
Abstract
Mitochondria fulfil multiple tasks in nutrient metabolism, energy production, redox homeostasis and stress response, and are essential for pancreatic beta cell function. The dynamism and health of the mitochondrial network is regulated by fission- and fusion-triggering factors and by a quality control system that removes dysfunctional organelles. Alongside the role of mitochondria in regulating apoptotic cell death mediated primarily via production of reactive oxygen species and release of cytochrome c, there is evidence of other links between mitochondria and inflammation that have implications for cell viability. This review briefly outlines two pathways that are potentially vital for pancreatic beta cell function. The first concerns the regulation of Parkin, a protein that acts, not only as a central player in regulating mitophagy, but also as an activator of the NF-ĸB pathway. The fact that expression of optic atrophy protein 1 (OPA1), a mitochondrial fusion inducer and master mitochondrial cristae biogenetic factor, is increased following NF-ĸB activation highlights a point of mitochondrial control that might be influenced by TNFα signalling. A second axis of interest is suggested by IL-6-mediated upregulation of the fission inducer FIS1 alongside downregulation of mitofusin 2 (MFN2), a guard of mitochondrial fusion and metabolism and an inhibitor of apoptosis. This review summarises a presentation given at the ‘Islet inflammation in type 2 diabetes’ symposium at the 2015 annual meeting of the EASD. It is accompanied two other reviews on topics from this symposium (by Marc Donath, DOI: 10.1007/s00125-016-3873-z, and Jerry Nadler and colleagues, DOI: 10.1007/s00125-016-3890-y) and a commentary by the Session Chair, Piero Marchetti (DOI: 10.1007/s00125-016-3875-x).
Similar content being viewed by others
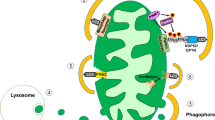


Mitochondrial shape and function
Mitochondria are crucial cell organelles. They fulfil a varied range of tasks in nutrient metabolism, energy production, redox homeostasis and stress response. Using Newton’s second law, Kaufman et al have described mitochondrial energy output as the product of mitochondrial mass and metabolic rate [1]. Loss of mitochondrial mass in pancreatic beta cells results in functional impairment and ultimately in the manifestation of type 2 diabetes mellitus [1, 2]. Because mitochondria form a cell type-specific, highly dynamic network, it is important to take into account not only their total mass but also their structure and mobility as health-determining factors [1, 3–5]. Mitochondrial fission and fusion occur constantly and, along with the removal of damaged mitochondria by mitophagy, these processes maintain organelle vitality and the physiological state of the cell (Fig. 1) [1, 3–5]. Because mitochondrial function is crucial for pancreatic beta cell stimulus–secretion coupling, mitochondrial network failure is capable of promoting hyperglycaemia [1, 6].

Inflammatory signals can affect the mitochondrial life cycle by modulating fusion, fission and mitophagy
Regulators of the mitochondrial network
Fission protein 1 (FIS1), which initiates fission, and dynamin-related protein 1 (DRP1), whose recruitment and ring-shaped oligomerisation on a mitochondrion result in its splitting, are two key regulators of the mitochondrial network [7–9]. The interactions of both proteins are mediated by mitochondrial fission factor (MFF) and the mitochondrial dynamics proteins (MID49/MID51) via a mechanism that is incompletely understood [7]. Viable mitochondria are able to fuse by a process that is conducted by the two mitofusins (MFN1 and MFN2) for the outer mitochondrial membrane and by optic atrophy protein 1 (OPA1) for the inner mitochondrial membrane and its cristae structure [10, 11]. Reintegration of a single mitochondrion into the network is dependent on respiratory chain activity, which determines mitochondrial membrane potential (MMP) [12, 13]. Organelles unable to recover their MMP fall prey to quality control processes and are removed by mitophagy [12, 13].
Parkin: a protein managing many tasks
Mutations in the PARK2 gene are responsible for hereditary Parkinson’s disease, a fact that accounts for the protein being named Parkin [4, 14]. In tandem with its well-known role in dopaminergic neurons in the ubiquitination of damaged proteins, targeting them for proteasomal degradation, the E3 ubiquitin ligase Parkin cooperates with PTEN-induced putative kinase 1 (PINK1) to orchestrate the removal of unhealthy mitochondria [15–17]. PINK1 is continuously degraded from the surface of mitochondria with high MMP, but as soon as there is a reduction in MMP, PINK1 is stabilised and binds Parkin, eventually leading to mitophagy [15–17].
Excursion: Overlap of mitophagy with autophagic turnover and nutrient supply
Ultimately, however, this specifically initiated pathway becomes fully integrated into the autophagic turnover process, rendering mitophagy dependent on overall cellular quality control mechanisms [18]. Accordingly, pancreatic beta cells from autophagy-related protein 7 (ATG7) mutant mice show mitochondrial swelling [19]. The regulation of autophagy in pancreatic beta cells is currently a topic of lively debate [20–25]. Autophagy is constitutively active at a low level in nearly all cells and is upregulated in response to stress [18]. Starvation, along with activation of AMP-dependent kinase (AMPK), initiates subsequent inactivation of mammalian target of rapamycin (mTOR), which in turn actuates autophagy [18]. The resulting reallocation of nutrients rescues cell viability [18]. This mechanism is also accepted for pancreatic beta cells, and downregulation of autophagic flux during nutrient abundance has been suggested to contribute to endoplasmic reticulum stress, inflammation and apoptosis, eventually supporting beta cell failure [23, 24]. Conversely, various studies have demonstrated the upregulation of autophagy in pancreatic beta cells in response to hyperglycaemia, diabetes mellitus and obesity [20, 22, 25, 26]. Goginashvili et al recently reported that starvation induces lysosomal degradation of nascent secretory insulin granules in pancreatic beta cells [21]. The resultant available nutrients, mainly amino acids, activate mTOR sufficiently to inhibit autophagy [21]. The authors concluded that there is a positive correlation between autophagy and nutrient-induced insulin secretion [21].
These new findings raise the question as to whether active pancreatic beta cells secreting insulin are more susceptible to mitophagy as a result of upregulation of autophagic flux. It can be speculated that this is plausible, because only mitochondria with high MMP remain in the network, thereby guaranteeing perfect stimulus–secretion coupling. However, assuming that this has a major influence on mitochondrial quality control, a continuous high glucose supply would provoke a dramatic loss of mitochondrial mass, which is at odds with the finding that mitochondrial fragmentation—but not mitochondrial loss—occurs in response to nutrient excess [27].
Parkin: active at the interface between mitochondrial quality control and inflammation
Recent observations suggest that there is tight cellular control of Parkin in pancreatic beta cells. Genome-wide studies have identified C-type lectin domain family 16, member A (CLEC16A), not only as a gene locus associated with multiple sclerosis and adrenal dysfunction, but also with type 1 diabetes mellitus [28]. Soleimanpour et al reported that C-type lectin domain family 16, member A (CLEC16A) is a membrane-associated endosomal protein and regulator of mitophagy [29]. They demonstrated that CLEC16A deficiency impairs glucose tolerance by reducing beta cell function [29]. CLEC16A functions as an activator of the E3 ubiquitin ligase known as neuregulin receptor degradation protein (NRDP1), which regulates the proteasomal degradation of Parkin. Accordingly, loss of CLEC16A results in increased Parkin expression [1, 29]. Parkin regulation seems to be a prerequisite for maintaining beta cell function. Both the overexpression and downregulation of Parkin result in significantly diminished glucose-induced insulin secretion [30, 31]. Parkin gene expression shows a glucose-dependent pattern. In both primary mouse islets and insulin-secreting MIN6 cells, Parkin gene expression is lower after incubation at high glucose compared with low glucose, suggesting that different mechanisms operate to keep Parkin in check [30].
This aspect also appears central to the recently identified role of Parkin in activating the NF-ĸB pathway [32]. In response to cellular stress, Parkin interacts with the linear ubiquitin assembly complex (LUBAC) in human neuroblastoma SH-SY5Y cells and mouse embryonic fibroblasts [33]. In this process, linear ubiquitination of NF-ĸB essential modulator (NEMO) is increased, thus inducing NF-ĸB signalling [33]. OPA1, a key regulator of inner mitochondrial membrane integrity and fusion that is associated with prevention of mitochondrial cytochrome c release [5, 10, 34], contains NF-ĸB-responsive promoter elements [33, 35]. Thus, the upregulation of OPA1 might explain the stress-protective and anti-apoptotic functions of Parkin that have to date been difficult to reconcile with its long-known task as an E3 ubiquitin ligase. Another recent study has questioned the involvement of the canonical NF-ĸB pathway and favours the non-classical pathway [36]. However, even that study does not doubt that the NF-ĸB pathway controls mitochondrial dynamics and OPA1 regulation [33, 36]. Further work to elucidate this pathway will clearly be important, because there is increasing evidence for OPA1 as a master mitochondrial cristae biogenetic factor [37–39]. In a recent study, moderate OPA1 overexpression was able to ameliorate the phenotype of two mouse models with defective mitochondrial bioenergetics [37]. Other research has demonstrated that insulin acts to stimulate the NF-ĸB–OPA1 pathway via mTOR signalling in cardiomyocytes [40], highlighting the cell type-specific nature of this regulation and the importance of beta cell-specific investigations regarding OPA1 regulation.
The TNFα–NF-ĸB–OPA1 pathway
Whereas cytokine-induced activation of the NF-ĸB pathway and its crosstalk with the unfolded protein response has been well studied in pancreatic beta cells [41–44], the regulation of mitochondrial health and its responsiveness to TNFα has not—to my knowledge—been addressed in this context. However, this interface might be vital for the balance between pancreatic beta cell survival and induction of mitochondrial outer membrane permeabilisation and cytochrome c release, culminating in apoptotic cell loss. Mitochondrial fragmentation is known to be increased in response to various stressors, rendering individual organelles more susceptible to mitophagy and enabling efficient clearance of the network. To avoid critical loss of mitochondrial mass, the TNFα–NF-ĸB–OPA1 pathway is apparently essential for re-balancing the system by increasing mitochondrial fusion and improving mitochondrial cristae structure and, hence, respiratory chain efficiency [37–39]. We have shown in the laboratory that the TNFα–NF-ĸB–OPA1 pathway plays a role in regulating mitochondrial integrity in primary mouse islets and insulin-secreting MIN6 cells (S. Baltrusch, unpublished results). In addition, we have detected an increase in TNFα expression alongside an increase in OPA1 following induction of mitochondrial stress. Interestingly, a significant increase in IL-6 was also detected (S. Baltrusch, unpublished results).
IL-6–FIS1 and IL-6–PGC1α–MFN2 pathways
IL-6 has been shown to be expressed in human islets and—to a somewhat greater extent—in those from patients with type 2 diabetes mellitus [45]. Furthermore, induction of IL-6 expression by palmitate, but not oleate or high glucose, has been demonstrated in human islets [45]. Another study has revealed phosphorylation of the signal transducer and activator of transcription 3 (STAT3) in response to treatment of human islets with IL-6 [46]. STAT3 activation is well-known to be involved in the regulation of autophagy and also affects mitophagy [47]. Direct IL-6-induced effects on mitochondrial remodelling have been demonstrated in C2C12 myotubes [48], a finding in accordance with our own preliminary results on FIS1 and MFN2 expression in mouse pancreatic islets (S. Baltrusch, unpublished results). IL-6 treatment has been shown to increase FIS1 gene expression, which induces mitochondrial fission. In addition, downregulation of peroxisome proliferator-activated receptor γ co-activator 1-α (PGC1α) diminishes the induction of MFN2, resulting in reduced mitochondrial fusion and metabolism [48–51]. Thus, it is legitimate to speculate a possible imbalance in mitochondrial dynamics, with higher fission and reduced fusion in response to IL-6. It is also noteworthy that MFN2 has an anti-apoptotic effect in B cell lymphoma 2 (Bcl-2)-dependent stabilisation of Bcl-2 homologous antagonist killer (BAK) and prevents the interaction between MFN1 and BAK [50].
Summary and perspective
In summary, alongside the known autophagy–inflammation–cell death axis [35, 52, 53], there is increasing evidence of direct interplay between mitochondrial remodelling and health control and inflammatory pathways. The PINK1–Parkin, TNFα–NF-ĸB–OPA1 and IL-6–FIS1/IL-6–PGC1α–MFN2 pathways delineated here still need improvement and revision, but have been shown to be important in recent work. Future studies are necessary to establish (1) whether these pathways are active in pancreatic beta cells in the same way as has been shown for other cell types; (2) whether they play a role in the manifestation of pancreatic beta cell dysfunction and/or loss in type 2 diabetes; and (3) whether inflammatory responsiveness is increased following mitochondrial stress.
Abbreviations
- Bcl-2:
-
B cell lymphoma 2
- CLEC16A:
-
C-type lectin domain family 16, member A
- FIS1:
-
Fission protein 1
- MFN1:
-
Mitofusin 1
- MFN2:
-
Mitofusin 2
- MMP:
-
Mitochondrial membrane potential
- mTOR:
-
Mammalian target of rapamycin
- OPA1:
-
Optic atrophy protein 1
- PGC1α:
-
Peroxisome proliferator-activated receptor gamma co-activator 1-alpha
- PINK1:
-
PTEN-induced putative kinase 1
- STAT3:
-
Signal transducer and activator of transcription 3
References
Kaufman BA, Li C, Soleimanpour SA (2015) Mitochondrial regulation of beta-cell function: maintaining the momentum for insulin release. Mol Asp Med 42:91–104
Yoon Y, Galloway CA, Jhun BS, Yu T (2011) Mitochondrial dynamics in diabetes. Antioxid Redox Signal 14:439–457
Chan DC (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125:1241–1252
de Castro IP, Martins LM, Loh SH (2011) Mitochondrial quality control and Parkinson’s disease: a pathway unfolds. Mol Neurobiol 43:80–86
Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11:872–884
Stiles L, Shirihai OS (2012) Mitochondrial dynamics and morphology in beta-cells. Best Pract Res Clin Endocrinol Metab 26:725–738
Loson OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24:659–667
Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–2256
Yoon Y, Krueger EW, Oswald BJ, McNiven MA (2003) The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 23:5409–5420
Chen H, Chan DC (2009) Mitochondrial dynamics—fusion, fission, movement, and mitophagy—in neurodegenerative diseases. Hum Mol Genet 18:R169–R176
Westermann B (2008) Molecular machinery of mitochondrial fusion and fission. J Biol Chem 283:13501–13505
Twig G, Elorza A, Molina AJ et al (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446
Twig G, Hyde B, Shirihai OS (2008) Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta 1777:1092–1097
Kitada T, Asakawa S, Hattori N et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803
Narendra DP, Jin SM, Tanaka A et al (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8, e1000298
Winklhofer KF (2014) Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol 24:332–341
Watada H, Fujitani Y (2015) Minireview: autophagy in pancreatic beta-cells and its implication in diabetes. Mol Endocrinol 29:338–348
Jung HS, Chung KW, Won Kim J et al (2008) Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 8:318–324
Chu KY, O’Reilly L, Ramm G, Biden TJ (2015) High-fat diet increases autophagic flux in pancreatic beta cells in vivo and ex vivo in mice. Diabetologia 58:2074–2078
Goginashvili A, Zhang Z, Erbs E et al (2015) Insulin granules. Insulin secretory granules control autophagy in pancreatic beta cells. Science 347:878–882
Han D, Yang B, Olson LK et al (2010) Activation of autophagy through modulation of 5'-AMP-activated protein kinase protects pancreatic beta-cells from high glucose. Biochem J 425:541–551
Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS (2011) Fatty acids suppress autophagic turnover in beta-cells. J Biol Chem 286:42534–42544
Stienstra R, Haim Y, Riahi Y, Netea M, Rudich A, Leibowitz G (2014) Autophagy in adipose tissue and the beta cell: implications for obesity and diabetes. Diabetologia 57:1505–1516
Ebato C, Uchida T, Arakawa M et al (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab 8:325–332
Masini M, Bugliani M, Lupi R et al (2009) Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 52:1083–1086
Molina AJ, Wikstrom JD, Stiles L et al (2009) Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes 58:2303–2315
Hakonarson H, Grant SF, Bradfield JP et al (2007) A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 448:591–594
Soleimanpour SA, Gupta A, Bakay M et al (2014) The diabetes susceptibility gene Clec16a regulates mitophagy. Cell 157:1577–1590
Hofmeister-Brix A, Kollmann K, Langer S, Schultz J, Lenzen S, Baltrusch S (2013) Identification of the ubiquitin-like domain of midnolin as a new glucokinase interaction partner. J Biol Chem 288:35824–35839
Jin HS, Kim J, Lee SJ et al (2014) The PARK2 gene is involved in the maintenance of pancreatic β-cell functions related to insulin production and secretion. Mol Cell Endocrinol 382:178–189
Sha D, Chin LS, Li L (2010) Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-κB signaling. Hum Mol Genet 19:352–363
Muller-Rischart AK, Pilsl A, Beaudette P et al (2013) The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell 49:908–921
Olichon A, Baricault L, Gas N et al (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278:7743–7746
Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ (2015) Redox homeostasis and mitochondrial dynamics. Cell Metab 22:207–218
Laforge M, Rodrigues V, Silvestre R et al (2016) NF-κB pathway controls mitochondrial dynamics. Cell Death Differ 23:89–98
Civiletto G, Varanita T, Cerutti R et al (2015) Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab 21:845–854
Varanita T, Soriano ME, Romanello V et al (2015) The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab 21:834–844
Zhang Z, Wakabayashi N, Wakabayashi J et al (2011) The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell 22:2235–2245
Parra V, Verdejo HE, Iglewski M et al (2014) Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFkappaB-Opa-1 signaling pathway. Diabetes 63:75–88
Azevedo-Martins AK, Lortz S, Lenzen S, Curi R, Eizirik DL, Tiedge M (2003) Improvement of the mitochondrial antioxidant defense status prevents cytokine-induced nuclear factor-kappaB activation in insulin-producing cells. Diabetes 52:93–101
Cardozo AK, Heimberg H, Heremans Y et al (2001) A comprehensive analysis of cytokine-induced and nuclear factor-κB-dependent genes in primary rat pancreatic beta-cells. J Biol Chem 276:48879–48886
Chan JY, Biden TJ, Laybutt DR (2012) Cross-talk between the unfolded protein response and nuclear factor-kappaB signalling pathways regulates cytokine-mediated beta cell death in MIN6 cells and isolated mouse islets. Diabetologia 55:2999–3009
Eizirik DL, Cardozo AK, Cnop M (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29:42–61
Igoillo-Esteve M, Marselli L, Cunha DA et al (2010) Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 53:1395–1405
Russell MA, Cooper AC, Dhayal S, Morgan NG (2013) Differential effects of interleukin-13 and interleukin-6 on Jak/STAT signaling and cell viability in pancreatic beta-cells. Islets 5:95–105
You L, Wang Z, Li H et al (2015) The role of STAT3 in autophagy. Autophagy 11:729–739
White JP, Puppa MJ, Sato S et al (2012) IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the Apc Min/+ mouse. Skelet Muscle 2:14
Martinez-Redondo V, Pettersson AT, Ruas JL (2015) The hitchhiker’s guide to PGC-1α isoform structure and biological functions. Diabetologia 58:1969–1977
Waxman AB, Kolliputi N (2009) IL-6 protects against hyperoxia-induced mitochondrial damage via Bcl-2-induced Bak interactions with mitofusins. Am J Respir Cell Mol Biol 41:385–396
Zorzano A, Liesa M, Palacin M (2009) Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol 41:1846–1854
Barazzoni R (2012) Modulating mitochondrial fission to lower diabetic oxidative stress. Diabetes 61:1915–1917
Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333:1109–1112
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Duality of interest
The author declares that there is no duality of interest associated with this manuscript.
Contribution statement
SB is the sole author of this manuscript.
Rights and permissions
About this article

Cite this article
Baltrusch, S. Mitochondrial network regulation and its potential interference with inflammatory signals in pancreatic beta cells. Diabetologia 59, 683–687 (2016). https://doi.org/10.1007/s00125-016-3891-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-3891-x