
Abstract
Aims/hypothesis
Pre-adipocytes and adipocytes are responsive to the acute phase protein serum amyloid A (SAA). The combined effects triggered by SAA encompass an increase in pre-adipocyte proliferation, an induction of TNF-α and IL-6 release and a decrease in glucose uptake in mature adipocytes, strongly supporting a role for SAA in obesity and related comorbidities. This study addressed whether SAA depletion modulates weight gain and insulin resistance induced by a high-fat diet (HFD).
Methods
Male Swiss Webster mice were fed an HFD for 10 weeks under an SAA-targeted antisense oligonucleotide (ASOSAA) treatment in order to evaluate the role of SAA in weight gain.
Results
With ASOSAA treatment, mice receiving an HFD did not differ in energy intake when compared with their controls, but were prevented from gaining weight and developing insulin resistance. The phenotype was characterised by a lack of adipose tissue expansion, with low accumulation of epididymal, retroperitoneal and subcutaneous fat content and decreased inflammatory markers, such as SAA3 and toll-like receptor (TLR)-4 expression, as well as macrophage infiltration into the adipose tissue. Furthermore, a metabolic status similar to chow-fed mice counterparts could be observed, with equivalent levels of leptin, adiponectin, IGF-I, SAA, fasting glucose and insulin, and remarkable improvement in glucose and insulin tolerance test profiles. Surprisingly, the expected HFD-induced metabolic endotoxaemia was also prevented by the ASOSAA treatment.
Conclusions/interpretation
This study provides further evidence of the role of SAA in weight gain and insulin resistance. Moreover, we also suggest that beyond its proliferative and inflammatory effects, SAA is part of the lipopolysaccharide signalling pathway that links inflammation to obesity and insulin resistance.
Similar content being viewed by others


Introduction
Chronic and low-grade inflammation is a hallmark of obesity and a key factor for the development of obesity comorbidities [1]. More than simply a consequence of obesity, low-grade chronic inflammation, especially that triggered by the endotoxaemia derived from intestinal microbiota, has been identified as a cause of weight gain, obesity and insulin resistance [2, 3]. Indeed, high-fat feeding modulates gut microbiota and the plasma concentration of endotoxin, correlating to the occurrence of metabolic diseases [3].
One point addressed in this study was the identification of additional factors linking endotoxaemia and weight gain. Biochemical changes favouring obesity include cytokines and several inflammatory factors, as well as the involvement of toll-like receptors (TLRs) [4, 5]. Herein, we addressed whether serum amyloid A (SAA), an acute phase protein and TLR ligand, links inflammation to the onset of weight gain and comorbidities.
In mice, three functional and distinct inducible isoforms of SAA have been identified, SAA1.1, SAA2.1 and SAA3 [6, 7]. In response to a proinflammatory stimulus, the liver produces the isoforms 1.1 and 2.1, while SAA3 is mainly expressed in extrahepatic tissues [8–10]. More specifically, SAA3 does not contribute to circulating levels of SAA and has been related mostly to the promotion of local inflammation [9, 11, 12].
However, liver-derived SAA has been implicated not only in the regulation of inflammatory responses [13–15] but also in the control of cell proliferation [16–18] and metabolic profile [18–20]. Although mainly secreted by the liver, SAA has also been reported as being produced by adipocytes, macrophages and several other cell types [21, 22], with levels positively correlated with obesity grade, as well as increased expression under hypoxic conditions [22, 23].
SAA is an endogenous ligand to TLR-4 [13] and TLR-2 [24] and was recently described as causing impairment to the scavenger receptor BI (SR-BI) [20]. All these receptors are admittedly associated with the inflammatory response and also related to lipopolysaccharide (LPS) responsiveness [13, 24, 25].
In order to evaluate the role of SAA in weight gain, SAA-targeted antisense oligonucleotide (ASOSAA) was used in a high-fat diet (HFD)-induced experimental model of obesity. Although metabolic endotoxaemia is considered one of the early factors in HFD-induced metabolic diseases [2], in this study we identified SAA as an additional trigger driving weight gain and insulin resistance.
Methods
Animals
Male Swiss Webster mice (21 days of age) were obtained from the Animal Facility at the Faculty of Pharmaceutical Sciences, University of São Paulo, Brazil, under approval by its ethics committee (number 297) and in accordance with international guidelines on animal experimentation. The animals were housed inside standard polypropylene cages in a room maintained at 22 ± 2°C with a 12:12 h light/dark cycle (lights on at 07:00 hours and off at 19:00 hours) and 55 ± 10% relative humidity. Body weight was measured once a week during the entire protocol. Food and water intake were ad libitum and measured every 2 days. No death was recorded and no mice were dropped out of the analysis during the course of the experimental protocol. Euthanasia occurred by anaesthesia overdose (i.p. administration of a combination of ketamine [100 mg/kg] and xylazine [15 mg/kg]), and ensured by cervical dislocation.
Diet-induced obesity protocol and SAA depletion
The experiments were performed using a custom-designed diet. The chow diet was produced following the American Institute of Nutrition recommendations AIN-93M (total energy: 75.8% carbohydrate, 9.3% fat and 14.9% protein) [26], while the HFD group received an AIN-93M-based diet that was enriched with lard (total energy: 24.2% carbohydrate, 60.9% fat and 14.9% protein) [27]. The diet composition is listed in Table 1. For the experimental protocol, mice were first randomly assigned to two different groups in order to receive chow or the HFD for 10 weeks. Each group was then split again to receive antisense oligonucleotide in vivo treatment for Saa mRNA inhibition (kindly provided by Ionis Pharmaceuticals, Carlsbad, CA, USA). The following groups were formed: control (no antisense oligonucleotide [ASO] treatment), ASO not specific to any murine transcript (ASOscramble) and ASOSAA (specific for the murine Saa1 and Saa2 transcripts). Both ASOs are based on generation 2.5 chemistry, which consisted of 2-O, 4-C-[(S)-ethylidene]-d-ribose (‘cEt’) modified sugars for the three terminal 5′ and 3′ nucleotides that flank a central ten-nucleotide DNA gap [28]. The ASO treatment was performed via i.p. administration at a dose of 25 mg/kg of the animal, once a week. The ASO sequences and chemistries are listed in Table 2.
Glucose and insulin tolerance tests and measurements of serum leptin, adiponectin, insulin, IGF-I, SAA and endotoxin
Glucose and insulin tolerance tests (GTTs and ITTs) were performed as described previously [29]. Serum concentrations for the following proteins were determined using ELISA following the manufacturer’s instructions: leptin (Millipore, Billerica, MA, USA, catalogue number EZML-82K), adiponectin (Millipore, EZMAD-60K), and insulin (Millipore, EZRMI-13K), SAA (Tridelta Development, Maynooth, Ireland, catalogue number TP802M) and IGF-I (R&D Systems, Minneapolis, MN, USA, catalogue number SMG100). Endotoxin was measured with the limulus amoebocyte lysate (LAL) chromogenic endpoint assay (Lonza, Allendale, NJ, USA, catalogue number QCL-1000).
Histological analysis
Paraffin-embedded sections (5 μm thick) from epididymal adipose tissue were stained with haematoxylin and eosin in order to assess morphology. Immunofluorescence for F4/80 (macrophage identification), SAA and perilipin (a marker of lipid droplet structures, also useful to identify crown-like structures) was performed using a rat anti-mouse F4/80 antibody and rabbit anti-mouse perilipin (both at 1:100 dilution; Abcam, Cambridge, UK, catalogue numbers ab6640 and ab3526), and a rabbit anti-mouse SAA (1:200 dilution, kindly provided by the de Beer laboratory, University of Kentucky, Lexington, KY, USA). They were subsequently incubated with the appropriate secondary fluorescent antibody (Invitrogen, Camarillo, CA, USA) and the slides mounted using Vectashield set mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA, catalogue number H-1200) for nuclei staining. An isotype control was used to ensure antibody specificity in each staining (data not shown). Tissue sections were observed with a Nikon Eclipse 80i microscope (Nikon, Melville, NY, USA), and digital images were captured with NIS-Element AR software (Nikon).
In vivo peripheral fat area quantification
Two x-ray images were taken at different energy levels, thus allowing us to circumscribe the adipose tissue on the animals as described previously [29].
Quantitative real-time PCR
Total RNA from epididymal adipose tissue was isolated using Qiagen RNeasy Lipid Tissue Mini kit (Qiagen, Hilden, Germany, catalogue number 74804). cDNA was synthesised from 1 μg of RNA using the High Capacity cDNA Reverse Transcription kit (Life Technologies, Grand Island, NY, USA, catalogue number 4368814). Real-time PCR was performed using SyBr Green Master Mix (Life Technologies, catalogue number 4309155). The primer sequences are detailed in Table 3. Real-time PCR for Saa3 was performed using the TaqMan assay (Applied Biosystems, Grand Island, NJ, USA, catalogue number Mm00441203_m1), with β-actin (ACTB) (catalogue number 4552933E) as an endogenous housekeeping gene control. Relative gene expression was determined using the \( {2}^{-\Delta \Delta {\mathrm{C}}_{\mathrm{t}}} \) method [30].
Statistical analysis
Results are presented as mean ± SEM. Statistical analysis was performed with Graph Pad Prism4 (Graph Pad Software, San Diego, CA, USA). Data with two independent factors were tested by two-way analysis of variance (two-way ANOVA, a parametric test), followed by Bonferroni post hoc test. The level of significance was set at p < 0.05.
Results
SAA depletion prevents weight gain induced by HFD
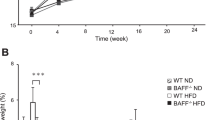
In order to address the involvement of the SAA protein in weight gain and insulin resistance, mice fed a chow diet or HFD for 10 weeks and concurrently subjected to ASOSAA administration were analysed regarding adipose tissue composition and architecture. SAA levels tend to increase when an HFD is introduced, but the ASOSAA mRNA inhibitor was effective in decreasing the serum concentration of SAA in mice receiving the HFD (Table 4). Considering the type of diet used, no effect was observed on the energy intake when ASOscramble or ASOSAA were administered (Fig. 1a). The growth curves for mice fed a chow diet did not show any developmental differences from control, ASOscramble or ASOSAA groups, demonstrating no toxic or side effects from the ASO treatment (Fig. 1b). However, the ASOSAA group was protected from the effects of the HFD. Moreover, the growth curve, the visceral fat depot and subcutaneous fat depot weight were comparable with those of a chow diet-fed animal (Fig. 1c–f). These data were confirmed using x-ray images highlighting the subcutaneous fat area, showing that HFD + ASOSAA mice have a minor peripheral fat area when compared with HFD or HFD + ASOscramble mice (Fig. 1g, h).

SAA depletion prevents weight gain triggered by a diet-induced obesity protocol. (a) Daily energy intake. (b, c) Weight gain curves of (b) chow-fed mice, and (c) HFD-fed mice. Control, black circles/solid line; ASOscramble, white squares/solid line; and ASOSAA, grey triangles/dashed line. (d) Epididymal, (e) retroperitoneal and (f) subcutaneous fat pad weight. (g) Subcutaneous fat area quantification, and (h) representative subcutaneous fat area in mice under chow or high-fat diets submitted to ASOscramble or ASOSAA. Data are means ± SEM from six mice per group. For all variables with the same letter, the difference between the means is not statistically significant. Where two variables have different letters, they are significantly different (p < 0.05). Retro, retroperitoneal
SAA depletion prevents HFD-induced adipose tissue expansion and macrophage infiltration into the adipose tissue
After 10 weeks on an HFD, it was possible to identify several crown-like structures, defined as a perilipin-free adipocyte surrounded by macrophage (F4/80+), in the adipose tissue from control and ASOscramble mice (Fig. 2, middle column). Moreover, the HFD significantly increased the staining for SAA in the adipose tissue in control and ASOscramble mice (Fig. 2, right column). When mice receiving the HFD were concomitantly treated with ASOSAA, no significant staining for F4/80 and SAA was observed (Fig. 2, middle and right column), indicating the absence of both macrophage infiltration and local inflammation. Furthermore, ASOSAA treatment also prevented adipocyte hypertrophy, demonstrating the involvement of SAA in adipose tissue expansion and remodelling (Fig. 2, left column and Fig. 3a). These effects derived from SAA depletion were not observed in chow-fed mice (Fig. 2, middle and right column).

SAA depletion prevents adipose tissue hypertrophy and macrophage infiltration in adipose tissue induced by an HFD. Histological sections of epididymal fat pads from mice under chow or HFD, submitted to ASOscramble or ASOSAA. Haematoxylin and eosin staining for adipose tissue morphology analysis (left column). Scale bars represent 100 μm. Immunofluorescence for perilipin (red), F4/80 (green) and nuclear staining with DAPI (blue) (middle column) and SAA (red) immunostaining (right column). Crown-like structures, defined as a perilipin-free adipocyte surrounded by macrophage (F4/80+), and an increase in SAA staining are indicated by white arrows in both control and ASOscramble groups under HFD. H&E, haematoxylin and eosin

SAA depletion prevents the metabolic imbalance induced by HFD. (a) Adipocyte size in epididymal adipose tissue. (b–h) Serum levels of (b) endotoxin, (c) leptin, (d) adiponectin, (e) IGF-I, (f) SAA, (g) fasting glucose and (h) insulin. (i, j) GTTs and (k, l) ITTs in (i, k) chow-fed animals and (j, l) HFD-fed animals. Control, black circles/solid line; ASOscramble, white squares/solid line; and ASOSAA, grey triangles/dashed line. (m, n) AUCs for the (m) GTT and (n) ITT assays. The mRNA expression of (o) Saa1/2, (p) Saa3, (q) Tlr2, (r) Tlr4 and (s) Cd14 in epididymal adipose tissue. Data are means ± SEM from six mice per group. White bars, standard chow-fed mice; black bars, HFD-fed mice. For all variables with the same letter, the difference between the means is not statistically significant. Where two variables have different letters, they are significantly different (p < 0.05). AU, arbitrary units. EU, endotoxin units
SAA depletion prevents the metabolic imbalance induced by an HFD
The metabolic status of the animals treated with ASOSAA was also assessed. Remarkably, endotoxin levels from mice treated with ASOSAA under an HFD were comparable with those from a chow-fed animal (Fig. 3b). Moreover, the trend to maintain the metabolic balance is clear, as no changes were observed in serum levels of leptin (Fig. 3c), adiponectin (Fig. 3d), IGF-I (Fig. 3e), SAA (Fig. 3f), fasting glucose (Fig. 3g) or insulin (Fig. 3h). Thus, treatment with ASOSAA enables an improvement in glucose tolerance and insulin resistance, notably affected when an HFD is introduced (Fig. 3i–n). As expected, an HFD resulted in an increase in some of the inflammatory markers (Saa3 [Fig. 3p] and Tlr4 [Fig. 3r]); however, no changes in Saa1/2 (Fig. 3o), Tlr2 (Fig. 3q) or Cd14 (Fig. 3s) were observed. Interestingly, SAA depletion also prevented the elevation of Saa3 and Tlr4 expression in adipose tissue from mice receiving an HFD (Fig. 3p, r, respectively).
Discussion
In order to define the contribution of SAA in obesity, the effect of SAA depletion in a diet-induced obesity protocol using ASOSAA was evaluated. Under an HFD, the serum levels of SAA doubled; ASOSAA treatment efficiently reduced SAA to its basal level. The treatment with ASOSAA was characterised by a striking reduction of deleterious alterations caused by an HFD, underlying the importance of SAA in obesity.
The role of SAA in adipose tissue began to be unravelled in recent years with a number of in vitro studies. When not associated with HDL, SAA is a stimulus for the production of cytokines, reactive oxygen species and NO [13–15, 31]. SAA is also able to activate the inflammasome pathway, considered a mediator of the innate and adaptive immune system [32, 33]. Furthermore, SAA is induced by hypoxia [22], a common event in fat expansion. Its production may affect adipose tissue homeostasis, such as inducing pre-adipocyte proliferation and inhibiting adipocyte glucose uptake [14, 17, 18, 31, 34]. SAA also induces lipolysis through an extracellular signal-regulated kinase (ERK)-dependent pathway [35] and, despite the fact that lipolysis results in a decrease in adiposity, it also contributes to an elevation of circulating levels of NEFA, which impair glucose uptake by muscle and hepatic cells found in metabolic disorders [36].
Also, the fact that neutrophils and monocytes of diabetic patients are more responsive to SAA regarding cytokine production and cell migration [37] further strengthens the need to study the potential effect of SAA in obesity and insulin resistance. Increased levels of SAA may contribute to a sustained accumulation and activation of inflammatory cells, as well as to the vascular complications observed in diabetic patients [37].
Although no receptor dedicated solely to SAA has yet been identified, SAA can bind and activate several cell surface receptors, including TLR-4 and TLR-2 [13, 23], both of which have known roles in the development of obesity. For instance, the activation of TLR-4 and its co-receptor cluster of differentiation 14 (CD14) is associated with obesity and insulin resistance in a diet-induced obesity protocol in mice [2, 38, 39]. The molecular mechanisms involving the activation of TLRs are associated with endoplasmic reticulum stress and activation of the c-Jun N-terminal protein kinases (JKNs) both in liver and in fat tissue, all associated with the promotion of inflammation [4, 39].
It is also important to consider the complex interplay that leads to the production of SAA. It has been shown that adipocytes stimulated by recombinant human (rh)SAA increase the expression of SAA3 [18, 35]. In this panorama, not only the HFD may act in the adipose tissue driving the production of SAA3, but also the elevation of SAA1.1 and SAA2.2 in plasma would stimulate SAA3 expression in adipose tissue [9]. The interplay of SAA isoforms could be the reason for the reduction in SAA3 expression in the adipose tissue of mice submitted to an HFD under ASOSAA treatment.
This study, in an in vivo approach, confirms the expected beneficial effect of SAA depletion on weight gain and its comorbidities. It is important to highlight that although no evidence was found for a role of SAA in the development of obesity and insulin resistance in a transgenic mouse model expressing human SAA1 in the adipose tissue [40], a recent study with Saa-knockout mice for isoform 3 showed an attenuated weight gain and macrophage infiltration into the adipose tissue after an obesogenic diet [41]. It is interesting to note that these effects were exclusive for female knockout mice. Moreover, Saa-knockout mice lacking SAA1.1 and SAA2.1 gained less weight when compared with their wild-type counterparts during their lifespan [42]. Taken together, the studies with Saa-knockout mice and the data presented herein with ASOSAA consolidate the participation of SAA in obesity through complementary experimental strategies.
Moreover, in an unprecedented manner, this study correlates SAA with metabolic endotoxaemia. It has found that ASOSAA prevented the elevation of endotoxin levels in serum, an increase that is expected when mice are submitted to an HFD. Changes observed in endotoxaemia levels under the ASOSAA treatment may be due to the effects of SAA on gut permeability or on LPS clearance. SAA is produced by colonic epithelium in response to the gut microbiota [11] and it is expected that inflammatory mediators cause an increase in gut permeability [43], and therefore, endotoxaemia. After reaching the blood stream, virtually all LPS molecules are rapidly complexed with circulating proteins and lipoproteins. When complexed with HDL [44], LPS is cleared by SR-BI in hepatocytes [45] and in other cell types such as macrophages and monocytes, as recently described [46]. The clearance of LPS through SR-BI efficiently determines the magnitude of the inflammatory response, and it has already been shown that SAA is associated with impairment of SR-BI [20]. Thus, the authors suggest that the reduction in SAA serum levels due to the ASOSAA treatment could decrease gut permeability to LPS and also improve SR-BI-mediated LPS clearance, leading to decreased endotoxaemia.
Figure 4 summarises the main findings of the authors’ previous [18] and current study outlining the possible relationship between LPS and SAA as factors driving obesity. The present study does not claim it has exhausted the subject and future studies should be addressed in order to fully unveil the mechanism underlying the role of SAA in diet-induced obesity. Accordingly, the authors think the focus should be on intestinal permeability and also on neuronal control of energy homeostasis, as hypothalamic injury and the activation of inflammatory signalling are associated with obesity in both rodents and humans [47].

Schematic pathways linking SAA and endotoxaemia to obesity and insulin resistance. (A) Endotoxaemia (increased serum LPS) caused by HFD. Endotoxaemia induces the release of a large amount of inflammatory mediators, such as SAA. This protein acts directly on different cells in adipose tissue promoting weight gain and insulin resistance. SAA induces pre-adipocyte proliferation [18], allowing further differentiation to adipocyte when an adipogenic factor, such as an HFD, is subsequently added. SAA impairs glucose uptake by decreasing GLUT-4 expression in mature adipocytes. In adipocytes, macrophages and leucocytes, SAA stimulates the production of cytokines promoting inflammation and a positive feedback on SAA production. An additional hypothesis from our data suggests that SAA also participates in (B) the LPS passage from the gut lumen into the bloodstream, increasing gut permeability, and in (C) LPS maintenance in serum, impairing SR-BI [18, 20], one of the receptors responsible for LPS clearance
Abbreviations
- ASO:
-
Antisense oligonucleotide
- ASOSAA :
-
SAA-targeted antisense oligonucleotide
- ASOscramble :
-
ASO not specific to any murine transcript
- CD14:
-
Cluster of differentiation 14
- cEt:
-
2-O, 4-C-[(S)-ethylidene]-d-ribose
- HFD:
-
High-fat diet
- ITT:
-
Insulin tolerance test
- LPS:
-
Lipopolysaccharide
- SAA:
-
Serum amyloid A
- SR-BI:
-
Scavenger receptor BI
- TLR:
-
Toll-like receptor
References
Ouchi N, Parker JL, Lugus JJ, Walsh K (2011) Adipokines in inflammation and metabolic disease. Nat Rev Immunol 11:85–97
Cani PD, Amar J, Iglesias MA et al (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56:1761–1772
Cani PD, Bibiloni R, Knauf C, Neyrinck AM, Delzenne NM, Burcelin R (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57:1470–1481
Caricilli AM, Picardi PK, de Abreu LL et al (2011) Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol 9:21
Tsukumo DML, Carvalho-Filho MA, Carvalheira JBC et al (2007) Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 56:1986–1998
Uhlar CM, Whitehead AS (1999) Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem 265:501–523
Sipe J (1999) Revised nomenclature for serum amyloid A (SAA). Nomenclature Committee of the International Society of Amyloidosis. Part 2. Amyloid 6:67–70
Meek RL, Benditt EP (1986) Amyloid A gene family expression in different mouse tissues. J Exp Med 164:2006–2017
Scheja L, Heese B, Zitzer H et al (2008) Acute-phase serum amyloid a as a marker of insulin resistance in mice. Exp Diabetes Res. doi:10.1155/2008/230837
Sommer G, Weise S, Kralisch S et al (2008) The adipokine SAA3 is induced by interleukin-1 beta in mouse adipocytes. J Cell Biochem 104:2241–2247
Reigstad CS, Lunden GO, Felin J, Backhed F (2009) Regulation of serum amyloid A3 (SAA3) in mouse colonic epithelium and adipose tissue by the intestinal microbiota. PLoS One 4:e5842
Chiba T, Han CY, Vaisar T et al (2009) Serum amyloid A3 does not contribute to circulating SAA levels. J Lipid Res 50:1353–1362
Sandri S, Rodriguez D, Gomes E, Monteiro HP, Russo M, Campa A (2008) Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Biol 83:1174–1180
Furlaneto CJ, Campa A (2000) A novel function of serum amyloid A: a potent stimulus for the release of tumor necrosis factor-alpha, interleukin-1 beta, and interleukin-8 by human blood neutrophil. Biochem Biophys Res Commun 268:405–408
Hatanaka E, Ribeiro FP, Campa A (2003) The acute phase protein serum amyloid A primes neutrophils. Fems Immunol Med Microbiol 38:81–84
Hatanaka E, Dermargos A, Armelin HA, Curi R, Campa A (2011) Serum amyloid A induces reactive oxygen species (ROS) production and proliferation of fibroblast. Clin Exp Immunol 163:362–367
Knebel FH, Albuquerque RC, Massaro RR, Maria-Engler SS, Campa A (2013) Dual effect of serum amyloid A on the invasiveness of glioma cells. Mediators Inflamm. doi:10.1155/2013/509089
Filippin-Monteiro FB, de Oliveira EM, Sandri S, Knebel FH, Albuquerque RC, Campa A (2012) Serum amyloid A is a growth factor for 3T3-L1 adipocytes, inhibits differentiation and promotes insulin resistance. Int J Obes 36:1032–1039
Liu LR, Lin SP, Chen CC et al (2011) Serum amyloid A induces lipolysis by downregulating perilipin through ERK1/2 and PKA signaling pathways. Obesity 19:2301–2309
Tsun JGS, Shiu SWM, Wong Y, Yung S, Chan TM, Tan KCB (2013) Impact of serum amyloid A on cellular cholesterol efflux to serum in type 2 diabetes mellitus. Atherosclerosis 231:405–410
Upragarin N, Landman WJM, Gaastra W, Gruys E (2005) Extrahepatic production of acute phase serum amyloid A. Histol Histopathol 20:1295–1307
de Oliveira EM, Sandri S, Knebel FH, Iglesias Contesini CG, Campa A, Filippin-Monteiro FB (2013) Hypoxia increases serum amyloid A3 (SAA3) in differentiated 3T3-L1 adipocytes. Inflammation 36:1107–1110
Eklund KK, Niemi K, Kovanen PT (2012) Immune functions of serum amyloid A. Crit Rev Immunol 32:335–348
Cheng N, He R, Tian J, Ye PP, Ye RD (2008) Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol 181:22–26
Guo L, Zheng Z, Ai J, Huang B, Li X-A (2014) Hepatic scavenger receptor BI protects against polymicrobial-induced sepsis through promoting LPS clearance in mice. J Biol Chem 289:14666–14673
Reeves PG, Nielsen FH, Fahey GC (1993) AIN-93 purified diets for laboratory rodents - final report of the American Institute of Nutrition Ad Hoc Writing Committee on the reformulation of the AIN-76a rodent diet. J Nutr 123:1939–1951
Pang J, Choi Y, Park T (2008) Ilex paraguariensis extract ameliorates obesity induced by high-fat diet: potential role of AMPK in the visceral adipose tissue. Arch Biochem Biophys 476:178–185
Donner AJ, Yeh ST, Hung G, Graham MJ, Crooke RM, Mullick AE (2015) CD40 generation 2.5 antisense oligonucleotide treatment attenuates doxorubicin-induced nephropathy and kidney inflammation. Mol Ther Nucleic Acids 4:e265
de Oliveira EM, Visniauskas B, Sandri S et al (2015) Late effects of sleep restriction: potentiating weight gain and insulin resistance arising from a high-fat diet in mice. Obesity 23:391–398
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-∆∆CT method. Methods 25:402–408
Franco AG, Sandri S, Campa A (2011) High-density lipoprotein prevents SAA-induced production of TNF-α in THP-1 monocytic cells and peripheral blood mononuclear cells. Mem Inst Oswaldo Cruz 106:986–992
Niemi K, Teirila L, Lappalainen J et al (2011) Serum amyloid A activates the NLRP3 inflammasome via P2X(7) receptor and a cathepsin B-sensitive pathway. J Immunol 186:6119–6128
Ather JL, Ckless K, Martin R et al (2011) Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol 187:64–73
Sandri S, Hatanaka E, Franco AG, Pedrosa AMC, Monteiro HP, Campa A (2008) Serum amyloid A induces CCL20 secretion in mononuclear cells through MAPK (p38 and ERK1/2) signaling pathways. Immunol Lett 121:22–26
Faty A, Ferre P, Commans S (2012) The acute phase protein serum amyloid A induces lipolysis and inflammation in human adipocytes through distinct pathways. PLoS One 7:e34031
Arner P (1988) Control of lipolysis and its relevance to development of obesity in man. Diabetes Metab Rev 4:507–515
Hatanaka E, Monteagudo PT, Marrocos MSM, Campa A (2007) Interaction between serum amyloid A and leukocytes—a possible role in the progression of vascular complications in diabetes. Immunol Lett 108:160–166
Song MJ, Kim KH, Yoon JM, Kim JB (2006) Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem Biophys Res Commun 346:739–745
Kim K-A, Gu W, Lee I-A, Joh E-H, Kim D-H (2012) High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS One 7:e47713
Ahlin S, Olsson M, Olsson B, Svensson P-A, Sjoholm K (2013) No evidence for a role of adipose tissue-derived serum amyloid A in the development of insulin resistance or obesity-related inflammation in hSAA1(+/)- transgenic mice. PLoS One 8:e72204
den Hartigh LJ, Wang SR, Goodspeed L et al (2014) Deletion of serum amyloid A3 improves high fat high sucrose diet-induced adipose tissue inflammation and hyperlipidemia in female mice. Plos One 9:e108564
De Beer MC, Wroblewski JM, Noffsinger VP et al (2014) Deficiency of endogenous acute phase serum amyloid A does not affect atherosclerotic lesions in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 34:255–261
Arrieta MC, Bistritz L, Meddings JB (2006) Alterations in intestinal permeability. Gut 55:1512–1520
Ulevitch RJ, Johnston AR, Weinstein DB (1979) New function for high-density lipoproteins—their participation in intra-vascular reactions of bacterial lipopolysaccharides. J Clin Investig 64:1516–1524
Guo L, Ai J, Zheng Z et al (2013) High density lipoprotein protects against polymicrobe-induced sepsis in mice. J Biol Chem 288:17947–17953
Li B, Yu M, Pan X et al (2014) Artesunate reduces serum lipopolysaccharide in cecal ligation/puncture mice via enhanced LPS internalization by macrophages through increased mRNA expression of scavenger receptors. Int J Mol Sci 15:1143–1161
Thaler JP, Yi C-X, Schur EA et al (2012) Obesity is associated with hypothalamic injury in rodents and humans. J Clin Investig 122:153–162
Acknowledgements
The authors would like to thank E. Wilson Cavalcante Oliveira Santos and A. Froes Marchi from the Departamento de Análises Clínicas e Toxicológicas, Faculdade de Ciências Farmacêuticas, Universidade de São Paulo, Brazil for technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (grant number 2011/24052-4 and 2010/18498-7 [doctoral scholarship]), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (grant number 47510/2010-6).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
EMO and SS contributed to the study conception and design, acquisition, analysis and interpretation of data, and writing of the article. TPA, JCS and SM contributed to the acquisition and interpretation of data and also to the final critical revision of the manuscript. RAF and AC contributed to the study conception and design, analysis and interpretation of data, and writing and editing of the manuscript. All authors approved the final version of the manuscript to be published. AC is the guarantor of this work and, as such, has had full access to all data in the study and takes responsibility for both the integrity of the data and the accuracy of the data analysis.
Rights and permissions
About this article

Cite this article
de Oliveira, E.M., Ascar, T.P., Silva, J.C. et al. Serum amyloid A links endotoxaemia to weight gain and insulin resistance in mice. Diabetologia 59, 1760–1768 (2016). https://doi.org/10.1007/s00125-016-3970-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3970-z