
Abstract
Aims/hypothesis
Glucose-dependent insulinotropic polypeptide (GIP) is a peptide hormone released from gut K cells. While the predominant form is GIP(1–42), a shorter form, GIP(1–30), is produced by pancreatic alpha cells and promotes insulin secretion in a paracrine manner. Here, we elucidated whether GIP(1–30) expression is modulated in mouse models of diabetes. We then investigated whether PEGylated GIP(1–30) can improve islet function and morphology as well as suppress the progression to hyperglycaemia in mice treated with low-dose streptozotocin (LD-STZ).
Methods
We examined pancreatic GIP immunoreactivity in rodent diabetic models. We synthesised [d-Ala2]GIP(1–30) and modified the C-terminus with polyethylene glycol (PEG) to produce a dipeptidyl peptidase-4 (DPP-4)-resistant long-acting GIP analogue, [d-Ala2]GIP(1–30)-PEG. We performed i.p.GTT and immunohistochemical analysis in non-diabetic and LD-STZ diabetic mice, with or without administration of [d-Ala2]GIP(1–30)-PEG.
Results
Pancreatic GIP expression was concomitantly enhanced with alpha cell expansion in rodent models of diabetes. Treatment with DPP-4 inhibitor decreased both the GIP- and glucagon-positive areas and preserved the insulin-positive area in LD-STZ diabetic mice. Body weight was not affected by [d-Ala2]GIP(1–30)-PEG in LD-STZ or non-diabetic mice. Treatment with GIP significantly ameliorated chronic hyperglycaemia and improved glucose excursions in LD-STZ mice. Treatment with GIP also reduced alpha cell expansion in the islets and suppressed plasma glucagon levels compared with non-treated LD-STZ mice. Additionally, [d-Ala2]GIP(1–30)-PEG preserved beta cell area via inhibition of apoptosis in LD-STZ mice.
Conclusions/interpretation
Our data suggest that GIP(1–30) expression is upregulated in diabetes, and PEGylated GIP(1–30) can suppress the progression to STZ-induced hyperglycaemia by inhibiting beta cell apoptosis and alpha cell expansion.
Similar content being viewed by others

Introduction
Glucose-dependent insulinotropic polypeptide (GIP) is an incretin hormone, released from the gut endocrine K cells, that promotes insulin secretion from pancreatic beta cells in a glucose-dependent manner. After secretion, GIP is rapidly inactivated by dipeptidyl peptidase-4 (DPP-4) by cleavage into truncated forms that are no longer insulinotropic [1–6]. The polypeptide was originally discovered as a 42 amino acid peptide [7–9] and pro-GIP is processed to GIP(1–42) by prohormone convertase (PC) 1/3 co-localised in the gut K cells [10]. Glucagon-like peptide 1 (GLP-1) and GIP are occasionally found in the same cells (so-called K/L or L/K cells); thus pro-GIP is presumably partially co-expressed with GLP-1 in the gut [11]. An antibody specific to the C-terminus of GIP(1–42) failed to detect GIP immunoreactivity in the pancreas [12]. However, Gip mRNA expression was confirmed by RT-PCR and in situ hybridisation in the periphery of pancreatic islets in mice [12]. Moreover, pancreatic GIP immunoreactivity can be found in alpha cells of mouse, human and python using a GIP antibody recognising the mid-portion of GIP(1–42) [12]. The pro-GIP molecule appears to be processed to short-form GIP(1–30) in pancreatic alpha cells via PC2 [12]. The short-form GIP(1–30) possesses insulinotropic activity similar to GIP(1–42) ex vivo [12].
Dipeptidyl peptidase-4 inhibitors and GLP-1 analogues are new classes of glucose-lowering treatment widely accepted to improve glycaemic control in patients with type 2 diabetes mellitus. In contrast, it is unclear whether GIP analogues are useful therapies for diabetes because GIP is largely considered to induce obesity. The GIP receptor (GIPR) is expressed in pancreatic beta cells, bone, central nervous system and adipose tissues [13, 14]. One of the extrapancreatic actions proposed for GIP is the promotion of fat deposition in adipocytes [13–16]. Indeed, genetic ablation of GIPR and GIPR blockage by GIPR antagonist or GIP neutralising antibody can protect mice from diet-induced obesity and improve glucose tolerance [17–21]. Conversely, recent studies have shown that GIP overexpression in transgenic mice or administration of a GIPR agonist to diabetic rodents ameliorates glycaemic control and insulin secretion, and these effects are accompanied by suppression of beta cell apoptosis without increases in body weight or food intake [22, 23].
Currently, it is unclear whether pancreatic GIP(1–30) expression is altered in diabetic conditions. Therefore, we first investigated whether GIP(1–30) expression is modulated in models of diabetes and could be influenced by treatment with a glucose-lowering agent. To elucidate the therapeutic potential of GIP(1–30), we synthesised a long-acting GIP(1–30) analogue. We conducted a study to assess whether the GIP analogue could suppress the progression to hyperglycaemia and improve glucose tolerance in STZ-induced diabetic mice. We also examined whether the short-form GIP(1–30) analogue could improve islet function and morphology in this model. In addition, we examined whether the GIP analogue promotes increased weight gain in mice fed a high-fat diet (HFD).
Methods
Animals and diet
Six-week-old male C57BL/6 mice were purchased from CLEA Japan (Tokyo, Japan) and housed under a 12 h light/dark cycle with free access to food and water. All procedures were performed in accordance with Asahikawa Medical University guidelines for the use of animals, with the approval of the university animal research committee. In the HFD feeding study, we used a commercial diet consisting of 60% energy from fat, 20% from protein and 20% from carbohydrate (D12492, Research Diets, New Brunswick, NJ, USA).
Peptide synthesis
We synthesised a DPP-4-resistant human GIP(1–30) analogue. The N-terminal alanine in position 2 was replaced with a d-alanine (d-Ala2) to confer DPP-4 resistance ([d-Ala2]GIP[1–30]). The C-terminal end was modified with 40 kDa polyethylene glycol (PEG) ([d-Ala2]GIP[1–30]-PEG). The purity of [d-Ala2]GIP(1–30) and [d-Ala2]GIP(1–30)-PEG was determined by HPLC to be 98.2% and 95.7%, respectively, and the molecular mass was confirmed by MALDI-MS (Peptide Institute, Osaka, Japan).
Cells, cell culture and measurement of GIP and glucagon by bioassay
We used HEK293 cell lines stably co-transfected with human forms of GIPR or glucagon receptor, and a cyclic AMP-inducible luciferase expression construct. The HEK293 cell lines were cultured in DMEM (25 mmol/l glucose) with 10% (vol./vol.) FBS (Gibco, Tokyo, Japan), 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen, Tokyo, Japan) at 37°C in 5% CO2. We incubated the cells overnight in 96-well plates (100,000 cells per well). The next day, we replaced media with samples or synthetic peptide standards (GIP[1–42]: 10−13 to 10−7 mol/l, glucagon: 10−13 to 10−10 mol/l) diluted in KRB (pH 7.4) containing 0.5% (wt/vol.) BSA (Sigma-Aldrich, Tokyo, Japan) and incubated for 5 h at 37°C in 5% CO2. After incubation, we measured luciferase activity with the Bright-Glo Assay Kit (Promega, Madison, WI, USA) using a Thermo Scientific Appliskan (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions, and calculated the hormone concentrations as previously described [12, 24].
Research design and generation of STZ-induced diabetic mice
We divided mice into four groups: non-STZ; non-STZ + GIP; STZ; and STZ + GIP. We injected STZ (50 mg/kg; Sigma Chemical, St Louis, MO, USA) dissolved in sodium citrate buffer or vehicle i.p. on days 1–5. We injected [d-Ala2]GIP(1–30)-PEG s.c. every 3 days (initial dose 100 pmol/animal and maintenance dose 70 pmol/animal) from day 1 of STZ injection. We chose the injection doses of [d-Ala2]GIP(1–30)-PEG based on our previous results [24] and current observations (see electronic supplementary material [ESM] Methods and ESM Fig. 1). In the group receiving DPP-4 inhibitor intervention, mice were treated with des-F-sitagliptin (provided by Merck, Kenilworth, NJ, USA) 2 weeks prior to LD-STZ treatment, as described previously [24].
Blood glucose, body weight and glycated haemoglobin
Non-fasted blood was collected from the tail vein, and blood glucose levels were measured with a Onetouch Ultra glucometer (Lifescan Japan, Tokyo, Japan) every 3 days until day 30. Body weight was determined at the time of glucose measurement. Levels of HbA1c were measured using a DCA Vantage Analyzer (Siemens, Munich, Germany) in blood collected from the tail vein on day 33.
Intraperitoneal GTT, insulin and glucagon measurement
We performed an i.p.GTT on day 34. Mice were fasted for 16–18 h before the i.p.GTT. Blood glucose levels were measured at 0, 10, 20, 30, 60 and 120 min after a glucose load (2 g/kg body weight). For plasma insulin and glucagon determinations, blood samples from the tail vein were collected into heparinised tubes at 0 and 10 min. The blood samples collected were mixed with a 10% volume of cooled NaCl (154 mmol/l) solution containing 12.5 mg/ml EDTA-2Na, 5,000 U/ml aprotinin and 1% (vol./vol.) DPP-4 inhibitor (Millipore, Billerica, MA, USA). These samples were separated by centrifugation at 4°C and stored at −80°C until assay. Plasma insulin levels were measured using a mouse insulin ELISA kit (Morinaga Institute of Biological Science, Yokohama, Japan) according to the instructions. Plasma glucagon levels were determined via cell-based, receptor-mediated bioassay as described above. This glucagon assay does not cross-react with at least up to 10−8 mol/l of either GIP or GLP-1 (ESM Fig. 2).
Immunohistochemistry and islet morphometry
Pancreases were resected immediately after the mice were killed on day 35, and fixed in 4% (wt/vol.) paraformaldehyde overnight at 4°C. Further procedures were performed as described elsewhere [24] and the antibodies used are described in ESM Methods. Stained sections were observed using an all-in-one fluorescence microscopy system (BZ-8100; Keyence, Osaka, Japan), and digital images were collected. We measured the insulin- and glucagon-positive cell areas using commercial software (BZ Analyzer; Keyence, Osaka, Japan), and assigned these as beta cell area and alpha cell area, respectively. We also counted the number of insulin-positive cells according to DAPI (nuclear) staining, and estimated islet area from the beta and alpha cell staining and DAPI staining. All morphometry studies were conducted in a blinded fashion.
Determination of cell proliferation and cell death in vivo
Insulin- and PCNA-double-positive cells were assigned as beta cell proliferation. Apoptotic beta cells were determined by TUNEL-positive staining and adjusted for beta cell area in serial sections stained with insulin. A commercial apoptotic cell detection kit (In Situ Cell Death Detection Kit, TMR Red; Roche Diagnostics Japan, Tokyo, Japan) was used for TUNEL staining.
HFD feeding study
Mice were treated with [d-Ala2]GIP(1–30)-PEG or PBS as in the LD-STZ study. Non-fasted blood glucose and body weight were monitored up to day 30. We measured HbA1c levels at day 33.
Statistical analysis
Data are expressed as mean ± SEM. Statistical analysis was performed using the unpaired Student’s t test, one-way ANOVA followed by Tukey post hoc test or two-way repeated ANOVA followed by the Bonferroni post hoc test. Data were analysed using commercial software (Prism 5; GraphPad, San Diego, CA, USA). A value of p < 0.05 was considered significant.
Results
Pancreatic GIP expression is enhanced in diabetic rodents
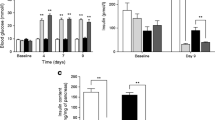
To detect pancreatic GIP, we used a GIP antibody that recognises the mid-portion of GIP. In previous studies, it was reported that this antibody can detect both GIP(1–42) and GIP(1–30), while an antibody specific to the C-terminus of GIP reacted only with GIP(1–42) and did not label any cells in islets of wild-type animals [10, 12]. Therefore, we chose the antibody capable of recognising GIP(1–30) for our current studies. We observed that pancreatic GIP immunoreactivity was enhanced concomitantly with alpha cell expansion and decreased beta cell area in rodent models of type 1 and type 2 diabetes, including NOD, db/db, Akita and LD-STZ (Fig. 1). In LD-STZ mice, the GIP-positive area per islet increased 3.6-fold compared with non-diabetic controls (p < 0.05; Fig. 2a, ESM Fig. 3). Pharmacological intervention via DPP-4 inhibition significantly decreased the GIP-positive area and ameliorated alpha cell expansion as well as preserving beta cell area in LD-STZ mice (Fig. 2a–c, ESM Fig. 3).

Pancreatic GIP expression was enhanced with alpha cell expansion concomitantly in diabetic rodent models (LD-STZ, Akita, db/db and NOD mice). Pancreatic sections were stained by GIP (green), glucagon (red) and insulin (blue), with representative islets shown (scale bars, 30 μm, magnification ×800)

Treatment via DPP-4 inhibitor reduced the GIP/alpha-positive area in LD-STZ mice. (a) GIP-positive alpha cell area per islet area. (b) Glucagon-positive alpha cell area per islet area. (c) Insulin-positive beta cell area per islet area (n = 4 in each group). Data are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Synthetic GIP(1–30) analogue can increase cAMP via GIPR in vitro
We synthesised [d-Ala2]GIP(1–30)-PEG to investigate the therapeutic potential of short-form GIP. Luciferase activity was increased by GIP(1–42), GIP(1–30) and synthetic GIP analogues in HEK293 cells stably co-transfected with GIPR and a cAMP-inducible luciferase expression construct in a concentration-dependent manner (ESM Fig. 4). Almost equivalent luciferase activity was produced by GIP(1–42) and GIP(1–30), indicative of comparable activation of the GIPR by these two forms of GIP. In contrast, [d-Ala2]GIP(1–30) and [d-Ala2]GIP(1–30)-PEG induced lower luciferase activity than GIP(1–42) or GIP(1–30) (ESM Fig. 4).
Single administration of [d-Ala2]GIP(1–30) lowered glucose excursion
Next, to assess glucose-lowering actions and the circulating kinetics of [d-Ala2]GIP(1–30), we conducted i.p.GTTs in normal mice given various s.c. doses of [d-Ala2]GIP(1–30) (7,000 pmol/animal, 1,400 pmol/animal, 350 pmol/animal) or vehicle (PBS). Treatment with GIP lowered glucose excursions during i.p.GTTs, even at the minimum dose (ESM Fig. 5a). The peak plasma concentration of [d-Ala2]GIP(1–30) appeared at 1 h after administration and was approximately 80 pmol/l, equivalent to levels following injection of 350 pmol/animal GIP(1–42). We did not detect any activity of GIP at 4 h after administration (ESM Fig. 5b).
[d-Ala2]GIP(1–30)-PEG is pharmaceutically long-acting in vivo
To further evaluate the pharmacokinetics of the peptides, we performed single s.c. injections of [d-Ala2]GIP(1–30) or [d-Ala2]GIP(1–30)-PEG in normal mice at 7,000 pmol/animal and collected blood samples at 1 h to 7 days after the injection. After a single administration, [d-Ala2]GIP(1–30) had rapidly increased by 1 h, but disappeared from the blood within 24 h. In contrast, [d-Ala2]GIP(1–30)-PEG was cleared very slowly from plasma, with an estimated half-life of approximately 60 h. Furthermore, [d-Ala2]GIP(1–30)-PEG was still detectable in the blood at day 7 when we injected 7,000 pmol/animal (ESM Fig. 5c).
Chronic [d-Ala2]GIP(1–30)-PEG treatment did not affect body weight, but ameliorated blood glucose levels in LD-STZ mice
In the intervention study, chronic GIP treatment did not affect body weight in STZ or non-STZ mice (Fig. 3a), or food intake (STZ vs STZ + GIP, 3.8 ± 0.1 g/day; 3.7 ± 0.1 g/day). In contrast, [d-Ala2]GIP(1–30)-PEG treatment significantly ameliorated non-fasted blood glucose levels in LD-STZ mice at days 27 and 30 (STZ + GIP vs STZ, p < 0.05; Fig. 3b). Treatment with GIP treatment also reduced HbA1c levels at day 33 in LD-STZ mice (STZ + GIP 4.9 ± 0.2% (29.7 ± 2.3 mmol/l) vs STZ 5.7 ± 0.4% (38.5 ± 4.3 mmol/l), p < 0.05; Fig. 3c). During i.p.GTTs, chronic GIP treatment did not alter the glucose excursion in non-diabetic mice, but improved glucose tolerance in LD-STZ mice. The AUC during i.p.GTTs was also significantly decreased by [d-Ala2]GIP(1–30)-PEG in STZ mice (AUC: STZ + GIP 2,465 ± 114 vs STZ 2,961 ± 166 mmol/l × min, p < 0.05; Fig. 3d, e) in association with suppressed plasma glucagon levels and increased plasma insulin levels (Fig. 4a–d).

Chronic GIP administration did not increase body weight in STZ or non-STZ mice, but improved fed blood glucose levels, HbA1c and glucose tolerance in LD-STZ mice. Body weight (a) and fed blood glucose (b) were measured every 3 days. (c) HbA1c levels at day 33. To convert values for HbA1c in DCCT % into mmol/mol, subtract 2.15 and multiply by 10.929. (d) Blood glucose levels were measured after a 2 g/kg i.p. glucose load. (e) Blood glucose AUC in i.p.GTT (n = 8–13 in each group). Data are presented as means ± SEM. *p < 0.05 vs PBS. Triangles, non-STZ; circles, non-STZ + GIP; inverted triangles, STZ; squares, STZ + GIP; black bars, PBS; white bars, GIP; grey horizontal bars, treatment with STZ or vehicle

In i.p.GTT (2 g/kg), insulin and glucagon levels were significantly improved by GIP treatment. (a) Plasma insulin levels at 0 min. (b) Plasma insulin levels at 10 min. (c) Plasma glucagon levels at 0 min. (d) Plasma glucagon levels at 10 min (n = 8–13 in each group). Data are presented as means ± SEM. *p < 0.05 vs PBS. Black bars, PBS; white bars, GIP
Chronic [d-Ala2]GIP(1–30)-PEG treatment suppressed alpha cell expansion in LD-STZ mice
To evaluate the effect of [d-Ala2]GIP(1–30)-PEG treatment on the islet architecture, we examined insulin and glucagon double immunofluorescence staining in the pancreas (Fig. 5a). In non-treated STZ mice, the beta cell area was decreased by about 40% and the alpha cell area was increased 4.2-fold compared with non-STZ mice. However, in GIP-treated STZ mice, the beta cell area was increased 1.47-fold and the alpha cell area was decreased by 48% compared with STZ mice (STZ + GIP vs STZ, p < 0.05; Fig. 5b, c). In GIP-treated STZ mice the insulin/glucagon ratio was 2.1-fold higher than in non-treated LD-STZ mice (STZ + GIP vs STZ, p < 0.05; Fig. 5d). Therefore, [d-Ala2]GIP(1–30)-PEG treatment partially restored islet morphology in LD-STZ mice.

Chronic GIP treatment reduced beta cell loss and alpha cell expansion in LD-STZ mice. Immunochemical analysis was investigated at day 35. (a) Pancreatic sections were stained with insulin (green), glucagon (red) and DAPI (blue), with representative islets shown (scale bars, 30 μm, magnification ×800). (b) Insulin-positive cell area per islet area. (c) Glucagon-positive cell area per islet area. (d) Beta cell area/alpha cell area ratio per islet (n = 5–6 in each group). Data are presented as means ± SEM. **p < 0.01, ***p < 0.001 vs PBS. Black bars, PBS; white bars, GIP
Beta cell apoptosis is suppressed by [d-Ala2]GIP(1–30)-PEG treatment in LD-STZ mice
We observed that GIP treatment partially suppressed the reduction in beta cell area in LD-STZ mice, so we next assessed whether beta cell apoptosis and/or proliferation contributed to the morphological changes. With respect to beta cell apoptosis, TUNEL-positive beta cells were significantly increased in non-treated STZ mice (Fig. 6a), while GIP treatment significantly reduced the number of TUNEL/insulin-double-positive cells in LD-STZ mice (Fig. 6b). There were no significant differences in the proliferation of beta cells between GIP-treated and non-treated LD-STZ mice assessed by the number of PCNA/insulin-double-positive cells (Fig. 6c). In addition, 8-OHdG immunostaining was not different between the groups, suggesting that oxidative stress was not involved in the reduction of beta cell area (ESM Fig. 6).

Chronic GIP treatment reduced beta cell death in LD-STZ mice. (a) TUNEL staining (red, TUNEL positive; blue, DAPI). Scale bars, 30 μm, magnification ×800. (b) TUNEL-positive beta cell number per islet. (c) PCNA-positive beta cell number per islet (n = 5–6 in each group). Data are presented as means ± SEM. *p < 0.05 vs PBS. Black bars, PBS; white bars, GIP
Chronic [d-Ala2]GIP(1–30)-PEG treatment did not enhance obesity in HFD-fed mice
We examined whether the GIP analogue might promote excessive weight gain and thereby exacerbate glucose intolerance in HFD-fed mice. In the HFD feeding study, chronic GIP treatment did not affect non-fasted glycaemia (data not shown) or HbA1c levels at day 33 (Fig. 7a). Interestingly, chronic GIP treatment significantly suppressed body weight gain in HFD mice compared with the non-treated group (Fig. 7b).

Chronic [d-Ala2]GIP(1–30)-PEG treatment did not enhance obesity in HFD-fed mice. (a) Levels of HbA1c at day 33. To convert values for HbA1c in DCCT % into mmol/mol, subtract 2.15 and multiply by 10.929. (b) Body weight gain from day 0 to 30 (Δ body weight) in mice fed a normal diet or HFD (n = 8–10 in each group). Data are presented as means ± SEM. *p < 0.05, ***p < 0.001. Black bars, PBS; white bars, GIP. HF, high fat; ND, normal diet
Discussion
In the current study, we observed that pancreatic GIP expression was enhanced with alpha cell expansion in various diabetic rodent models. It was previously demonstrated that the transcription factor paired box 6 (PAX6) induces pro-GIP and proglucagon promoter activity [11]. Therefore, it is possible that PAX6 is upregulated in diabetes and thereby increases transcription of the two genes, resulting in enhanced GIP(1–30) and glucagon expression. We previously observed that alpha cell area was increased with enhanced alpha cell proliferation [24]. Therefore, both increased cell number and GIP content per cell might contribute to our observation. Despite the increase in islet GIP expression in the diabetic setting, the insulinotropic effect of GIP may be diminished [25, 26], perhaps because GIPR expression in beta cells is reduced in the hyperglycaemic state [27, 28]. We speculate that pancreatic GIP expression may be upregulated to compensate for reduced GIP signalling in beta cells in diabetic animals. Additionally, GIPR signalling in beta cells is believed to be important to maintain islet architecture. Mice with Gipr knockout have abnormal islet structure [29, 30] and GIP is expressed in alpha cells and beta cells of human fetal islets [12]. Therefore, increased expression of pancreatic GIP may contribute to maintaining and protecting beta cells against various challenges in diabetes.
With regard to the therapeutic potential of short-form GIP(1–30), the GIP analogue successfully ameliorated chronic hyperglycaemia in LD-STZ mice. Frequent injections of GIP(1–30) also improved beta cell mass and function together with glucose tolerance in Zucker diabetic fatty (ZDF) rats [23]. The GIP(1–42) and GIP(1–30) forms are rapidly cleaved in the circulation by DPP-4 [31]. While [D-Ala2]GIP(1–30) is DPP-4 resistant, it is not sufficiently long acting, as GIP activity disappeared within 4 h after a single administration in vivo. In contrast, [d-Ala2]GIP(1–30)-PEG activity was still detectable on day 7 after administration, illustrating the advantage of the PEG moiety in permitting maintenance of an effective plasma concentration with infrequent administration.
Notably, in [d-Ala2]GIP(1–30)-PEG-treated LD-STZ mice, glucagon secretion was significantly suppressed both before and after a glucose load. However, GIP has been reported to increase glucagon release in both rodents and humans [32, 33], but may do so only at low glucose levels [32, 34]. The effect of [d-Ala2]GIP(1–30)-PEG on glucagon secretion might be modulated by GIPR downregulation in alpha cells. Nevertheless, while we are not aware of reports of alpha cell desensitisation to GIP, we cannot discount the possibility that this occurred, thereby alleviating the glucagonotropic action of GIP. Here, we administrated a GIP(1–30) analogue rather than GIP(1–42), and it is possible that GIP(1–30) acts differently from GIP(1–42) on glucagon secretion from alpha cells. However, our preliminary data suggest that [d-Ala2]GIP(1–30)-PEG enhances glucagon release from rat isolated islets at low (3 mmol/l) glucose (data not shown). Consistent with our in vivo findings, we have previously reported that pharmaceutical intervention via a DPP-4 inhibitor that boosts active GIP and GLP-1 levels can also suppress glucagon secretion, in addition to alpha cell proliferation, in LD-STZ mice [24]. We speculate that exogenous GIP administration might indirectly suppress fasting glucagon secretion through enhanced basal insulin release.
We have previously observed that treatment with a DPP-4 inhibitor suppresses beta cell apoptosis in LD-STZ mice [24]. By inhibiting apoptosis, GIP reportedly has a protective effect on pancreatic beta cells [22, 23]. In our histological analysis, [d-Ala2]GIP(1–30)-PEG treatment suppressed both the loss of beta cells and expansion of alpha cells in LD-STZ mice. Treatment with GIP significantly decreased TUNEL-positive beta cells but did not increase PCNA-positive beta cells in LD-STZ mice. These results suggest that GIP(1–30) has the potential for conferring beta cell protection rather than proliferation. The mechanisms by which GIP inhibits islet beta cell apoptosis appear to involve decreasing the expression of the pro-apoptotic Bax gene and increasing expression of the anti-apoptotic Bcl2 gene [23, 34–36]. It is possible that GIP expression in islets may be enhanced to block several apoptotic factors in diabetes.
Despite the restoration of beta cell area in LD-STZ mice to almost that of control mice following [d-Ala2]GIP(1–30)-PEG treatment, insulin levels were elevated only in the fasting state, compared with non-treated LD-STZ mice. We have observed similar results with LD-STZ mice treated with DPP-4 inhibitor, whereby beta cell numbers were similar to non-STZ controls, yet the treatment only partially improved the glucose excursion during an OGTT [24]. Therefore, restoration of beta cell mass is not necessarily sufficient to normalise insulin output and glycaemia. While in control mice, acute treatment improved glucose tolerance (ESM Fig. 5a), chronic treatment with [d-Ala2]GIP(1–30)-PEG had no effect on glucose tolerance and insulin secretion (Fig. 4). Widenmaier et al also reported that [d-Ala2]GIP(1–30) can acutely induce insulin secretion [23]. Chronic treatment may induce downregulation of GIP receptor expression and/or desensitisation of downstream signalling pathways, such that the enhancement of insulin secretion is not maintained; this would thereby limit the long-term effect on glucose tolerance. However, it is notable that heterozygous mice with zinc-inducible overexpression of GIP (Tg-GIP) maintained GIP sensitivity and displayed improved glucose tolerance, along with increased postprandial insulin secretion, preserved beta cell area and reduced beta cell apoptosis [22]. In contrast, zinc-treated homozygous Tg-GIP mice with even greater overexpression of GIP (approximately four fold higher plasma GIP levels than in zinc-treated heterozygous mice) developed GIP resistance [22]. Thus, the ability of exogenous PEG-GIP to have sustained insulinotropic effects without inducing GIP resistance is likely to be concentration dependent and further studies will be required to determine the optimal PEG–GIP dosing.
The GIP(1–30) and GIP(1–42) forms display approximately equivalent insulinotropic action [12, 23, 37, 38] and anti-apoptotic effects on pancreatic beta cells [23, 34–36]. In contrast to pancreatic actions, the two isoforms may show dissimilar effects on extrapancreatic tissues. For example, in 3T3-L1 adipocytes, [d-Ala2]GIP(1–30) induces significantly lower lipoprotein lipase (LPL) activity than [d-Ala2]GIP(1–42) [23]. Our study also suggests that the GIP(1–30) analogue does not induce fat accumulation and body weight gain, possibly because of the limited action of GIP(1–30) on adipose tissue.
It has been proposed that simultaneous targeting of GIP, GLP-1 and glucagon receptors may be beneficial in the treatment of obesity and metabolic disorders [39]. Indeed, administration of a tri-agonist improved glucose intolerance in rodents [40]. Additionally, body weight, food intake, fat mass, plasma cholesterol and hepatic lipid accumulation were reduced in mice fed a high-sucrose diet [40]. Interestingly, the tri-agonist reduced body weight more intensively than liraglutide alone or a GLP-1/glucagon co-agonist, suggesting that agonistic action of GIP may not promote weight gain, but rather weight loss.
In conclusion, our data suggest that expression of pancreatic GIP, likely GIP(1–30), is upregulated in diabetes and PEGylated GIP(1–30) can suppress the progression to STZ-induced hyperglycaemia by inhibiting beta cell apoptosis and alpha cell expansion. Therefore, GIP(1–30) may play an important role in glucose metabolism, and further studies are warranted to explore the therapeutic potential of this peptide.
Abbreviations
- DPP-4:
-
Dipeptidyl peptidase-4
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GLP-1:
-
Glucagon-like peptide-1
- HFD:
-
High-fat diet
- LD-STZ:
-
Low-dose streptozotocin
- PC:
-
Prohormone convertase
- PCNA:
-
Proliferating cell nuclear antigen
- PEG:
-
Polyethylene glycol
- STZ:
-
Streptozotocin
- Tg-GIP:
-
Zinc-inducible overexpression of GIP
References
Dupre J, Ross SA, Watson D, Brown JC (1973) Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab 37:826–828
Schirra J, Katschinski M, Weidmann C et al (1996) Gastric emptying and release of incretin hormones after glucose ingestion in humans. J Clin Invest 97:92–103
Pauly RP, Rosche F, Wermann M, McIntosh CH, Pederson RA, Demuth HU (1996) Investigation of glucose-dependent insulinotropic polypeptide(1-42) and glucagon-like peptide-1(7-36) degradation in vitro by dipeptidyl peptidase IV using matrix-assisted laser desorption/ionization-time of flight mass spectrometry. J Biol Chem 271:23222–23229
Van Ginneken C, Weyns A (2004) A stereological evaluation of secretin and gastric inhibitory peptide-containing mucosal cells of the perinatal small intestine of the pig. J Anat 205:267–275
Kieffer TJ, Habener JF (1999) The glucagon-like peptides. Endocr Rev 20:876–913
Deacon CF, Nauck MA, Meier J, Hücking K, Holst JJ (2000) Degradation of endogenous and exogenous gastric inhibitory polypeptide in healthy and in type 2 diabetic subjects as revealed using a new assay for the intact peptide. J Clin Endocrinol Metab 85:3575–3581
Jörnvall H, Carlquist M, Kwauk S et al (1981) Amino acid sequence and heterogeneity of gastric inhibitory polypeptide (GIP). FEBS Lett 123:205–210
Carlquist M, Maletti M, Jörnvall H, Mutt V (1984) A novel form of gastric inhibitory polypeptide (GIP) isolated from bovine intestine using a radioreceptor assay. Fragmentation with staphylococcal protease results in GIP1-3 and GIP4-42, fragmentation with enterokinase in GIP1-16 and GIP17-42. Eur J Biochem 145:573–577
Moody AJ, Thim L, Valverde I (1984) The isolation and sequencing of human gastric inhibitory peptide (GIP). FEBS Lett 172:142–148
Fujita Y, Asadi A, Yang GK, Kwok YN, Kieffer TJ (2010) Differential processing of pro-glucose-dependent insulinotropic polypeptide in gut. Am J Physiol Gastrointest Liver Physiol 298:G608–G614
Fujita Y, Chui JY, King DS et al (2008) Pax6 and Pdx1 are required for production of glucose-dependent insulinotropic polypeptide in proglucagon-expressing L cells. Am J Physiol Endocrinol Metab 295:E648–E657
Fujita Y, Wideman RD, Asadi A et al (2010) Glucose-dependent insulinotropic polypeptide is expressed in pancreatic islet α-cells and promotes insulin secretion. Gastroenterology 138:1966–1975
Baggio LL, Drucker DJ (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology 132:2131–2157
Kim W, Egan JM (2008) The role of incretins in glucose homeostasis and diabetes treatment. Pharmacol Rev 60:470–512
Irwin N, Flatt PR (2009) Evidence for beneficial effects of compromised gastric inhibitory polypeptide action in obesity-related diabetes and possible therapeutic implications. Diabetologia 52:1724–1731
Flatt PR (2008) Dorothy Hodgkin Lecture 2008 Gastric inhibitory polypeptide (GIP) revisited: a new therapeutic target for obesity–diabetes? Diabet Med 25:759–764
Miyawaki K, Yamada Y, Ban N et al (2002) Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med 8:738–742
Gault VA, McClean PL, Cassidy RS, Irwin N, Flatt PR (2007) Chemical gastric inhibitory polypeptide receptor antagonism protects against obesity, insulin resistance, glucose intolerance and associated disturbances in mice fed high-fat and cafeteria diets. Diabetologia 50:1752–1762
McClean PL, Irwin N, Cassidy RS, Holst JJ, Gault VA, Flatt PR (2007) GIP receptor antagonism reverses obesity, insulin resistance, and associated metabolic disturbances induced in mice by prolonged consumption of high-fat diet. Am J Physiol Endocrinol Metab 293:E1746–E1755
McClean PL, Irwin N, Hunter K, Gault VA, Flatt PR (2008) (Pro3) GIP [mPEG]: novel, long-acting, mPEGylated antagonist of gastric inhibitory polypeptide for obesity-diabetes (diabesity) therapy. Br J Pharmacol 155:690–701
Montgomery IA, Irwin N, Flatt PR (2010) Active immunization against (Pro3) GIP improves metabolic status in high-fat-fed mice. Diabetes Obes Metab 12:744–751
Kim SJ, Nian C, Karunakaran S, Clee SM, Isales CM, McIntosh CH (2012) GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PLoS ONE 7, e40156
Widenmaier SB, Kim SJ, Yang GK et al (2010) A GIP receptor agonist exhibits β-cell anti-apoptotic actions in rat models of diabetes resulting in improved β-cell function and glycemic control. PLoS ONE 5, e9590
Takeda Y, Fujita Y, Honjo J et al (2012) Reduction of both beta cell death and alpha cell proliferation by dipeptidyl peptidase-4 inhibition in a streptozotocin-induced model of diabetes in mice. Diabetologia 55:404–412
Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W (1993) Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest 91:301–307
Vilsbøll T, Krarup T, Madsbad S, Holst JJ (2002) Defective amplification of the late phase insulin response to glucose by GIP in obese type II diabetic patients. Diabetologia 45:1111–1119
Piteau S, Olver A, Kim SJ et al (2007) Reversal of islet GIP receptor down-regulation and resistance to GIP by reducing hyperglycemia in the Zucker rat. Biochem Biophys Res Commun 362:1007–1012
Shu L, Matveyenko AV, Kerr-Conte J, Cho JH, McIntosh CH, Maedler K (2009) Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum Mol Genet 18:2388–2399
Pamir N, Lynn FC, Buchan AM et al (2003) Glucose-dependent insulinotropic polypeptide receptor null mice exhibit compensatory changes in the enteroinsular axis. Am J Physiol Endocrinol Metab 284:E931–E939
Herbacha N, Goeke B, Schneider M, Hermanns W, Wolf E, Wanke R (2005) Overexpression of a dominant negative GIP receptor in transgenic mice results in disturbed postnatal pancreatic islet and beta-cell development. Regul Pept 125:103–117
Kieffer TJ, McIntosh CH, Pederson RA (1995) Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 136:3585–3596
Christensen M, Vedtofte L, Holst JJ, Vilsbøll T, Knop FK (2011) Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes 60:3103–3131
Pederson RA, Brown JC (1978) Interaction of gastric inhibitory polypeptide, glucose, and arginine on insulin and glucagon secretion from the perfused rat pancreas. Endocrinology 103:610–615
Kim SJ, Winter K, Nian C, Tsuneoka M, Koda Y, McIntosh CH (2005) Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic beta-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor FOXO1, and down-regulation of bax expression. J Biol Chem 280:22297–22307
Kim SJ, Nian C, Widenmaier S, McIntosh CH (2008) Glucose-dependent insulinotropic polypeptide-mediated up-regulation of beta-cell antiapoptotic Bcl-2 gene expression is coordinated by cyclic AMP (cAMP) response element binding protein (CREB) and cAMP-responsive CREB coactivator 2. Mol Cell Biol 28:1644–1656
Widenmaier SB, Ao Z, Kim SJ, Warnock G, McIntosh CH (2009) Suppression of p38 MAPK and JNK via Akt-mediated inhibition of apoptosis signal-regulating kinase 1 constitutes a core component of the beta-cell pro-survival effects of glucose-dependent insulinotropic polypeptide. J Biol Chem 284:30372–30382
Hinke SA, Manhart S, Pamir N et al (2001) Identification of a bioactive domain in the amino-terminus of glucose-dependent insulinotropic polypeptide (GIP). Biochim Biophys Acta 1547:143–155
Gault VA, Porter DW, Irwin N, Flatt PR (2011) Comparison of sub-chronic metabolic effects of stable forms of naturally occurring GIP(1-30) and GIP(1-42) in high-fat fed mice. J Endocrinol 208:265–271
Cho YM, Merchant CE, Kieffer TJ (2012) Targeting the glucagon receptor family for diabetes and obesity therapy. Pharmacol Ther 135:247–278
Finan B, Yang B, Ottaway N et al (2015) A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med 21:27–36
Acknowledgements
We sincerely thank S. Covey (University of British Columbia, Canada) for proofreading.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research was supported in part by a research grant from Merck Investigator Initiated Studies Program. YF is supported by grants from the Japan Society for the Promotion of Science (Grant-in-Aid for Scientific Research [C] number 23591291), Japan Diabetes Foundation (Incretin Research) and Insulin Research Foundation (Novo Nordisk Pharma, Japan). YF also received scholarships from Eli Lilly, Pfizer and MSD.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
TY, YF, Y Takeda, JH, KKA, Y Takiyama, AA, YM, TJK and MH contributed to the study concept and design. TJK produced the HEK293 cell lines co-transfected with human forms of GIPR or glucagon receptor, and a cyclic AMP-inducible luciferase expression construct. TY, YF, Y Takeda and KKA acquired the data. TY, YF, Y Takeda, JH, KKA, Y Takiyama, AA, YM, and MH analysed and interpreted the data. TY and YF drafted the manuscript. TY, YF, Y Takeda, KKA, JH, Y Takiyama, AA, YM, TJK and MH reviewed the manuscript for important intellectual content. All authors have approved the final version of this manuscript. YF and MH are the guarantors of this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 49.4 kb)
ESM Fig. 1
(PDF 35.5 kb)
ESM Fig. 2
(PDF 57 kb)
ESM Fig. 3
(PDF 440 kb)
ESM Fig. 4
(PDF 58 kb)
ESM Fig. 5
(PDF 53.1 kb)
ESM Fig. 6
(PDF 2,333 kb)
Rights and permissions
About this article

Cite this article
Yanagimachi, T., Fujita, Y., Takeda, Y. et al. Pancreatic glucose-dependent insulinotropic polypeptide (GIP) (1–30) expression is upregulated in diabetes and PEGylated GIP(1–30) can suppress the progression of low-dose-STZ-induced hyperglycaemia in mice. Diabetologia 59, 533–541 (2016). https://doi.org/10.1007/s00125-015-3842-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3842-y