
Abstract
Aims/hypothesis
Hypertriacylglycerolaemia is a hallmark of diabetic dyslipidaemia with increased concentrations of triacylglycerol (TG)-rich VLDL1 particles. However, whether VLDL1 secretion or removal is abnormal in type 2 diabetes remains unclear. The aim of this study was to compare basal and insulin-mediated VLDL1- and VLDL2-TG kinetics in men with type 2 diabetes and healthy men using a novel direct VLDL1- and VLDL2-TG labelling method.
Methods
Twelve men with type 2 diabetes and 12 healthy men matched for age and BMI were recruited. VLDL1- and VLDL2-TG turnover were measured during a 4 h basal period and a 3.5 h hyperinsulinaemic clamp period using a primed-constant infusion of ex vivo labelled VLDL1-TG and VLDL2-TG.
Results
Basal VLDL1-TG and VLDL2-TG secretion rates were similar in men with diabetes and healthy men. During hyperinsulinaemia, VLDL1-TG secretion rates were suppressed significantly in both groups, whereas no suppression of VLDL2-TG secretion rate was observed. VLDL1-TG to VLDL2-TG transfer rate was not significantly different from zero in both groups, while VLDL1-TG fatty acid oxidation rate was substantial, with a contribution to total energy expenditure of approximately 15% during postabsorptive conditions. VLDL1 and VLDL2 particle size (TG/apolipoprotein B [apoB] ratio) and apoB-100 concentration were unaltered by hyperinsulinaemia in men with type 2 diabetes, but significantly reduced in healthy men.
Conclusions/interpretation
Insulin inhibits VLDL1-TG secretion rate similarly in age- and BMI-matched men with type 2 diabetes and healthy men, while VLDL2-TG secretion is unaltered by hyperinsulinaemia. However, VLDL1- and VLDL2-apoB levels are not lowered by hyperinsulinaemia in men with type 2 diabetes, which is indicative of a diminished hepatic response to insulin.
Trial registration
ClinicalTrials.gov NCT01564550
Similar content being viewed by others

Introduction
Dyslipidaemia is an established atherogenic risk factor associated with obesity and type 2 diabetes [1], comprising postprandial lipidaemia, hypertriacylglycerolaemia, low levels of HDL-cholesterol and increased levels of small, dense LDL particles. An elevated concentration of VLDL appears to be central to the development of dyslipidaemia [2–5]. However, there is still uncertainty regarding VLDL regulation. Although chronic hyperinsulinaemia in obesity and type 2 diabetes is associated with increased VLDL production [6], this is not the case in insulinoma patients, indicating that the former primarily reflects an association with insulin resistance. In fact, acute experimental hyperinsulinaemia decreases VLDL secretion in both insulin-sensitive and insulin-resistant individuals [7–9]. Insulin inhibits hepatic VLDL secretion both directly and indirectly, by decreasing hepatic delivery of NEFA [8, 10, 11].
Plasma VLDL can be separated into large triacylglycerol (TG)-rich VLDL1 (Svedberg flotation units [Sf] 60–400) and smaller VLDL2 (Sf 20–60) particles. VLDL1 is solely secreted from the liver, while VLDL2 is formed either directly in the liver or from conversion of plasma VLDL1. Previously, the contribution from VLDL1 to VLDL2 has been estimated to be more than twice the hepatic contribution [12, 13]. In addition, studies suggest that solely VLDL1 production is suppressed by insulin; therefore, abnormal insulin suppression of VLDL1 seems to promote diabetic dyslipidaemia [4, 12, 14]. Presently, however, there are still uncertainties regarding VLDL1 and VLDL2 regulation [4, 14, 15], perhaps due to methodological shortcomings. Most earlier studies have used bolus infusions of stable isotope precursors in combination with multicompartmental modelling to assess VLDL1- and VLDL2-TG kinetics [15], whereas direct measurements of VLDL1- and VLDL2-TG kinetics using traditional tracer dilution techniques under steady-state conditions have not been published.
The aim of this study was to assess VLDL1- and VLDL2-TG kinetics during postabsorptive and hyperinsulinaemic conditions, by use of a previously validated ex vivo labelling technique [16], allowing simultaneous assessment of VLDL-TG subclass turnover from isotope dilution measurements. We hypothesised that men with type 2 diabetes had greater VLDL1-TG secretion rates and that an impaired VLDL1-TG oxidation rate or VLDL1-TG to VLDL2-TG transfer rate could also contribute to increased VLDL1-TG concentrations.
Methods
The study was approved by the local ethics committee, and informed consent was obtained from all participants.
Participants
Twelve men with type 2 diabetes and 12 healthy men, matched for BMI and age, were recruited through local advertisements. All had stable weight, were non-smokers and did not suffer from other diseases. A normal haematology and chemistry panel was documented. Additional inclusion criteria were age 35–65 years, BMI 25–35 kg/m2 and HbA1c ≤9% (75 mmol/mol). Diabetes treatments were lifestyle modifications in two patients, metformin in seven, sitagliptin in one, sulfonylurea in one, and sitagliptin/metformin and sulfonylurea in one patient. Glucose-lowering medication was discontinued 3 weeks before the study day. Five men with diabetes received antihypertensive treatment and four received simvastatin treatment, which was discontinued 2 weeks before the study day. One healthy individual was excluded because of insufficient VLDL kinetic data.
Protocol
Four days before the study day a 108 ml blood sample was drawn after a 10 h overnight fast for ex vivo labelling of VLDL1-TG and VLDL2-TG. A whole-body dual-energy x-ray absorptiometry (DXA) scan and an abdominal computed tomography (CT) scan were performed. To ensure comparable macronutrient intake, participants were interviewed by a dietitian, and a weight-maintaining diet (55% carbohydrate, 15% protein, 30% fat) was provided by the hospital kitchen for 3 days prior to the study day. Participants were also instructed not to perform high-intensity exercise or drink alcohol in this period.
Study day
Participants were admitted to the clinical research centre at 22:00 hours the evening before the study day. From this time they remained in bed until the end of the study (with the exception of voiding) and fasted (with the exception of tap water). The following morning, catheters were placed in an antecubital vein for infusion and in a contralateral heated hand vein for sampling of arterialised blood.
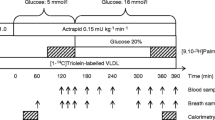
The protocol is illustrated in Fig. 1. At 07:30 hours (t = 0), a primed-constant infusion (20% priming dose) of [1-14C]VLDL1-TG and [9,10-3H]VLDL2-TG tracers was initiated. At t = 240 min, a 4 h infusion of insulin (1.0 mU kg FFM−1 min−1) (Actrapid; Novo Nordisk, Bagsvaerd, Denmark) was commenced. Plasma glucose was measured every 10 min and clamped at 5 mmol/l by variable infusion of 20% glucose. Glucose infusion rate during the last hour of the clamp was used as an index of insulin sensitivity (M value). Blood samples were drawn to determine VLDL1- and VLDL2-TG specific activity (SA) at t = 0, 180, 200, 220 and 240 min in the basal period and at t = 360, 390, 410, 430 and 450 min in the clamp period. Insulin and NEFA concentrations were determined every 60 min and apolipoprotein B-100 (apoB-100) concentration at t = 220 and 430 min. Breath and blood samples to determine 14CO2 and 3H2O SA were obtained at t = 0, 150, 180, 200, 220, 240, 360, 390, 410, 420 and 450 min. Indirect calorimetry was performed at t = 60–90 min and t = 420–450 min. At t = 450 min, catheters were removed and the participants were discharged when blood glucose had stabilised.

Study protocol
VLDL1- and VLDL2-TG tracer preparation
The ex vivo labelling technique used in this study has previously been validated and described in detail [16, 17]. Each half of plasma obtained from the 108 ml blood sample was mixed with either 1.11 MBq [1-14C]triolein or 1.48 MBq [9,10-3H]triolein and sonicated at 4°C for 2 h.
Lipoprotein fractioning was then performed by density gradient ultracentrifugation, as described by Redgrave and Carlson [18], with minor modification [19]. Cumulative rate ultracentrifugation times were calculated from g/min from VLDL subfractioning experiments by Lindgren et al [20], adjusted for differences in temperature [21, 22]. Plasma was adjusted to density 1.10 kg/l with solid NaCl. A density gradient consisting of 4 ml of 1.10 kg/l labelled plasma and 3 ml each of 1.065, 1.020 and 1.006 kg/l NaCl solutions was layered in 13.4 ml polyallomer tubes (Beckman Coulter, Brea, CA, USA). The tubes containing [1-14C]triolein-labelled plasma were then centrifuged in an SW 40 Ti Rotor (Beckman Coulter) for 22 min at 285,000 g at 10°C and the 0.5 ml supernatant fraction was discarded to ensure removal of chylomicrons and free triolein (Sf >400). The supernatant fraction was replaced with 1.006 kg/l saline solution, and samples were centrifuged for another 3 h and 55 min to isolate the [1-14C]VLDL1-TG-containing supernatant fraction. The 1 ml top fraction containing labelled VLDL1 was then isolated and filtered into sterile tubes and stored at 4°C for later reinfusion. Tubes with [9,10-3H]triolein-labelled plasma were centrifuged for 3 h and 55 min at 285,000 g at 10°C to isolate the VLDL1-containing top fraction (1 ml), which was discarded. The supernatant fraction was replaced with 1.006 kg/l saline solution, and samples were centrifuged for another 17 h and 51 min to isolate the [9.10-3H]VLDL2-TG-containing supernatant fraction. The 1 ml top fraction with labelled VLDL2 was then isolated and filtered into sterile tubes and stored at 4°C for later reinfusion. All VLDL isolation procedures were performed under sterile conditions and the labelled VLDL subfractions were tested for bacterial growth to ensure sterility.
Plasma VLDL1- and VLDL2-TG concentration and SA
VLDL1 and VLDL2 from study day plasma samples were isolated as described above. The VLDL1 and VLDL2 subfractions were transferred into scintillation vials. A 300 μl sample from each vial was analysed for TG content, after which the plasma concentration of VLDL1- and VLDL2-TG was calculated. Scintillation fluid (Optiphase HiSafe 2; PerkinElmer, Waltham, MA, USA) was added to the remaining sample, and 3H and 14C activity, expressed as disintegrations per min (dpm), was assessed using dual-channel counting to a <2% counting error, with a ratio of 3H/14C activity of approximately 3/1.
14CO2 and 3H2O activity
Breath samples were collected in IRIS breath bags (Wagner Analysen Technik, Bremen, Germany) and 14CO2 activity was determined as previously described [17]. Plasma 3H2O activity was also measured as previously described [23], but since the activity was not sufficiently greater than background, calculations of VLDL2-TG fatty acid (FA) oxidation could not be performed.
Indirect calorimetry
Resting energy expenditure (REE), respiratory exchange rates and net substrate oxidation rates were measured by indirect calorimetry using a Deltatrac monitor (Datex Instruments, Baldwin Park, CA, USA) in combination with protein oxidation rates estimated from urinary urea excretion [24].
Body composition
Body composition was assessed by DXA scan (QDR-2000; Hologic, Bedford, MA, USA). Upper-body fat and visceral fat mass were calculated from CT scan of intra-abdominal and subcutaneous adipose tissue combined with abdominal fat mass measured from the DXA scan, as previously described [25]. Abdominal subcutaneous fat was taken as upper-body fat (DXA) minus visceral fat. Leg fat was measured using the ‘region of interest’ program in the DXA instrument.
Laboratory procedures
Plasma glucose was measured using an YSI 2300 STAT Plus glucose analyser (YSI, Yellow Springs, OH, USA). Blood samples were cooled and separated as quickly as possible by centrifugation (2,753 g at 4°C for 10 min). Aliquots of plasma were stored at 4°C for isolation of VLDL subfractions after study day completion. Remaining samples were stored at −80°C for later analysis. TG concentrations were analysed using a cobas c 111 analyser (Roche, Basel, Switzerland). ApoB-100 concentrations were measured by immunoassay (Mabtech, Nacka Strand, Sweden). Serum insulin was measured using an AutoDELFIA immunoassay (PerkinElmer), and serum NEFA by a colorimetric method (Wako, Neuss, Germany).
Calculations
VLDL1 and VLDL2-TG SA (dpm/μmol) steady state was effectively reached during the basal and hyperinsulinaemic periods.
VLDL1- and VLDL2-TG rate of appearance (RA) was calculated by dividing tracer infusion rate with the steady-state SA of VLDL1- or SA VLDL2-TG, respectively:
VLDL1-TG secretion rate was corrected for [9,10-3H]VLDL2-TG in the isolated VLDL1-TG pool:
and VLDL2-TG secretion rate for [1-14C]VLDL1-TG in the isolated VLDL2-TG pool:
Net VLDL1 to VLDL2 transfer rate was calculated from the following equation:
VLDL1-TG and VLDL2-TG clearance rate was calculated by dividing the VLDL1- and VLDL2-TG secretion rate by the VLDL1- and VLDL2-TG concentration (CVLDL), respectively:
In participants where VLDL1-TG concentration was suppressed below the detection limit (0.01 mmol/l) during the clamp a conservative value of 0.009 mmol/l was used for kinetic calculations.
Fractional oxidation (%) of the infused [1-14C]VLDL1-TG was calculated as follows:
Where k is the volume of 1 mol of CO2 at 20°C and 1 atm. pressure (22.4 l/mol) and Ar is the fractional acetate carbon recovery factor in breath CO2, and F is the tracer infusion rate. Ar has been estimated to be 0.56 during basal resting conditions [26].
Total VLDL1-TG FA oxidation rate (μmol/min) rate was calculated by multiplying fractional VLDL1-TG oxidation by VLDL1-TG secretion rate. Energy production (kJ/day) from VLDL1-TG FA oxidation was calculated as described previously [17].
Statistics
Data are mean ± SD or median (range). Where appropriate, data were log transformed to obtain normal distribution. Differences in degree of insulin suppression between groups were accessed by two-way ANOVA or by Student’s t test for paired comparisons of delta values. Comparisons between groups were performed using Student’s t test or the Mann–Whitney U test. Within groups, comparisons were performed using Student’s t test for paired comparisons or Wilcoxon’s test. A p value below 0.05 was considered significant.
Results
Participant characteristics
The groups were well matched for age and BMI, and there were no difference in body composition apart from a significantly greater visceral fat mass in men with type 2 diabetes (Table 1). The healthy men had significantly higher total-cholesterol and LDL-cholesterol at screening compared with the men with type 2 diabetes, several of whom was treated with lipid-lowering agents (discontinued 2 weeks before the study day).
Metabolites and metabolic variables
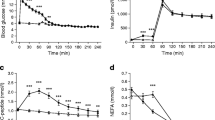
Basal study day plasma glucose and serum insulin were significantly greater in men with type 2 diabetes (Fig. 2, Table 2). No significant differences were found in concentrations of serum NEFA, plasma TG, VLDL1-TG or VLDL2-TG. During the clamp, plasma TG was significantly and equally suppressed and a near-complete suppression of serum NEFA was observed in both groups. Plasma VLDL1-TG was only significantly reduced in healthy men (p < 0.01), but the level of decrease was not significantly different between groups (ANOVA). Plasma VLDL2-TG was significantly and similarly reduced in both groups. Men with type 2 diabetes were less insulin sensitive.

Concentration of glucose (a), insulin (b), NEFA (c), TG (d), VLDL1-TG (e), VLDL2-TG (f). Black circles, healthy men; white circles, men with type 2 diabetes. Data are mean ± SEM (a), mean ± SD (b) or median (25th–75th percentile) (c, d, e, f). *p < 0.05 between groups (last hour of each period)
VLDL1- and VLDL2-TG kinetics
Basal VLDL1-TG secretion rate was comparable in men with diabetes and healthy men (34 ± 25 vs 43 ± 36 μmol/min; NS) (Fig. 3). During hyperinsulinaemia, VLDL1-TG secretion rate was suppressed significantly and similarly in both groups. VLDL1-TG secretion rate decreased to 17 ± 32 μmol/min (p < 0.05) in men with type 2 diabetes and to 14 ± 26 μmol/min (p < 0.001) in healthy men. VLDL1-TG clearance rates were comparable in groups during the basal state (90 [34–334] vs 118 [33–614] ml/min; NS), while clearance rate during hyperinsulinaemia could not be calculated, due to complete suppression of plasma VLDL1-TG concentrations in three men with diabetes and five healthy men, which, when entered in the formula for calculation of clearance rate, resulted in abnormally high values.

VLDL1-TG secretion rate (a), VLDL2-TG secretion rate (b), VLDL2-TG clearance rate (c), VLDL1-TG FA oxidation rate (d), VLDL1-TG FA oxidation, fraction of energy expenditure (EE) (e). Black bars, healthy men; white bars, men with type 2 diabetes. Data are mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 within-group comparisons basal vs clamp
Basal VLDL2-TG secretion rate was not significantly different in the groups (men with type 2 diabetes 15.5 ± 14.3 μmol/min; healthy men 12.0 ± 11.8 μmol/min) and did not change significantly during hyperinsulinaemia. However, the secretion rate tended to increase in healthy men (p = 0.05). Basal VLDL2-TG clearance rate was also comparable (men with type 2 diabetes 64 ± 75; healthy men 69 ± 68 ml/min; NS) and increased similarly during the clamp (men with type 2 diabetes 97 ± 76 ml/min, p < 0.05; healthy men 163 ± 150, p = 0.06; NS). While basal VLDL1- and VLDL2-TG clearance rates were comparable in men with diabetes, VLDL1-TG clearance rate was significantly greater than VLDL2-TG clearance rate in healthy men (p < 0.05). VLDL1 to VLDL2 transfer rate was negligible and not significantly different from zero in either group.
VLDL1 oxidation
Basal VLDL1-TG FA oxidation rate was comparable in men with diabetes and healthy men (17 ± 13 vs 22 ± 21 μmol/min; NS), and decreased similarly during hyperinsulinaemia (NS) (Fig. 3). Levels during the clamp were reduced to 10 ± 22 μmol/min (NS) in men with type 2 diabetes and to 8.3 ± 16.7 μmol/min (p < 0.01) in healthy men. The relative contribution from VLDL1-TG FA oxidation to total REE was decreased similarly (NS) from 17% to 10% in men with type 2 diabetes (NS) and from 13% to 5% in healthy men (p < 0.01).
VLDL1- and VLDL2-apoB and TG/apoB ratio
Basal and clamp VLDL1-apoB-100 concentrations and TG/apoB ratio were comparable between groups (Table 3). VLDL1-apoB concentration was lowered significantly more by hyperinsulinaemia in healthy men vs men with diabetes (p < 0.05, ANOVA), and was only significantly reduced in healthy men (p < 0.001). The level of reduction in VLDL1-TG/apoB ratio was not significantly different between groups (ANOVA), although VLDL1-TG/apoB ratio was only significantly reduced during the clamp in healthy men (p < 0.05).
Basal VLDL2-apoB concentration and VLDL2-TG/apoB ratio were comparable in men with type 2 diabetes and healthy men, and the level of reduction in both variables during hyperinsulinaemia was not significantly different between groups (ANOVA). However, VLDL2-apoB concentration was significantly reduced during the clamp in healthy men (p < 0.05), as was the VLDL2-TG/apoB ratio (p < 0.05).
Correlations
No significant correlations were observed in any variables in men with type 2 diabetes. By contrast, clamp VLDL1-TG secretion rate correlated inversely with the M value (r = −0.75, p = 0.008) and positively with visceral fat mass (r = 0.62, p = 0.04) in healthy men. Furthermore, inverse correlations were found between M value and basal VLDL1-apoB level (r = −0.72, p = 0.01) and VLDL2-apoB level (r = 0.61, p = 0.04), respectively.
Discussion
This study was undertaken to determine basal and insulin-regulated VLDL1- and VLDL2-TG secretion in men with type 2 diabetes and healthy men matched for age- and BMI. In addition, we wanted to quantify plasma VLDL1-TG to VLDL2-TG transfer rate and VLDL1-TG oxidation rate. Basal VLDL1- and VLDL2-TG secretions rates were similar in men with diabetes and healthy men, and experimental hyperinsulinaemia suppressed VLDL1-TG secretion significantly in both groups. Conversely, VLDL2-TG secretion remained unaffected by insulin. In addition, we found that net VLDL1-TG to VLDL2-TG transfer rate was negligible, whereas a significant proportion of VLDL1-TG turnover was accounted for by oxidation.
We compared men with type 2 diabetes with age- and BMI-matched healthy men in order to minimise the independent effects of sex and body weight on VLDL-TG kinetics. Using this design we found similar postabsorptive VLDL1-TG and VLDL2-TG secretion rates in the two groups. In two previous, partly overlapping, studies, VLDL1-TG and VLDL2-TG secretion rates were reported to be increased in men with type 2 diabetes [12, 27]. However, the healthy men had significantly lower body weight; therefore, the differences might not be invariably ascribed to the presence of type 2 diabetes but may equally be related to insulin resistance or other body weight-associated effects. In another study of men with type 2 diabetes and healthy men with similar BMI, postabsorptive VLDL1-apoB secretion rate was found to be comparable. Therefore, postabsorptive VLDL1- and VLDL2-TG secretion rates do not seem to be increased by type 2 diabetes per se, but by other overweight/obesity associated factors, e.g. insulin resistance or increased intrahepatic fat. The present observations may appear in contrast with findings of a previous study in which basal total VLDL-TG secretion rate was found to be greater in men with type 2 diabetes [9]. Compared with the present study, body composition, insulin concentrations and M values were equivalent, but lower baseline total TG in healthy men in the former study and differences in methodology may explain the different findings.
The present study is the first to compare the effects of insulin on both VLDL1-TG and VLDL2-TG secretion rates in men with type 2 diabetes and healthy men, matched for age and BMI. We found VLDL1-TG secretion rate to be equally and significantly reduced during hyperinsulinaemia in both groups. Only two previous studies have examined the effect of hyperinsulinaemia on VLDL1 turnover in type 2 diabetes, both using multicompartmental modelling of VLDL turnover. In the first study, insulin suppression of VLDL1-TG glycerol and VLDL1-apoB secretion were both blunted in a group of mainly men with type 2 diabetes with high liver fat content compared with non-diabetic men with normal liver fat [28]. However, men with high liver fat also had greater body weight. Therefore, conclusions regarding the effect of type 2 diabetes cannot be drawn with certainty, as hepatic steatosis and weight-associated insulin resistance both independently impact on lipid metabolism. In the second study, hyperinsulinaemia failed to suppress VLDL1-apoB secretion in men with type 2 diabetes compared with healthy men with similar BMI [29]. Surprisingly, the experimental protocol also failed to suppress plasma TG concentration despite 8.5 h of hyperinsulinaemia (1.0 mU kg−1 min−1) and plasma insulin concentrations of ∼550 pmol/l, where we used a lower insulin infusion rate of 1.0 mU kg FFM−1 min−1 (plasma insulin ∼300 pmol/l). Thus, it appears that the men with type 2 diabetes in the latter study were severely insulin resistant. Another explanation could be different regulation of VLDL-TG compared with VLDL-apoB [30]. VLDL2-TG secretion rates tended to increase during hyperinsulinaemia in both groups, as also reported by others [28, 31]. Since VLDL2-TG clearance rate also increased, VLDL2-TG concentration was, however, lowered during the clamp.
Interestingly, basal VLDL1-TG clearance rate was significantly greater than VLDL2-TG clearance rate in healthy men, but not in men with diabetes, suggesting that VLDL1 is removed more rapidly from the circulation in healthy men. In another study, impaired VLDL1 clearance was found to explain ∼48% of the increased serum TG in insulin-resistant obese individuals [32].
An important advantage of our experimental approach is the use of two different tracers, which allows independent tracing of VLDL1-TG and VLDL2-TG simultaneously, whereas studies relying on mathematical modelling only use one tracer to label both VLDL subclasses, e.g. leucine and/or glycerol. Such approaches may limit the ability to detect accurate exchange rates between VLDL1-TG and VLDL2-TG. Furthermore, labelling of the FA moiety of the TG molecule allows measurements of TG FA oxidation. The present study is the first to report data on VLDL1-TG oxidation. We found that the oxidation rate was comparable between groups in the basal state, accounting for ∼15% of total REE. During the clamp, VLDL1-TG oxidation was only significantly reduced in healthy men. We have previously reported impaired insulin suppression of total VLDL-TG oxidation in similar groups of men with type 2 diabetes compared with healthy men [9], suggestive of a metabolic inflexibility in the shift from lipid to glucose oxidation during hyperinsulinaemia.
Another novel finding was that net VLDL1-TG to VLDL2-TG conversion was negligible in both groups, suggestive of either whole particle catabolism or rapid turnover of VLDL1-TG into other lipoproteins, which is supported by previously published data [33, 34]. Other studies have reported the contribution from VLDL1-TG to VLDL2-TG production to be 2.3–3.1 times greater than the direct hepatic VLDL2-TG secretion [12, 27]. Our results, therefore, challenge the current understanding of VLDL kinetics, where significant net amounts of plasma VLDL1 are assumed to convert to VLDL2. As discussed above, a probable explanation for the discrepancy may be that previous research is based on multicompartmental modelling of the behaviour of an intravenous bolus of a single precursor tracer, including theoretical assumptions regarding the transfer rate between VLDL1 and VLDL2. In the present study we had the advantage of two independent tracers to directly label the two VLDL-TG subclasses, which allowed assessment of the presence of each tracer in both pools. Importantly, even though gold standard VLDL1 and VLDL2 isolation was used, we noticed that the VLDL1 fraction invariably contained a substantial amount of [9.10-3H]VLDL2 and, similarly, that the VLDL2 fraction invariably contained a substantial amount of [1-14C]VLDL1. Thus, the SA of the tracer in the opposing VLDL subfraction was around 50% of that present in the original subfraction. This may not be surprising for at least two reasons. First, VLDL1 and VLDL2 subfractions originate from a continuous lipoprotein fraction only separated by an arbitrarily defined difference in density. Thus, one can easily envision that the VLDL particles populating the zone around the density cut-off line cannot be 100% separated along a clear density scale. This suggests that current protocols are likely to result in incomplete separation of VLDL subfractions, as demonstrated when labelling with two different tracers is performed. We are unaware of other studies that have used independent labelling of the two subclasses for assessment of the feasibility of complete separation. Second, cholesterol ester transfer protein enzyme could, in theory, mediate exchange of radiolabelled TG FA between VLDL subfractions. To our knowledge such exchanges have only been assessed between VLDL and HDL [2]. However, the limited time of action including the cold environment during separation would seem insufficient for this mechanism to allow a pronounced effect.
Both VLDL1- and VLDL2-apoB concentration and TG/apoB ratio decreased significantly during hyperinsulinaemia in healthy men, reflecting a reduction in both size and amount of VLDL1 and VLDL2 particles. Previous studies have found that VLDL1-apoB, but not VLDL2-apoB, concentration was reduced [10, 35]. Our findings suggest that de novo VLDL1- and VLDL2-apoB secretion is similarly regulated by insulin. In men with diabetes, a lack of VLDL1- and VLDL2-apoB inhibition was found, likely reflecting reduced hepatic insulin inhibition in the insulin-resistant state, as previously demonstrated [10].
This study has limitations. First, it is possible that another insulin dose during the clamp could have resulted in different responses [36]. However, the dose of 1.0 mU kg FFM−1 min−1 has previously been shown to suppress VLDL-TG in men with type 2 diabetes and matched healthy men [9] and to allow simultaneous assessments of insulin-mediated glucose and NEFA turnover [37]. Second, the relatively low number of participants could have introduced a type II error in statistical comparisons of groups. Third, the methodology of this study does not allow simultaneous labelling of apoB-100.
In conclusion, postprandial hepatic VLDL1- and VLDL2-TG secretion rate is comparable in age- and BMI-matched men with type 2 diabetes and healthy men. Hyperinsulinaemia suppresses VLDL1-TG, but not VLDL2-TG, secretion in both groups. However, VLDL1- and VLDL2-apoB concentrations are unaltered by hyperinsulinaemia in men with type 2 diabetes, indicating a decreased hepatic response to insulin. A novel finding of this study is a negligible net VLDL1-TG to VLDL2-TG transfer rate, suggesting that VLDL1-TG removal is determined by other pathways such as oxidation, which was found to be substantial. Further studies that determine the secretion and fate of VLDL1- and VLDL2-TG are warranted to understand the mechanisms leading to atherogenic dyslipidaemia.
Abbreviations
- apoB-100:
-
Apolipoprotein B-100
- CT:
-
Computed tomography
- dpm:
-
Disintegrations per min
- DXA:
-
Dual-energy x-ray absorptiometry
- FA:
-
Fatty acid
- RA:
-
Rate of appearance
- REE:
-
Resting energy expenditure
- SA:
-
Specific activity
- Sf:
-
Svedberg flotation units
- TG:
-
Triacylglycerol
References
Carr MC, Brunzell JD (2004) Abdominal obesity and dyslipidemia in the metabolic syndrome: importance of type 2 diabetes and familial combined hyperlipidemia in coronary artery disease risk. J Clin Endocrinol Metab 89:2601–2607
Guerin M, Lassel TS, Le Goff W, Farnier M, Chapman MJ (2000) Action of atorvastatin in combined hyperlipidemia: preferential reduction of cholesteryl ester transfer from HDL to VLDL1 particles. Arterioscler Thromb Vasc Biol 20:189–197
Adiels M, Olofsson SO, Taskinen MR, Boren J (2006) Diabetic dyslipidaemia. Curr Opin Lipidol 17:238–246
Taskinen MR (2003) Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia 46:733–749
Packard CJ, Shepherd J (1997) Lipoprotein heterogeneity and apolipoprotein B metabolism. Arterioscler Thromb Vasc Biol 17:3542–3556
Duvillard L, Florentin E, Pont F et al (2011) Endogenous chronic hyperinsulinemia does not increase the production rate of VLDL apolipoprotein B: proof from a kinetic study in patients with insulinoma. J Clin Endocrinol Metab 96:2163–2170
Durrington PN, Newton RS, Weinstein DB, Steinberg D (1982) Effects of insulin and glucose on very low density lipoprotein triglyceride secretion by cultured rat hepatocytes. J Clin Invest 70:63–73
Lewis GF, Uffelman KD, Szeto LW, Weller B, Steiner G (1995) Interaction between free fatty acids and insulin in the acute control of very low density lipoprotein production in humans. J Clin Invest 95:158–166
Sorensen LP, Andersen IR, Sondergaard E et al (2011) Basal and insulin mediated VLDL-triglyceride kinetics in type 2 diabetic men. Diabetes 60:88–96
Lewis GF, Uffelman KD, Szeto LW, Steiner G (1993) Effects of acute hyperinsulinemia on VLDL triglyceride and VLDL apoB production in normal weight and obese individuals. Diabetes 42:833–842
Vatner DF, Majumdar SK, Kumashiro N et al (2015) Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc Natl Acad Sci U S A 112:1143–1148
Adiels M, Boren J, Caslake MJ et al (2005) Overproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemia. Arterioscler Thromb Vasc Biol 25:1697–1703
Taskinen MR, Packard CJ, Shepherd J (1990) Effect of insulin therapy on metabolic fate of apolipoprotein B-containing lipoproteins in NIDDM. Diabetes 39:1017–1027
Gill JM, Brown JC, Bedford D et al (2004) Hepatic production of VLDL1 but not VLDL2 is related to insulin resistance in normoglycaemic middle-aged subjects. Atherosclerosis 176:49–56
Adiels M, Packard C, Caslake MJ et al (2005) A new combined multicompartmental model for apolipoprotein B-100 and triglyceride metabolism in VLDL subfractions. J Lipid Res 46:58–67
Gormsen LC, Jensen MD, Nielsen S (2006) Measuring VLDL-triglyceride turnover in humans using ex vivo-prepared VLDL tracer. J Lipid Res 47:99–106
Sorensen LP, Gormsen LC, Nielsen S (2009) VLDL-TG kinetics: a dual isotope study for quantifying VLDL-TG pool size, production rates, and fractional oxidation in humans. Am J Physiol Endocrinol Metab 297:E1324–E1330
Redgrave TG, Carlson LA (1979) Changes in plasma very low density and low density lipoprotein content, composition, and size after a fatty meal in normo- and hypertriglyceridemic man. J Lipid Res 20:217–229
Karpe F, Steiner G, Olivecrona T, Carlson LA, Hamsten A (1993) Metabolism of triglyceride-rich lipoproteins during alimentary lipemia. J Clin Invest 91:748–758
Lindgren FT, Jensen LC, Hatch FT (1972) Blood lipids and lipoproteins. Wiley, New York, pp 221–248
Mills GL, Lane PA, Weech PK (1984) A guidebook to lipoprotein technique. Amsterdam, pp 18-116
Tong H, Knapp HR, VanRollins M (1998) A low temperature flotation method to rapidly isolate lipoproteins from plasma. J Lipid Res 39:1696–1704
Heiling VJ, Miles JM, Jensen MD (1991) How valid are isotopic measurements of fatty acid oxidation? Am J Physiol 261:E572–E577
Frayn KN (1983) Calculation of substrate oxidation rates in vivo from gaseous exchange. J Appl Physiol 55:628–634
Sorensen LP, Sondergaard E, Nellemann B, Christiansen JS, Gormsen LC, Nielsen S (2011) Increased VLDL-triglyceride secretion precedes impaired control of endogenous glucose production in obese, normoglycemic men. Diabetes 60:2257–2264
Sidossis LS, Coggan AR, Gastaldelli A, Wolfe RR (1995) A new correction factor for use in tracer estimations of plasma fatty acid oxidation. Am J Physiol 269:E649–E656
Adiels M, Taskinen MR, Packard C et al (2006) Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 49:755–765
Adiels M, Westerbacka J, Soro-Paavonen A et al (2007) Acute suppression of VLDL1 secretion rate by insulin is associated with hepatic fat content and insulin resistance. Diabetologia 50:2356–2365
Malmstrom R, Packard CJ, Caslake M et al (1997) Defective regulation of triglyceride metabolism by insulin in the liver in NIDDM. Diabetologia 40:454–462
Melish J, Le NA, Ginsberg H, Steinberg D, Brown WV (1980) Dissociation of apoprotein B and triglyceride production in very-low-density lipoproteins. Am J Physiol 239:E354–E362
Malmstrom R, Packard CJ, Caslake M et al (1998) Effects of insulin and acipimox on VLDL1 and VLDL2 apolipoprotein B production in normal subjects. Diabetes 47:779–787
Taskinen MR, Adiels M, Westerbacka J et al (2011) Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler Thromb Vasc Biol 31:2144–2150
Beltz WF, Kesaniemi YA, Howard BV, Grundy SM (1985) Development of an integrated model for analysis of the kinetics of apolipoprotein B in plasma very low density lipoproteins, intermediate density lipoproteins, and low density lipoproteins. J Clin Invest 76:575–585
Gaw A, Packard CJ, Lindsay GM et al (1995) Overproduction of small very low density lipoproteins (Sf 20-60) in moderate hypercholesterolemia: relationships between apolipoprotein B kinetics and plasma lipoproteins. J Lipid Res 36:158–171
Malmstrom R, Packard CJ, Watson TD et al (1997) Metabolic basis of hypotriglyceridemic effects of insulin in normal men. Arterioscler Thromb Vasc Biol 17:1454–1464
Sondergaard E, Sorensen LP, Rahbek I, Gormsen LC, Christiansen JS, Nielsen S (2012) Postprandial VLDL-triacylglycerol secretion is not suppressed in obese type 2 diabetic men. Diabetologia 55:2733–2740
Jensen MD, Nielsen S (2007) Insulin dose response analysis of free fatty acid kinetics. Metabolism 56:68–76
Acknowledgements
The authors acknowledge the excellent technical assistance of D. Wulff, H. Petersen and L. Buus, Medical Research Laboratories, Aarhus University Hospital, Aarhus, Denmark.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was supported by a grant from the Danish Council for Independent Research, the Novo Nordisk Foundation, the Riisfort Foundation and the Aase og Ejnar Danielsens Foundation.
Duality of interest
There were no potential conflicts of interest related to this study.
Contribution statement
RFJ collected and analysed the data and wrote the manuscript. ES contributed to the analysis and interpretation of data and reviewed and edited the manuscript. LPS advised on method development and reviewed the manuscript. AGJ provided CT scan data analysis and reviewed the manuscript. JSC contributed to the analysis and interpretation of the data and reviewed the manuscript. SN designed the study, wrote the protocol, and reviewed and edited the manuscript. SN is the guarantor of the study, as he had full access to all data, and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors approved the final version to be published.
Additional information
Jens S. Christiansen died on 16 December 2015.
Rights and permissions
About this article

Cite this article
Johansen, R.F., Søndergaard, E., Sørensen, L.P. et al. Basal and insulin-regulated VLDL1 and VLDL2 kinetics in men with type 2 diabetes. Diabetologia 59, 833–843 (2016). https://doi.org/10.1007/s00125-015-3856-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3856-5