
Abstract
Aims/hypothesis
Progression to type 1 diabetes in children and adolescents is not uniform. Based on individual genetic background and environment, islet autoimmunity may develop at variable age, exhibit different autoantibody profiles and progress to clinical diabetes at variable rates. Here, we aimed to quantify the qualitative dynamics of sequential islet autoantibody profiles in order to identify longitudinal patterns that stratify progression rates to type 1 diabetes in multiple-autoantibody-positive children.
Methods
Qualitative changes in antibody status on follow-up and progression rate to diabetes were analysed in 88 children followed from birth in the prospective BABYDIAB study who developed multiple autoantibodies against insulin (IAA), GAD (GADA), insulinoma-associated antigen-2 (IA-2A) and/or zinc transporter 8 (ZnT8A). An algorithm was developed to define similarities in sequential autoantibody profiles and hierarchical clustering was performed to group children with similar profiles.
Results
We defined nine clusters that distinguished children with respect to their sequential profiles of IAA, GADA, IA-2A and ZnT8A. Progression from first autoantibody appearance to clinical diabetes between clusters ranged from 6% (95% CI [0, 16.4]) to 73% (28.4, 89.6) within 5 years. Delayed progression was observed in children who were positive for only two autoantibodies, and for a cluster of 12 children who developed three or four autoantibodies but were IAA-negative in their last samples, nine of whom lost IAA positivity during follow-up. Among all children who first seroconverted to IAA positivity and developed at least two other autoantibodies (n = 57), the 10 year risk of diabetes was 23% (0, 42.9) in those who became IAA-negative during follow-up compared with 76% (58.7, 85.6) in those who remained IAA-positive (p = 0.004).
Conclusions/interpretation
The novel clustering approach provides a tool for stratification of islet autoantibody-positive individuals that has prognostic relevance, and new opportunities in elucidating disease mechanisms. Our data suggest that losing IAA reactivity is associated with delayed progression to type 1 diabetes in multiple-islet-autoantibody-positive children.
Similar content being viewed by others


Introduction
Type 1 diabetes is a chronic autoimmune disease with increasing incidence, particularly in young children [1]. The development of type 1 diabetes is associated with humoral and cellular immune responses against antigens expressed in the insulin-producing beta cells of the islets of Langerhans within the pancreas [2]. Although the aetiology of the disease is still uncertain [3], an interplay of genetic predisposition, causing a defective immune regulation, and environmental influences, such as infant diet [4], viruses [5] and/or other still poorly defined factors [6], is thought to contribute to the initiation and continuation of an autoimmune process that leads to destruction of beta cells and, consequently, clinical diabetes [2]. For more than two decades, prospective cohort studies starting at birth have examined the development of islet autoimmunity and diabetes and amassed invaluable data on the natural history of type 1 diabetes [7–10]. We are now at the stage where we can apply refined mathematical modelling using longitudinal data to elucidate complex interactions and mechanisms in the pathogenesis of the disease.
Recent findings suggest that children who develop autoantibodies against multiple beta cell autoantigens will almost inevitably develop clinical diabetes, although the progression rate may vary from months up to many years [11]. Identifying features that stratify this progression could be of benefit for prediction and prevention studies, and may help to identify mechanisms accelerating or delaying diabetes onset. With respect to islet autoantibodies, some of these features are known [12–21]. However, the analysis of longitudinal profiles, which might picture disease pathogenesis more precisely, remains challenging. In particular, there is a need for methods addressing complex temporal interactions between multiple disease-associated factors (e.g. autoimmune, genetic, metabolic and environmental factors) as well as their qualitative and quantitative changes over time. Towards this aim, we provide a mathematical model quantifying similarities of sequential islet autoantibody profiles in multiple-autoantibody-positive children from the prospective BABYDIAB study cohort [7, 22]. This method allowed us to cluster children with similar longitudinal profiles and identify a subgroup with markedly delayed progression of islet autoimmunity to clinical type 1 diabetes among those developing a broad autoantibody response against three or more beta cell autoantigens.
Methods
Study cohort, participants and samples
The study was performed in children from the BABYDIAB study, a longitudinal study examining the natural history of islet autoimmunity and type 1 diabetes in 1650 children born to a mother or a father with type 1 diabetes [7, 22]. Recruitment began in 1989 and ended in 2000. All children were recruited from Germany, and 97% of included families are German and white. Venous blood samples were obtained from children at study visits scheduled at 9 months and 2, 5, 8, 11, 14, 17 and 20 years of age. Islet autoantibodies (autoantibodies against insulin [IAA], GAD [GADA], insulinoma-associated antigen-2 [IA-2A], and zinc transporter 8 [ZnT8A]) were measured in samples taken at all scheduled visits. If a child had a positive autoantibody finding, the family was asked to provide a sample for confirmation of autoantibody status within 6 months and provide further samples yearly. An OGTT was performed yearly in islet autoantibody-positive children. Families were asked to report the occurrence of symptoms of diabetes. Diabetes onset was defined according to ADA criteria [23]. By August 2015, the median follow-up time from birth to the last sample was 14.1 years (interquartile range [IQR] 8.0–18.0 years). In total, 170 children developed IAA, GADA, IA-2A and/or ZnT8A that were positive in at least two consecutive blood samples. These children were followed from seroconversion for a median of 8.5 years (IQR 4.5–12.6 years). Of these 170 children, 88 (52%) had developed two or more of the islet autoantibodies and 60 (35%) had developed diabetes. Longitudinal autoantibody profiles of the 88 multiple-autoantibody-positive children were analysed in the current study. The BABYDIAB study was approved by the Ethics Committee of Bavaria, Germany (Bayerische Landesärztekammer number 95357). All families gave written informed consent to participate in the study. Investigations were carried out in accordance with the principles of the Declaration of Helsinki, as revised in 2008.
Islet autoantibody measurements
Levels of IAA, GADA, IA-2A and ZnT8A were determined using radio-binding assays as previously described [22, 24]. The upper limit of normal for each assay was determined using Q–Q plots and corresponded to the 99th percentile of 836 control children. Children were considered islet autoantibody-positive when two consecutive serum samples were positive. Autoantibody assays were evaluated by the Diabetes Antibody Standardization Program, and performances are shown as laboratory 121 in published reports [25–27].
Genotyping
HLA class II alleles HLA-DRB1 and HLA-DQB1 were determined using PCR-amplified DNA and non-radioactive sequence-specific oligonucleotide probes, as previously described [28].
Clustering algorithm for longitudinal autoantibody profiles
We developed an algorithm to define a distance measure between sequential autoantibody profiles that allows accounting for qualitative changes in antibody status over time (Fig. 1). The distance measure was used for clustering of prospectively followed children based on similarity of their longitudinal islet autoantibody profiles. The source code of the algorithm is provided in the electronic supplementary material (ESM).

Algorithmic workflow to define a distance measure for the clustering of longitudinal autoantibody profiles. (a) For each time point and each individual, quantitative autoantibody levels were transformed to binary data (positive = 1, negative = 0) and identical successive profiles deleted in order to generate a compressed matrix of qualitative changes in autoantibody profiles over time. (b) For each pair of children, the compressed matrices were compared and the number of identical binary entries in each pair of rows written into a matrix of row similarities (i.e. first row of child A vs rows of child B [purple lines/numbers], second row of child A vs rows of child B [blue lines/numbers], third row of child A vs rows of child B [green lines/numbers]). The highest-scoring path is defined from top left to bottom right of this matrix (red numbers with grey background colour). (c) Uniformly distributed weights in the interval [0.25, 1] are used to obtain a score (S) by calculating the weighted average of the similarity values on the highest-scoring path. Scores are further transformed into distance measures for all pairs of children
First, the algorithm transforms longitudinal quantitative autoantibody levels of IAA, GADA, IA-2A and ZnT8A into binary vectors based on thresholds for positivity of the autoantibody assays (Fig. 1a). Removing identical successive profiles then leads to one compressed matrix per child of qualitative changes in autoantibody profiles over time. Second, the compressed matrices are compared between all pairs of children by counting the number of identical binary entries in each pair of rows of the respective matrices, i.e. the non-normalised Hamming similarity [29] between the respective rows, and numbers/similarity values are written into a matrix of row similarities (Fig. 1b). All possible paths from top left of this matrix (representing the similarity between first autoantibody profiles of two children) to bottom right (representing similarity between last profiles) are then searched by going down and/or right only, and the path with the highest sum of entries is selected as the highest-scoring path. Third, the similarity values on the highest-scoring path are used to define a score (S) for the respective pair of children by taking a weighted average of the values on the highest-scoring path (Fig. 1c). Weights are allocated to the entries on the scoring path to ensure that similar endpoints are assigned higher scores. This is achieved by choosing uniformly distributed numbers from the interval [0.25, 1]. The latest similarity value (bottom right in the matrix of row similarities) is assigned the highest weight while the earliest value (top left in matrix) is assigned the lowest weight. Finally, the score S is transformed into a distance measure via the equation \( d\left({x}_i,{x}_j\right)=\sqrt{S\left({x}_i,{x}_i\right)+S\left({x}_j,{x}_j\right)-2S\left({x}_i,{x}_j\right)} \).
To determine distinct clusters of children based on their longitudinal autoantibody profiles, the distance measures calculated this way for all pairs of children were used for hierarchical clustering using the unweighted pair group method with arithmetic mean (UPGMA) method [30].
Statistical analysis
Kaplan–Meier survival analysis was used to examine progression from islet autoantibody seroconversion to type 1 diabetes. The period from the age of seroconversion to the age at diagnosis of diabetes or the age at last contact in non-diabetic children was used as the event time. Analysis considered censoring for losses to follow-up. The logrank and likelihood-ratio tests were used to compare progression to diabetes between groups. All algorithms were implemented in MATLAB version R2012b. Statistical analyses were performed using R version 3.0.2 (http://www.R-project.org) and the IBM SPSS Statistics software (version 22.0, Chicago, IL, USA).
Results
Clustering algorithm applied on BABYDIAB cohort
The clustering algorithm was applied to the longitudinal autoantibody profiles of 88 children from the prospective BABYDIAB cohort who developed multiple islet autoantibodies. Branches of the resulting dendrogram (Fig. 2a) were used to determine cluster sizes such that reasonable statistical comparisons between clusters were possible. Based on this approach, children were grouped into nine clusters of two to 18 children with similar sequential autoantibody profiles (Fig. 2b). Detailed autoantibody profiles are shown for each child in ESM Fig. 1. Characteristics of the clusters are shown in ESM Table 1.

Hierarchical clustering results. (a) Dendrogram showing the results of the hierarchical clustering analysis for 88 children who developed multiple islet autoantibodies. Each column of the heat map shows the percentage of positive probes on follow-up for the respective autoantibody for one child (red = 100%, white = 0%). (b) Results of the hierarchical clustering analysis after grouping children into nine autoantibody clusters. For each cluster, the compressed qualitative autoantibody profiles of all children are displayed. Sequential profiles are ordered by increasing age of the child, from bottom to top. For each autoantibody profile, the status of IAA, GADA, IA-2A and ZnT8A (order from left to right) is displayed. Autoantibody-positive status is indicated in black, and autoantibody-negative status in grey. The compressed profiles are framed indicating whether the child has developed diabetes on follow-up (red frame) or remained diabetes free (blue frame)
Autoantibody clusters and progression to diabetes
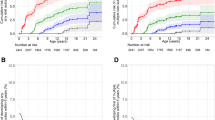
Progression from islet autoantibody seroconversion to clinical type 1 diabetes or last contact in non-diabetic children was effectively stratified by the autoantibody clusters (likelihood-ratio test, p = 0.003, compared with null model without predictors; Fig. 3). The 5 year risk of diabetes ranged from 6% (95% CI 0, 16.4) for children in cluster 1 up to 73% (28.4, 89.6) for children in cluster 6 (Fig. 3; ESM Table 1). Notably, two clusters (cluster 1 and cluster 4) showed considerably delayed progression compared with all remaining clusters (Fig. 3). While most children in cluster 1 (n = 14; 78%) were characterised by not developing more than two autoantibody specificities, all 12 children in cluster 4 developed at least three and the majority (75%) all four autoantibodies (Fig. 2b; ESM Fig. 1). The sequential autoantibody profiles of cluster 4 had in common a lack of IAA at the last observation. Nine (75%) children in this cluster developed IAA and then lost positivity during follow-up. The remaining three children in cluster 4 were IAA-negative throughout. Similar to the longitudinal change of IAA status in cluster 4, most children in cluster 3 (n = 7; 78%) developed all four autoantibodies and then lost GADA positivity during follow-up. Children in cluster 3 progressed faster to diabetes than those in cluster 4 (p = 0.02; Fig. 3), suggesting that in children with three or four positive islet autoantibodies, losing IAA positivity was more strongly associated with a delay in progression than losing GADA. Moreover, children in the closely related clusters 5, 6 and 7 who developed all four autoantibodies without losing IAA on follow-up progressed significantly faster to diabetes than those in cluster 4 (p = 0.004).

Progression to type 1 diabetes with respect to autoantibody clusters. Cumulative diabetes-free survival is shown for multiple-autoantibody-positive children stratified by clusters. Coloured curves display clusters including more than ten children, and grey curves display clusters including five to ten children. Cluster 5, including only two children, is not shown. Numbers below the x-axis indicate number of diabetes-free children remaining on follow-up
Loss of IAA positivity on follow-up and progression to diabetes
Based on the variation in progression to diabetes among clusters of children with three or four positive islet autoantibodies, and the observation that most children in the rather slowly progressing cluster 4 became IAA-negative during follow-up, we hypothesised that losing IAA positivity could be associated with a delay in the development of clinical type 1 diabetes among multiple-autoantibody-positive children. In order to test this hypothesis, all children who first seroconverted to IAA positivity and developed at least two other islet autoantibodies (n = 57) were stratified according to IAA status on follow-up and analysed for progression to diabetes (Fig. 4). Supporting our assumption, the 10 year risk of diabetes from seroconversion was 23% (0, 42.9) in those who became IAA-negative during follow-up compared with 76% (58.7, 85.6) in those who remained IAA-positive (logrank test, p = 0.004; Fig. 4a). Notably, HLA DR-DQ genotypes were similarly distributed between both groups (Fig. 4b). For comparison, when all 88 multiple-autoantibody-positive children were stratified according to the IAA status of their last available follow-up samples, the 10 year diabetes risks were 23% (6.3, 36.8) in IAA-negative and 75% (59.2, 84.1) in IAA-positive children (logrank test, p < 0.0001; ESM Fig. 2).

Progression to type 1 diabetes with respect to longitudinal IAA status. (a) Cumulative diabetes-free survival is shown for children who first seroconverted to IAA positivity and developed at least two other islet autoantibodies and either became IAA-negative on follow-up (dashed line; group consisting of children from cluster 4 [n = 8], cluster 1 [n = 2], cluster 8 [n = 2], and cluster 3 [n = 1]) or remained IAA-positive (solid line; group consisting of children from cluster 7 [n = 14], cluster 6 [n = 10], cluster 3 [n = 8], cluster 2 [n = 7], cluster 8 [n = 3], and cluster 9 [n = 2]) (logrank test, p = 0.004). Numbers below the x-axis indicate the number of diabetes-free children remaining on follow-up. (b) Distribution of HLA DR-DQ genotypes between groups of children with stable IAA or, respectively, loss of IAA on follow-up. ‘x’ indicates non-DR3 and non-DR4-DQ8
Discussion
Identifying characteristic patterns of progression in islet autoantibody-positive children may help to improve our understanding of the aetiology and pathophysiological mechanisms of type 1 diabetes and refine risk stratification. In this study, we developed a clustering algorithm to tackle the complexity of dynamic changes in longitudinal datasets with multiple variables. Specifically, we considered the sequence of appearance and longitudinal qualitative changes of islet autoantibodies, enabling the clustering of children from the prospective BABYDIAB cohort based on similarities in their sequential autoantibody profiles. This novel analytical approach revealed different progression rates to clinical diabetes between clusters of multiple-autoantibody-positive children. Moreover, the progression rates and sequential autoantibody profiles of clusters pointed to a longitudinal immune pattern, namely the loss of IAA reactivity in children with a broad autoantibody response against three or more beta cell autoantigens, being associated with delayed progression to type 1 diabetes.
Islet autoantibodies are currently the best established and most widely used immune markers for stratifying pre-clinical type 1 diabetes and selecting participants for secondary prevention trials [31]. It has long been recognised that diabetes risk is related to the number of positive autoantibody specificities [32, 33]; more recently, it has been demonstrated that, within a variable time course, multiple-autoantibody-positive children will progress to clinical type 1 diabetes [11]. Progression can be stratified at various degrees by antibody characteristics such as target specificity [14, 16, 17, 34], titre [13, 15] or age at seroconversion [35], as well as by genetic [36–39] and metabolic markers [40–43]. However, individual sequences of complex autoantibody profiles over time have so far rarely been analysed in prospective studies of type 1 diabetes [21, 44–47]. We here provide an unbiased data-driven algorithm to subdivide children positive for multiple islet autoantibodies based on their longitudinal autoantibody profiles. The analysis was performed using data from the longest-running prospective cohort studying the natural history of type 1 diabetes in the offspring of affected parents [7]. Additional strengths of this study lie in the consideration of all four major islet autoantibodies (IAA, IA-2A, GADA and ZnT8A), which were measured for all children on follow-up. Importantly, our approach was able to refine risk stratification among children with three or four positive autoantibodies, which was not possible using the common model based on the maximal number of positive islet autoantibodies developed on follow-up (ESM Fig. 3). Slower progression to diabetes was demonstrated in those with two positive autoantibodies, similar to cluster 1 in our approach. Furthermore, our novel approach is generally applicable to analyse other qualitative longitudinal data. We therefore believe that the proposed method provides new opportunities to stratify children into high- and low-risk clusters that could be linked to genotype, and environmental and/or other aetiological factors. In addition, longitudinal patterns could be recognised and investigated with respect to pathophysiological relevance.
At this stage, however, constraints have to be considered. The high complexity of the data means that any approach to analysis must aim at suitable reduction of dimensionality without losing valuable information. For this reason, the applied distance measure was chosen such that the complexity of the longitudinal autoantibody profiles was reduced to enable the quantification of differences in binary on and off patterns of sequential autoantibody profiles between children. The derived measure is based on Hamming distances [29], a commonly applied distance measure for comparisons of binary vectors. Furthermore, our approach abstracts from the concrete time intervals between successive probes. In order to deal with differences in sampling intervals and sampling times, we computed compressed matrices reflecting qualitative changes in autoantibody profiles only. We deliberately did not consider quantitative changes in autoantibody responses in the current analysis. In addition, the sample size within clusters was relatively small. The approach would therefore benefit from further refinement and validation in larger cohorts with shorter and more consistent time intervals between individual samples, such as The Environmental Determinants of Diabetes in the Young (TEDDY) study [21].
It is reassuring overall that our clustering approach was able to identify reasonable patterns of longitudinal autoantibody profiles and to reveal heterogeneity among multiple-autoantibody-positive children. Immunological heterogeneity has recently been shown in a combinatorial multivariable analysis of autoantibodies and autoreactive T cell responses in children and adolescents with newly diagnosed diabetes, suggesting that different immunopathological processes, or endotypes, may underlie type 1 diabetes [48]. Longitudinal profiles were not considered in this study. Importantly, our clustering approach revealed the loss of IAA reactivity in multiple-autoantibody-positive children as a longitudinal immune pattern that was indicative of delayed progression to clinical type 1 diabetes. We were able to recognise this relation because losing IAA immunogenicity was a main characteristic of children in autoantibody cluster 4, who also constituted the majority of children in the subgroup with delayed progression. In contrast, the loss of GADA reactivity was not associated with a delayed progression rate, suggesting that the beta cell autoantigen against which the autoantibody response is lost in the course of diabetes development could be of pathophysiological relevance. Insulin autoimmunity is a hallmark of childhood type 1 diabetes. High-affinity autoantibodies against insulin are usually the first to appear in young children, followed by the expansion of autoimmunity to other beta cell autoantigens before the onset of diabetes [13, 49]. It could therefore be possible that insulin autoimmunity is directing disease progression in the majority of affected children. Our current observation may support the hypothesis that loss of IAA reactivity in children reflects an insulin-specific immune regulation, which could have implications for the design of future intervention trials in type 1 diabetes and should be explored in future studies. Alternatively, the loss of IAA may simply be a matter of increasing age and therefore seen in children with slow progression. Nevertheless, we have recently shown that high frequencies of insulin-specific regulatory T cells are found in multiple-autoantibody-positive children who do not progress to clinical type 1 diabetes and therefore could indicate at least periods of successful ongoing immune regulation in such children [50].
In conclusion, our unbiased data-driven clustering approach provides a novel tool for stratification of islet autoantibody-positive children that has prognostic relevance. The refined stratification could provide new opportunities for elucidating complex disease mechanisms of type 1 diabetes. Furthermore, the approach could be applied to any dataset where multiple variables are measured over time. The observed association between the loss of humoral autoimmunity against insulin and the delay in progression to clinical type 1 diabetes among multiple-islet-autoantibody-positive children in our study demands further analyses of the underlying biological mechanisms.
Abbreviations
- GADA:
-
Autoantibodies against GAD
- IA-2A:
-
Autoantibodies against insulinoma-associated antigen-2
- IAA:
-
Autoantibodies against insulin
- IQR:
-
Interquartile range
- ZnT8A:
-
Autoantibodies against zinc transporter 8
References
Patterson CC, Dahlquist GG, Gyurus E, Green A, Soltesz G, EURODIAB Study Group (2009) Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-20: a multicentre prospective registration study. Lancet 373:2027–2033
Ziegler AG, Nepom GT (2010) Prediction and pathogenesis in type 1 diabetes. Immunity 32:468–478
Todd JA (2010) Etiology of type 1 diabetes. Immunity 32:457–467
Knip M, Virtanen SM, Akerblom HK (2010) Infant feeding and the risk of type 1 diabetes. Am J Clin Nutr 91:1506S–1513S
Laitinen OH, Honkanen H, Pakkanen O et al (2014) Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes 63:446–455
Knip M, Simell O (2012) Environmental triggers of type 1 diabetes. Cold Spring Harb Perspect Med 2:a007690
Ziegler AG, Hillebrand B, Rabl W et al (1993) On the appearance of islet associated autoimmunity in offspring of diabetic mothers: a prospective study from birth. Diabetologia 36:402–408
Kupila A, Muona P, Simell T et al (2001) Feasibility of genetic and immunological prediction of type I diabetes in a population-based birth cohort. Diabetologia 44:290–297
Rewers M, Bugawan TL, Norris JM et al (1996) Newborn screening for HLA markers associated with IDDM: diabetes autoimmunity study in the young (DAISY). Diabetologia 39:807–812
Honeyman MC, Coulson BS, Stone NL et al (2000) Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes 49:1319–1324
Ziegler AG, Rewers M, Simell O et al (2013) Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 309:2473–2479
Bonifacio E, Bingley PJ, Shattock M et al (1990) Quantification of islet-cell antibodies and prediction of insulin-dependent diabetes. Lancet 335:147–149
Achenbach P, Koczwara K, Knopff A, Naserke H, Ziegler AG, Bonifacio E (2004) Mature high-affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. J Clin Invest 114:589–597
Achenbach P, Warncke K, Reiter J et al (2004) Stratification of type 1 diabetes risk on the basis of islet autoantibody characteristics. Diabetes 53:384–392
Mayr A, Schlosser M, Grober N et al (2007) GAD autoantibody affinity and epitope specificity identify distinct immunization profiles in children at risk for type 1 diabetes. Diabetes 56:1527–1533
Achenbach P, Bonifacio E, Williams AJ et al (2008) Autoantibodies to IA-2β improve diabetes risk assessment in high-risk relatives. Diabetologia 51:488–492
De Grijse J, Asanghanwa M, Nouthe B et al (2010) Predictive power of screening for antibodies against insulinoma-associated protein 2 beta (IA-2β) and zinc transporter-8 to select first-degree relatives of type 1 diabetic patients with risk of rapid progression to clinical onset of the disease: implications for prevention trials. Diabetologia 53:517–524
Yu L, Miao D, Scrimgeour L, Johnson K, Rewers M, Eisenbarth GS (2012) Distinguishing persistent insulin autoantibodies with differential risk: nonradioactive bivalent proinsulin/insulin autoantibody assay. Diabetes 61:179–186
Sosenko JM, Skyler JS, Palmer JP et al (2013) The prediction of type 1 diabetes by multiple autoantibody levels and their incorporation into an autoantibody risk score in relatives of type 1 diabetic patients. Diabetes Care 36:2615–2620
Williams AJ, Lampasona V, Wyatt R et al (2015) Reactivity to N-terminally truncated GAD65(96-585) identifies GAD autoantibodies that are more closely associated with diabetes progression in relatives of patients with type 1 diabetes. Diabetes 64:3247–3252
Steck AK, Vehik K, Bonifacio E et al (2015) Predictors of progression from the appearance of islet autoantibodies to early childhood diabetes: The Environmental Determinants of Diabetes in the Young (TEDDY). Diabetes Care 38:808–813
Ziegler AG, Hummel M, Schenker M, Bonifacio E (1999) Autoantibody appearance and risk for development of childhood diabetes in offspring of parents with type 1 diabetes: the 2-year analysis of the German BABYDIAB Study. Diabetes 48:460–468
Puavilai G, Chanprasertyotin S, Sriphrapradaeng A (1999) Diagnostic criteria for diabetes mellitus and other categories of glucose intolerance: 1997 criteria by the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus (ADA), 1998 WHO consultation criteria, and 1985 WHO criteria. World Health Organization. Diabetes Res Clin Pract 44:21–26
Achenbach P, Lampasona V, Landherr U et al (2009) Autoantibodies to zinc transporter 8 and SLC30A8 genotype stratify type 1 diabetes risk. Diabetologia 52:1881–1888
Schlosser M, Mueller PW, Torn C, Bonifacio E, Bingley PJ, participating laboratories (2010) Diabetes antibody standardization program: evaluation of assays for insulin autoantibodies. Diabetologia 53:2611–2620
Torn C, Mueller PW, Schlosser M, Bonifacio E, Bingley PJ, participating laboratories (2008) Diabetes antibody standardization program: evaluation of assays for autoantibodies to glutamic acid decarboxylase and islet antigen-2. Diabetologia 51:846–852
Lampasona V, Schlosser M, Mueller PW et al (2011) Diabetes antibody standardization program: first proficiency evaluation of assays for autoantibodies to zinc transporter 8. Clin Chem 57:1693–1702
Walter M, Albert E, Conrad M et al (2003) IDDM2/insulin VNTR modifies risk conferred by IDDM1/HLA for development of type 1 diabetes and associated autoimmunity. Diabetologia 46:712–720
Hamming RW (1950) Error detecting and error correcting codes. Bell Syst Tech J 29:147–160
Sokal R, Michener C (1958) A statistical method for evaluating systematic relationships. Univ Kansas Sci Bull 38:1409–1438
Bingley PJ (2010) Clinical applications of diabetes antibody testing. J Clin Endocrinol Metab 95:25–33
Bingley PJ, Christie MR, Bonifacio E et al (1994) Combined analysis of autoantibodies improves prediction of IDDM in islet cell antibody-positive relatives. Diabetes 43:1304–1310
Verge CF, Gianani R, Kawasaki E et al (1996) Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 45:926–933
Decochez K, de Leeuw IH, Keymeulen B et al (2002) IA-2 autoantibodies predict impending type I diabetes in siblings of patients. Diabetologia 45:1658–1666
Hummel M, Bonifacio E, Schmid S, Walter M, Knopff A, Ziegler AG (2004) Brief communication: early appearance of islet autoantibodies predicts childhood type 1 diabetes in offspring of diabetic parents. Ann Intern Med 140:882–886
Achenbach P, Hummel M, Thumer L, Boerschmann H, Hofelmann D, Ziegler AG (2013) Characteristics of rapid vs slow progression to type 1 diabetes in multiple islet autoantibody-positive children. Diabetologia 56:1615–1622
Bonifacio E, Krumsiek J, Winkler C, Theis FJ, Ziegler AG (2014) A strategy to find gene combinations that identify children who progress rapidly to type 1 diabetes after islet autoantibody seroconversion. Acta Diabetol 51:403–411
Steck AK, Dong F, Wong R et al (2014) Improving prediction of type 1 diabetes by testing non-HLA genetic variants in addition to HLA markers. Pediatr Diabetes 15:355–362
Lempainen J, Laine AP, Hammais A et al (2015) Non-HLA gene effects on the disease process of type 1 diabetes: from HLA susceptibility to overt disease. J Autoimmun 61:45–53
Bingley PJ, Gale EA, European Nicotinamide Diabetes Intervention Trial Group (2006) Progression to type 1 diabetes in islet cell antibody-positive relatives in the European Nicotinamide Diabetes Intervention Trial: the role of additional immune, genetic and metabolic markers of risk. Diabetologia 49:881–890
Fourlanos S, Narendran P, Byrnes GB, Colman PG, Harrison LC (2004) Insulin resistance is a risk factor for progression to type 1 diabetes. Diabetologia 47:1661–1667
Truyen I, de Pauw P, Jorgensen PN et al (2005) Proinsulin levels and the proinsulin:c-peptide ratio complement autoantibody measurement for predicting type 1 diabetes. Diabetologia 48:2322–2329
Sosenko JM, Krischer JP, Palmer JP et al (2008) A risk score for type 1 diabetes derived from autoantibody-positive participants in the diabetes prevention trial-type 1. Diabetes Care 31:528–533
Decochez K, Tits J, Coolens JL et al (2000) High frequency of persisting or increasing islet-specific autoantibody levels after diagnosis of type 1 diabetes presenting before 40 years of age. The Belgian Diabetes Registry. Diabetes Care 23:838–844
Savola K, Laara E, Vahasalo P et al (2001) Dynamic pattern of disease-associated autoantibodies in siblings of children with type 1 diabetes: a population-based study. Diabetes 50:2625–2632
Achenbach P, Warncke K, Reiter J et al (2006) Type 1 diabetes risk assessment: improvement by follow-up measurements in young islet autoantibody-positive relatives. Diabetologia 49:2969–2976
Sosenko JM, Skyler JS, Palmer JP et al (2011) A longitudinal study of GAD65 and ICA512 autoantibodies during the progression to type 1 diabetes in Diabetes Prevention Trial-Type 1 (DPT-1) participants. Diabetes Care 34:2435–2437
Arif S, Leete P, Nguyen V et al (2014) Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes 63:3835–3845
Giannopoulou EZ, Winkler C, Chmiel R et al (2015) Islet autoantibody phenotypes and incidence in children at increased risk for type 1 diabetes. Diabetologia 58:2317–2323
Serr I, Fürst RW, Achenbach P et al (2016) Type 1 diabetes vaccine candidates promote human Foxp3+ Treg induction in humanized mice. Nat Commun 7:10991
Acknowledgements
The authors thank: S. Krause (Forschergruppe Diabetes, Klinikum rechts der Isar, Technische Universität München, Germany), C. Matzke, M. Scholz, S. Riethausen, L. Wendel, L. Schneider and C. Gezginci (Institute of Diabetes Research, Helmholtz Zentrum München, Germany) for data collection and expert technical assistance; R. Puff (Institute of Diabetes Research, Helmholtz Zentrum München, Germany) for laboratory management; M. Bhardwaj (Forschergruppe Diabetes e.V., Neuherberg, Germany) for analytical assistance; and M. Hummel, M. Harsunen, E. Giannopoulou and M. Krasmann (Forschergruppe Diabetes, Klinikum rechts der Isar, Technische Universität München) for clinical assistance. We also thank all paediatricians and family doctors in Germany for participating in the BABYDIAB study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
This study was supported in part by grants from the German Federal Ministry of Education and Research (BMBF) to the Competence Network for Diabetes mellitus (FKZ 01GI0805) and the German Center for Diabetes Research (DZD e.V.), and from the JDRF (JDRF 17-2012-16; 2-SRA-2015-13-Q-R). EB is supported by the DFG Research Center and Cluster of Excellence - Center for Regenerative Therapies Dresden (FZ 111).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
DE, WzC and PA researched data and wrote, reviewed and edited the manuscript. MH researched data and contributed to writing the manuscript. DE, MH, WzC and PA developed the clustering algorithm. CW, FH, SZ and AK contributed to acquisition, analysis and interpretation of data and writing the manuscript. EB and AGZ contributed to interpretation of data, and reviewed and edited the manuscript. AGZ is principal investigator of the BABYDIAB study. All listed authors approved the final version of the manuscript. PA takes responsibility for the integrity of the work as a whole.
Additional information
Wolfgang zu Castell and Peter Achenbach contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(PDF 3146 kb)
Rights and permissions
About this article

Cite this article
Endesfelder, D., Hagen, M., Winkler, C. et al. A novel approach for the analysis of longitudinal profiles reveals delayed progression to type 1 diabetes in a subgroup of multiple-islet-autoantibody-positive children. Diabetologia 59, 2172–2180 (2016). https://doi.org/10.1007/s00125-016-4050-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4050-0