulin secretion in pancreatic β cells by enhancing protein kinase Akt-dependent expression of SNARE proteins in mice. Proc Natl Acad Sci U S A 109:8919–8924
Ryu J, Galan AK, Xin X et al (2014) APPL1 potentiates insulin sensitivity by facilitating the binding of IRS1/2 to the insulin receptor. Cell Rep 7:1227–1238
Prudente S, Jungtrakoon P, Marucci A et al (2015) Loss-of-function mutations in APPL1 in familial diabetes mellitus. Am J Hum Genet 97:177–185
Wang C, Li X, Mu K et al (2013) Deficiency of APPL1 in mice impairs glucose-stimulated insulin secretion through inhibition of pancreatic β cell mitochondrial function. Diabetologia 56:1999–2009
Li X, Cheng KK, Liu Z et al (2016) The MDM2-p53-pyruvate carboxylase signalling axis couples mitochondrial metabolism to glucose-stimulated insulin secretion in pancreatic β-cells. Nat Commun 7:11740
Rossini AA, Williams RM, Appel MC, Like AA (1978) Complete protection from low-dose streptozotocin-induced diabetes in mice. Nature 276:182–184
Like AA, Rossini AA (1976) Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science 193:415–417
Tipoe GL, Leung T-M, Liong E et al (2006) Inhibitors of inducible nitric oxide (NO) synthase are more effective than an NO donor in reducing carbon-tetrachloride induced acute liver injury. Histol Histopathol 21:1157–1165
Boni-Schnetzler M, Thorne J, Parnaud G et al (2008) Increased interleukin (IL)-1β messenger ribonucleic acid expression in β-cells of individuals with type 2 diabetes and regulation of IL-1β in human islets by glucose and autostimulation. J Clin Endocrinol Metab 93:4065–4074
Yuan H-D, Chung S-H (2010) Protective effects of fermented ginseng on streptozotocin-induced pancreatic β-cell damage through inhibition of NF-κB. Int J Mol Med 25:53–58
Eldor R, Yeffet A, Baum K et al (2006) Conditional and specific NF-κB blockade protects pancreatic β cells from diabetogenic agents. Proc Natl Acad Sci U S A 103:5072–5077
Salem HH, Trojanowski B, Fiedler K et al (2014) Long-term IKK2/NF-κB signaling in pancreatic beta-cells induces immune-mediated diabetes. Diabetes 63:960–975
Mandrup-Poulsen T (2001) β-cell apoptosis: stimuli and signaling. Diabetes 50:S58
Flodstrom M, Tyrberg B, Eizirik DL, Sandler S (1999) Reduced sensitivity of inducible nitric oxide synthase-deficient mice to multiple low-dose streptozotocin-induced diabetes. Diabetes 48:706–713
Takamura T, Kato I, Kimura N et al (1998) Transgenic mice overexpressing type 2 nitric-oxide synthase in pancreatic β cells develop insulin-dependent diabetes without insulitis. J Biol Chem 273:2493–2496
Hupalowska A, Pyrzynska B, Miaczynska M (2012) APPL1 regulates basal NF-κB activity by stabilizing NIK. J Cell Sci 125:4090–4102
Chandrasekar B, Boylston WH, Venkatachalam K, Webster NJ, Prabhu SD, Valente AJ (2008) Adiponectin blocks interleukin-18-mediated endothelial cell death via APPL1-dependent AMP-activated protein kinase (AMPK) activation and IKK/NF-κB/PTEN suppression. J Biol Chem 283:24889–24898
Yeo JC, Wall AA, Luo L, Condon ND, Stow JL (2016) Distinct roles for APPL1 and APPL2 in regulating Toll-like receptor 4 signaling in macrophages. Traffic 17:1014–1026
Tian L, Luo N, Zhu X, Chung BH, Garvey WT, Fu Y (2012) Adiponectin-AdipoR1/2-APPL1 signaling axis suppresses human foam cell formation: differential ability of AdipoR1 and AdipoR2 to regulate inflammatory cytokine responses. Atherosclerosis 221:66–75
Huang B, Yang XD, Lamb A, Chen LF (2010) Posttranslational modifications of NF-κB: another layer of regulation for NF-κB signaling pathway. Cell Signal 22:1282–1290
Moore F, Naamane N, Colli ML et al (2011) STAT1 is a master regulator of pancreatic β-cell apoptosis and islet inflammation. J Biol Chem 286:929–941
Rakatzi I, Mueller H, Ritzeler O, Tennagels N, Eckel J (2004) Adiponectin counteracts cytokine- and fatty acid-induced apoptosis in the pancreatic β-cell line INS-1. Diabetologia 47:249–258
Jian L, Su YX, Deng HC (2013) Adiponectin-induced inhibition of intrinsic and extrinsic apoptotic pathways protects pancreatic β-cells against apoptosis. Horm Metab Res 45:561–566
Herold KC, Montag AG, Fitch FW (1987) Treatment with anti-T-lymphocyte antibodies prevents induction of insulitis in mice given multiple doses of streptozocin. Diabetes 36:796–801
Maier B, Ogihara T, Trace AP et al (2010) The unique hypusine modification of eIF5A promotes islet β cell inflammation and dysfunction in mice. J Clin Invest 120:2156–2170
Park M, Youn B, Zheng XL, Wu D, Xu A, Sweeney G (2011) Globular adiponectin, acting via AdipoR1/APPL1, protects H9c2 cells from hypoxia/reoxygenation-induced apoptosis. PLoS One 6:e19143
Wang YB, Wang JJ, Wang SH et al (2012) Adaptor protein APPL1 couples synaptic NMDA receptor with neuronal prosurvival phosphatidylinositol 3-kinase/Akt pathway. J Neurosci 32:11919–11929
Wen L, Yang Y, Wang Y, Xu A, Wu D, Chen Y (2010) Appl1 is essential for the survival of Xenopus pancreas, duodenum, and stomach progenitor cells. Dev Dyn 239:2198–2207
Schenck A, Goto-Silva L, Collinet C et al (2008) The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell 133:486–497
Tan Y, You H, Wu C, Altomare DA, Testa JR (2010) Appl1 is dispensable for mouse development, and loss of Appl1 has growth factor-selective effects on Akt signaling in murine embryonic fibroblasts. J Biol Chem 285:6377–6389
Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA (2001) Islet β cell expression of constitutively active Akt1/PKB α induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest 108:1631–1638
Chau TL, Goktuna SI, Rammal A et al (2015) A role for APPL1 in TLR3/4-dependent TBK1 and IKKepsilon activation in macrophages. J Immunol 194:3970–3983
Benomar Y, Amine H, Crepin D et al (2016) Central Resistin/TLR4 impairs adiponectin signaling contributing to insulin and FGF21 resistance. Diabetes 65:913–926
Sente T, Van Berendoncks AM, Fransen E, Vrints CJ, Hoymans VY (2016) Tumor necrosis factor-α impairs adiponectin signalling, mitochondrial biogenesis and myogenesis in primary human myotubes cultures. Am J Phys Heart Circ Phys 310:H1164–H1175
Prasad KM, Yang Z, Bleich D, Nadler JL (2000) Adeno-associated virus vector mediated gene transfer to pancreatic β cells. Gene Ther 7:1553–1561
Kolb H (1987) Mouse models of insulin dependent diabetes: low-dose streptozocin-induced diabetes and nonobese diabetic (NOD) mice. Diabetes Metab Rev 3:751–778
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Data availability
The data generated during and/or analysed during the current study are available from the corresponding authors on reasonable request.
Funding
This work was supported by the General Research Fund (782612) from the Research Grants Council of Hong Kong, the National Science Foundation of China (81270881) and a matching grant for State Key Laboratory of Pharmaceutical Biotechnology from the University of Hong Kong.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All the authors contributed substantially to the conception and design of this study and to the acquisition and analysis of data. All authors participated in drafting the article and gave final approval of the version to be published. KKYC is the guarantor of this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 2.15 mb)
Rights and permissions
About this article

Cite this article
Jiang, X., Zhou, Y., Wu, K.K.L. et al. APPL1 prevents pancreatic beta cell death and inflammation by dampening NFκB activation in a mouse model of type 1 diabetes. Diabetologia 60, 464–474 (2017). https://doi.org/10.1007/s00125-016-4185-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4185-z