
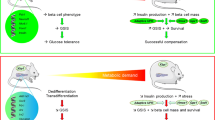
Abstract
Aims/hypothesis
‘Glucotoxicity’ is a term used to convey the negative effect of hyperglycaemia on beta cell function; however, the underlying molecular mechanisms that impair insulin secretion and gene expression are poorly defined. Our objective was to define the role of transcription factor v-ets avian erythroblastosis virus E26 oncogene homologue 1 (Ets-1) in beta cell glucotoxicity.
Methods
Primary islets and Min6 cells were exposed to high glucose and Ets-1 expression was measured. Recombinant adenovirus and transgenic mice were used to upregulate Ets-1 expression in beta cells in vitro and in vivo, and insulin secretion was assessed. The binding activity of H3/H4 histone on the Ets-1 promoter, and that of forkhead box (FOX)A2, FOXO1 and Ets-1 on the Pdx-1 promoter was measured by chromatin immunoprecipitation and quantitative real-time PCR assay.
Results
High glucose induced upregulation of Ets-1 expression and hyperacetylation of histone H3 and H4 at the Ets-1 gene promoter in beta cells. Ets-1 overexpression dramatically suppressed insulin secretion and biosynthesis both in vivo and in vitro. Besides, Ets-1 overexpression increased the activity of FOXO1 but decreased that of FOXA2 binding to the pancreatic and duodenal homeobox 1 (PDX-1) homology region 2 (PH2), resulting in inhibition of Pdx-1 promoter activity and downregulation of PDX-1 expression and activity. In addition, high glucose promoted the interaction of Ets-1 and FOXO1, and the activity of Ets-1 binding to the Pdx-1 promoter. Importantly, PDX-1 overexpression reversed the defect in pancreatic beta cells induced by Ets-1 excess, while knockdown of Ets-1 prevented hyperglycaemia-induced dysfunction of pancreatic beta cells.
Conclusions/interpretation
Our observations suggest that Ets-1 links glucotoxicity to pancreatic beta cell dysfunction through inhibiting PDX-1 expression in type 2 diabetes.
Similar content being viewed by others


Introduction
Under physiological conditions, pancreatic beta cells modulate insulin secretion to maintain blood glucose in the normal range [1]. Diabetes mellitus occurs when beta cells fail to maintain adequate insulin production to prevent hyperglycaemia. Currently, intrinsic beta cell dysfunction and decreased functional beta cell mass are thought to be two important components of diabetic beta cell failure [2]. Results from studies in db/db mice, the partial pancreatectomy rats and ZDF rats revealed that chronic hyperglycaemia, commonly called ‘glucotoxicity’, is a major determinant of causing beta cell failure [3–5]. Several complex mechanisms are involved in beta cell glucotoxicity, including beta cell overstimulation, oxidative stress, endoplasmic reticulum stress, hypoxic stress and protein glycation [6]. Among these, O-GlcNAc modification of transcription factors such as pancreatic and duodenal homeobox 1 (PDX-1), neurogenic differentiation 1 (NeuroD1), SP1 and p53 might play a key role in glucotoxicity [7].
PDX-1, which is expressed in beta cells and few delta cells of the adult islet of Langerhans, is a master regulator of beta cell growth and function [8]. PDX-1 regulates the expression of islet-specific genes through its interaction with their promoter regions, which include insulin, GLUT2, glucokinase (GCK), islet amyloid polypeptide (IAPP), somatostatin and PDX-1 itself [9–14]. Clinically, mutations in PDX-1 cause MODY4 and later-onset type 2 diabetes mellitus [15, 16]. In mice, selective inactivation of PDX-1 in beta cells leads to the development of diabetes with increasing age [3]. In addition, chronic hyperglycaemia causes loss of beta cell function by reducing PDX-1 expression and DNA binding activities [17].
Forkhead box (FOX)O1 is a transcription factor that negatively regulates PDX-1 expression and has also been implicated in beta cell glucotoxicity [18, 19]. FOXO1 inhibits PDX-1 expression by binding to the PDX-1 homology region 2 (PH2) of the PDX-1 promoter, which contains a FOXA2 binding site [18, 20]. As a multifunctional protein, FOXO1 regulates beta cell function, proliferation, differentiation and apoptosis [21]. Asada et al demonstrated that FOXO1 could interact with transcription factor v-ets avian erythroblastosis virus E26 oncogene homologue 1 (Ets-1) in bovine carotid artery endothelial cells, which we have also demonstrated in pancreatic beta cells [22]. As a result, Ets-1 might be involved in regulation of beta cell failure in diabetes mellitus.
In recent years, evidence has confirmed that the acetylation status of histones has been shown to be important in pancreatic beta cell function [23]. Histone acetylation promotes gene expression by increasing the approachability of promoters to the transcription machinery [24]. We found that transcription factor Ets-1 was significantly upregulated in high glucose-treated primary islets and Min6 cells as well as isolated islets from db/db mice. We also found the histone deacetylase inhibitor could upregulate Ets-1 expression in pancreatic beta cells. We hypothesised that epigenetic modification might be involved in regulation of Ets-1 expression in high glucose-treated beta cells. Our research group tested this hypothesis using the pancreatic beta cell line Min6, primary islets and transgenic mice that overexpress Ets-1.
Methods
Reagents
See Electronic Supplementary Material (ESM) Methods.
Cell culture
Mouse Min6 and rat RINm5F cell lines were established as described previously [25, 26]. See ESM Methods.
Islet isolation and culture
Male wild-type C57BL/BKS mice and mice functionally deficient for the long-form leptin receptor (db/db mice) were obtained from Nanjing University, China. Male 8-week-old Sprague-Dawley rats were purchased from Nanjing Medical University Laboratory Animal Centre, Nanjing, China. Islet isolation and culturing techniques were described previously [27]. See ESM Methods.
RNA interference, plasmid and recombinant adenoviruses construction
The Ets-1 expression plasmid used was as previously described [26]. Ets-1 overexpression adenovirus (Ad-Ets-1), PDX-1 overexpression adenovirus (Ad-Pdx-1) and small interfering (RNA)-Ets-1 adenovirus (si-Ets-1) were constructed and purified as previously described [28]. See ESM Methods.
Glucose-stimulated insulin secretion assay and insulin content extraction
Glucose-stimulated insulin secretion assays and insulin content extraction in Min6 cells and isolated islets were performed as previously reported [27]. See ESM Methods.
Real-time PCR
Total RNA was extracted from Min6 cells, RINm5F cells and isolated islets. Real-time PCR was used to determine the relative expression levels of mRNAs. See ESM Methods.
Western blot analysis
Total protein was isolated from Min6 cells, RINm5F cells and isolated islets. Western blotting was performed as described [29]. See ESM Methods.
Transient transfection and luciferase reporter assay
The plasmids were transfected into RINm5F cells. Transient transfections and luciferase reporter assays were performed as described [30]. See ESM Methods.
Electron microscopy analysis
Islets were fixed in 2.5% (vol./vol.) glutaraldehyde and embedded in Lowicryl K4M resin. See ESM Methods.
Chromatin immunoprecipitation and chromatin immunoprecipitation-quantitative PCR assay
Chromatin immunoprecipitation (ChIP) and ChIP-quantitative-PCR (ChIP-qPCR) assays were performed using lysates of Min6 and RINm5F cells as described previously [31]. See ESM Methods.
Immunoprecipitation assay
Immunoprecipitation (IP) assays were performed using extracts of RINm5F cells as described previously [32]. See ESM Methods.
Generation of Ets-1 transgenic mice
Ets-1 overexpression in beta cells of transgenic mice (C57BL/6J strain) was constructed (Model Animal Research Centre, Nanjing University, Nanjing, China) as described previously [33, 34]. Founder mice were identified by PCR of tail biopsies using two pairs of primers (ESM Table 1). Heterozygous transgenic mice were used throughout the present studies and non-transgenic littermates were used as controls. We studied 3- to 12-week-old male mice. All animals had free access to standard mouse chow and water, and were housed in a temperature-controlled facility with a 12 h light/dark cycle. All animal studies were performed according to guidelines established by the Research Animal Care Committee of Nanjing Medical University, China (Permit Number: IACUC-NJMU 1404075). The experimenters were not blind to group assignment and outcome assessment.
i.p. glucose tolerance test and insulin tolerance test in mice
At 8 weeks of age, control mice and transgenic mice were randomised to perform i.p. glucose tolerance tests and insulin tolerance tests [35]. See ESM Methods.
Statistical analysis
Comparisons were performed using Student’s t test between two groups or ANOVA in multiple groups. Results are presented as means ± SD. A p value of <0.05 was considered to be statistically significant. Results are representative of three individual experiments. No data, samples or animals were excluded or omitted from reporting.
Results
Ets-1 expression is elevated in db/db mice islets and in pancreatic beta cells exposed to hyperglycaemia
Although Ets-1 was reported to be related to pancreatic development [36], few studies have investigated its expression in pancreatic islets. We compared Ets-1 gene expression in pancreatic islets from 8-week-old db/db and control C57BL/BKS mice. Our results showed that Ets-1 mRNA and protein level was significantly increased in islets from db/db mice compared with control C57BL/BKS mice (Fig. 1a,b). Moreover, high glucose increased Ets-1 expression and Ets-1 protein level in Min6 cells, and isolated mouse and rat islets (Fig. 1c–f).

Ets-1 expression is elevated in db/db mouse islets and in pancreatic beta cells exposed to hyperglycaemia. The mRNA (a) and protein level (b) of Ets-1 was significantly elevated in islets isolated from 8-week-old db/db mice. (c, d) Primary islets isolated from mice and rats were treated with high glucose (25 mmol/l) for 48 h, and the expression of Ets-1 was measured. The mRNA (c) and protein level (d) of Ets-1 increased in islets treated with 25 mmol/l (black bars) compared with islets treated with 11.1 mmol/l (white bars). (e, f) Min6 cells were precultured in DMEM with 5.5 mmol/l glucose for 24 h, and then cells were incubated with different concentrations of glucose for an additional 48 h. The mRNA (e) and protein level (f) of Ets-1 was upregulated in a dose-dependent manner. Values are means ± SD and are representative of three individual experiments. *p < 0.05, **p < 0.01 vs control
High glucose mediates hyperacetylation of histone H3 and H4 at the Ets-1 gene promoter
To explore whether high glucose induced upregulation of Ets-1 expression by causing hyperacetylation of histone, we treated Min6 cells with different concentrations of the histone deacetylase inhibitor trichostatin A (TSA). We observed that TSA significantly upregulated Ets-1 expression both in mRNA and protein levels (Fig. 2a,b). Then, we performed a ChIP-qPCR assay to measure whether high glucose mediated hyperacetylation of histone at the Ets-1 gene promoter. Exposure of Min6 cells to high concentrations of glucose for 48 h led to ∼12.5-fold increase in histone H4 acetylation and a sixfold increase in histone H3 acetylation (Fig. 2c,d).

Glucose mediates hyperacetylation of histones H3 and H4 at the Ets-1 gene promoter. Min6 cells were treated with different concentrations of the histone deacetylase inhibitor TSA for 48 h, and the mRNA (a) and protein (b) levels of Ets-1 were measured. Min6 cells were precultured in DMEM with 5.5 mmol/l glucose for 24 h, and then cells were incubated with 5.5 mmol/l or 25 mmol/l glucose for an additional 48 h. ChIP-qPCR analysis of the capacity of acetylation of histone H3 (c) and H4 (d) binding to the Ets-1 promoter was performed. Values are means ± SD and are representative of three individual experiments. **p < 0.01 vs control
Overexpression of Ets-1 impairs insulin secretion and synthesis in vitro and in vivo
The finding that Ets-1 expression was significantly increased by high glucose prompted us to investigate the effect of Ets-1 on insulin secretion in Min6 cells and isolated primary mouse islets. Overexpression of Ets-1 in Min6 cells and islets significantly inhibited the glucose-stimulated index (GSI), which reflects the insulin secretion ratio of pancreatic beta cells stimulated by high and low glucose (Fig. 3a,b and ESM Fig. 1). Similar results for insulin content were obtained (Fig. 3c,d). Moreover, insulin secretion under high glucose, normalised to insulin content, was also repressed after overexpressing Ets-1 (Fig. 3e,f).

Overexpression of Ets-1 impaired insulin secretion in vitro and in vivo. Ad-Ets-1 (50 multiplicity of infection [MOI]) or control adenovirus (Ad-Gfp, 50 MOI) were infected into Min6 cells and mouse islets for 48 h, when GSIS assays were carried out and GSI was calculated. Infection caused an effective decrease of GSI in Min6 cells (a) and islets (b), insulin content in Min6 cells (c) and islets (d), and insulin secretion normalised to insulin content in Min6 cells (e) (2 mmol/l, white bars; 20 mmol/l, black bars) and islets (f) (3.3 mmol/l, white bars; 16.7 mmol/l, black bars). (g) Islets from 8-week-old male control mice (CON) and transgenic mice (TM) were fixed, pelleted and sectioned for EM (magnification ×12,000). Average total granule number per cell cross section is depicted and data are represented as means ± SD from 30 cells. (h) Hyperglycaemia at fasting in CON (white triangles) and TM (black triangles) at different weeks of age (n = 8 from each group). (i) Blood glucose levels after an i.p. injection of glucose (1.5 g/kg BW) in 8-week-old male CON and TM (n = 8 from each group). (j) Serum insulin concentrations after i.p. injection of glucose (3 g/kg BW) in 8-week-old male CON and TM (n = 8 from each group). (k) Insulin tolerance test (0.75 U/kg BW) in 8-week-old male CON and TM (n = 8 from each group). Values are means ± SD and are representative of three individual experiments. *p < 0.05, **p < 0.01 vs control
To investigate whether Ets-1 overexpression could impair insulin secretion in vivo, we next measured the effect of Ets-1 on pancreatic beta cell function using a transgenic mice (line 107) expressing the Ets-1 gene directed by an insulin promoter [37]. Immunofluorescent staining of islets revealed Ets-1 was specifically overexpressed in pancreatic beta cells and there was a distinct inverse relationship between Ets-1 and insulin protein levels (ESM Fig. 2a). Islet electron microscopy showed a decrease in the number of insulin secretory granules in transgenic mice compared with control mice (Fig. 3g). In addition, Ets-1 expression was unaffected in nonpancreatic tissues, including spleen and liver (ESM Fig. 2b).
The transgenic mice were born at the expected Mendelian ratio and maintained normal weight through our 12-week observational period (ESM Fig. 2c); however, they displayed fasting hyperglycaemia starting at 7 weeks of age (Fig. 3h). Besides, blood glucose levels of transgenic mice were higher at every time point after intravenous glucose loading (1.5 g/kg body weight [BW]) compared with wild-type controls (Fig. 3i). In addition, plasma insulin levels of transgenic mice were lower at every time point after intravenous glucose loading (3 g/kg BW) compared with littermate controls (Fig. 3j). However, peripheral glucose uptake was not significantly different between transgenic mice and control mice (Fig. 3k). This result indicated that the glucose intolerance of Ets-1 transgenic mice was due to defective insulin secretion but not insulin resistance. It is important to note that we also obtained three transgenic lines exhibiting high (line 107), medium (line 123) and low (line 79) levels of Ets-1 expression in islets. Both line 123 and line 79 displayed defective insulin secretion (ESM Fig. 2d–g).
Ets-1 overexpression exerts a suppressive effect on PDX-1 expression by inhibiting PDX-1 promoter activity
To evaluate the possible implications of Ets-1 overexpression for beta cell dysfunction, we examined the effects of Ets-1 on beta cell-associated gene expression. As shown in Fig. 4a,b, the amount of Pdx-1 mRNA was remarkably decreased in the Ets-1-overexpressing Min6 and RINm5F cells, whereas the expression levels of Neurod1, Mafa and Foxa2 were unchanged. Furthermore, Ets-1 overexpression led to similar reduction of PDX-1 protein in Min6 and RINm5F cells. The expression of PDX-1 in islets of transgenic mice was also significantly decreased compared with littermate control islets (Fig. 4c). Therefore, the expression of PDX-1 in vivo and in vitro was inhibited by Ets-1 overexpression. Next, we determined the mRNA levels of insulin and Gck, which are both directly regulated by PDX-1, to explore whether Ets-1 had an effect on PDX-1 transcriptional activity. ChIP-qPCR assay confirmed that DNA fragments pulled down by an anti-PDX antibody in the Ets-1 overexpressing cells were reduced to approximately 0.6-fold for the insulin promoter site and 0.55-fold for the Gck promoter site (Fig. 4d).

Ets-1 exerts a suppressive effect on PDX-1 expression by inhibiting Pdx-1 promoter activity. Min6 cells were infected with Ad-Ets-1 (or Ad-Gfp) and RINm5F cells were transfected with Ets-1 expression plasmid (or vector) for 48 h, when real-time PCR was carried out. Infection in Min6 cells (a) or transfection in RINm5F cells (b) caused an effective decrease in Pdx-1 mRNA level but had no effect on Mafa, Neurod and Foxa2 mRNA levels (Ad-Gfp or vector, white bars; Ad-Ets-1 or Ets-1 expression plasmid, black bars). (c) Infection or transfection caused an effective decrease in PDX-1 protein level. The PDX-1 protein level was significantly decreased in transgenic mice (TM) compared with control mice (CON). (d) ChIP-qPCR analysis of effect of Ets-1 on the capacity of PDX-1 binding to the Ins1, Ins2 and Gck promoters was performed in RINm5F cells transfected with Ets-1 expression plasmid for 24 h (vector, white bars; Ets-1 expression plasmid, black bars). (e) Ets-1 bound directly to the Pdx-1 promoter in a ChIP analysis in RINm5F cells transfected with Ets-1 expression plasmid for 24 h. ChIP-qPCR analysis of the capacity of FOXA2 (f), FOXO1 (g) and Ets-1 (h) binding to the PH2 domain of the Pdx-1 promoter was performed in RINm5F cells transfected with Ets-1 expression plasmid for 24 h. (i) RINm5F cells were transiently transfected with Ets-1 expression plasmid (or vector) and a pGL3-PDX-1-PH2 luciferase reporter construct. Luciferase activity was assayed 24 h after transfection. Values are means ± SD and are representative of three individual experiments. **p < 0.01 vs control
Further study showed that Ets-1 could bind to the PH2 region of the Pdx-1 promoter, which was the common DNA binding site of FOXO1 and FOXA2 (Fig. 4e). To investigate whether Ets-1 overexpression could affect the activity of FOXA2 and FOXO1 on the Pdx-1 promoters, a ChIP-qPCR assay was performed. As shown in Fig. 4f, DNA fragments pulled down by the anti-FOXA2 antibody in the Ets-1 overexpression group were reduced to approximately 0.46-fold for the PH2 region of the Pdx-1 promoter. However, the effect of Ets-1 overexpression on activity of FOXO1 binding to the PH2 region was elevated up to 2.05-fold (Fig. 4g). Additionally, in Ets-1 overexpression groups, DNA fragments pulled down by the anti-Ets-1 antibody were increased to twofold (Fig. 4h). More importantly, Ets-1 overexpression significantly decreased Pdx-1 promoter activity (Fig. 4i).
Overexpression of PDX-1 restored beta cell function impaired by Ets-1
To determine the role of PDX-1 in Ets-1-induced impairment of beta cell function, Min6 cells were infected with PDX-1 recombinant adenoviral vectors for 12 h, and then infected with Ets-1 recombinant adenoviral vectors for another 48 h. Western blot demonstrated that PDX-1 and Ets-1 were both overexpressed (Fig. 5a). Insulin secretion was reduced to 60% in Min6 cells treated with Ets-1 overexpression alone, while restored to 85% by co-overexpression of Ets-1 and PDX-1 (Fig. 5b). Besides, PDX-1 overexpression reversed the decrease of insulin content, and insulin secretion, under high glucose that was normalised to insulin content in Min6 cells overexpressing Ets-1 (Fig. 5c,d).

Overexpression of PDX-1 restored beta cell function impaired by Ets-1. After infection with Ad-Pdx-1 for 12 h, Min6 cells were infected with Ad-Ets-1 for another 48 h. The expression of Ets-1 and PDX-1 (a), GSI (b), insulin content (c) and insulin secretion normalised to insulin content (d) (2 mmol/l, white bars; 20 mmol/l, black bars) was measured. Values are means ± SD and are representative of three individual experiments. *p < 0.05, **p < 0.01 vs control
Hyperglycaemia promotes the interaction of Ets-1 and FOXO1, and the activity of Ets-1 binding to the Pdx-1 promoter
To explore whether high glucose promotes the interaction of Ets-1 and FOXO1, we performed an IP assay. As shown in Fig. 6a, endogenous interaction of Ets-1 and FOXO1 was significantly increased in RINm5F cells treated with high glucose for 48 h. Next, we performed a ChIP-qPCR assay to measure the activity of Ets-1 binding to the Pdx-1 promoter in high glucose-treated RINm5F cells. DNA fragments pulled down by the anti-Ets-1 antibody in the high glucose-treated group were increased to approximately 2.1-fold for the Pdx-1 promoter (Fig. 6b).

Hyperglycaemia promotes the interaction of Ets-1 and FOXO1, and the activity of Ets-1 binding to the Pdx-1 promoter. RINm5F cells were treated with 11.1 or 25 mmol/l glucose for 48 h. IP for interaction of Ets-1 and FOXO1 (a) and ChIP-qPCR analysis for the capacity of Ets-1 binding to the Pdx-1 promoter (b) was performed. Values are means ± SD and are representative of three individual experiments. **p < 0.01 vs control. IB, immunoblot
Inhibition of Ets-1 reverses hyperglycaemia-induced beta cell failure
Using the recombinant adenoviruses for knockdown of Ets-1 expression (si-Ets-1), we next analysed the effect of Ets-1 downregulation on glucose-stimulated insulin secretion (GSIS) and insulin content in isolated islets, and Min6 cells treated with high glucose for 72 h. As demonstrated, si-Ets-1 effectively silenced Ets-1 gene expression and co-ordinately increased PDX-1 protein level in high glucose (Fig. 7a). Knockdown of Ets-1 effectively restored GSI in islets and Min6 cells previously exposed to high glucose over a 72 h period (Fig. 7c,d), and increased insulin content (Fig. 7e,f) and insulin secretion under high glucose that was normalised to insulin content (Fig. 7g,h).

Inhibition of Ets-1 reversed hyperglycaemia-induced impairment in Min6 cells and mouse islets. Min6 cells and mouse islets were infected with adenovirus-control-siRNA (si-control) or si-Ets-1 for 24 h and then treated with high glucose (High G; 25 mmol/l) for 72 h. Ets-1 and PDX-1 protein levels (a), GSI in Min6 cells (c), GSI in mouse islets (d), insulin content in Min6 cells (e), insulin content in mouse islets (f), insulin secretion normalised to insulin content in Min6 cells (g) (2 mmol/l, white bars; 20 mmol/l, black bars), and insulin secretion normalised to insulin content in mouse islets (h) (3.3 mmol/l, white bars; 16.7 mmol/l, black bars) were measured. (b) Diagram depicting the role of Ets-1 in beta cell glucose toxicity and signalling pathway. Values are means ± SD and are representative of three individual experiments. **p < 0.01
Together, these results identified Ets-1 as a mediator of glucotoxicity, as depicted in Fig. 7b. Chronic high glucose treatment stimulated Ets-1 expression, which directly downregulated PDX-1 expression and activity. Following the inhibition of PDX-1 expression and activity, insulin expression was downregulated, which resulted in pancreatic beta cell dysfunction.
Discussion
Glucotoxicity is a major determinant of causing beta cell failure in type 2 diabetes. The current studies demonstrate that Ets-1 mediates pancreatic beta cell dysfunction, linking glucotoxicity to type 2 diabetes. In db/db mice islets as well as in high glucose-treated Min6 cells and primary islets, Ets-1 expression was significantly increased. Using newly generated transgenic mice with overexpression of Ets-1 in pancreatic beta cells, we demonstrate that Ets-1 excess induces glucose intolerance with defective insulin secretion. It is important to note that we obtained three transgenic lines exhibiting high (line 107), medium (line 123) and low (line 79) levels of Ets-1 expression in islets, which all displayed defective insulin secretion and PDX-1 expression (ESM Fig. 1d–h). Thus, the observed effect in transgenic mice was due to expression levels of Ets-1 and was not explained by insertional mutagenesis, i.e. random incorporation of the transgene leading to inactivation of another gene.
Histone modification has been shown to be important in pancreatic beta cell function [23]. Histone H4 acetylation is critical for activation of insulin gene expression [38]. However, it is unclear whether epigenetic modification is involved in manipulating Ets-1 expression in pancreatic beta cells treated with high glucose. We first analysed the promoter region of Ets-1, and found there were many CpG islands. However, 5-Aza-CdR, a specific inhibitor of DNA methylation, had no effect on Ets-1 expression (data not shown). Interestingly, the histone deacetylase inhibitor could upregulate Ets-1 expression in pancreatic beta cells. Meanwhile, high glucose increased histone H4 and H3 acetylation at the −1250 to −1054 regions of the Ets-1 promoter. These results clearly demonstrate the role of epigenetics in regulating Ets-1 transcription induced by high glucose.
It is important to recognise that Ets-1 overexpression caused a reduction in insulin content in pancreatic beta cells both in vivo and in cell-based models. It is therefore possible that decreased GSIS induced by Ets-1 was due to reduced insulin granule formation. Another possibility is that Ets-1 might impair insulin secretion from pancreatic beta cells. We found that overexpression of Ets-1 still decreased insulin secretion when normalised to insulin content. We also found that that overexpression of Ets-1 reduced ATP content under high glucose conditions (ESM Fig. 3a) and GCK expression (ESM Fig. 3b,c). Given ATP and GCK are major contributing factors involved in GSIS [39, 40], reduction of ATP content and GCK expression could participate in Ets-1 impairing insulin secretion.
We identified, for the first time, PDX-1 as the downstream target of Ets-1 in mediating its regulatory effects on pancreatic beta cell function. We demonstrated that Ets-1 could bind to the promoter of Pdx-1 (the PH2 region) shared with FOXO1 and FOXA2 [18]. When Ets-1 was overexpressed in beta cells, the activity of FOXO1 binding to the PDX-1 promoter increased, but that of FOXA2 binding to the same site decreased, which was accompanied by the reduction of activity of the Pdx-1 promoter. Previous studies demonstrated that FOXO1 negatively regulated PDX-1 expression through decreasing Pdx-1 promoter activity while FOXA2 positively regulated PDX-1 expression through increasing Pdx-1 promoter activity [20, 41]. We also found that FOXO1 could interact with Ets-1 in pancreatic beta cells (Fig. 6a). As shown here and previously, we propose that overexpression of Ets-1 enhanced FOXO1 occupying the area left vacant by FOXA2 on the Pdx-1 promoter, which resulted in inhibiting Pdx-1 promoter activity.
It should be noticed that Ets-1 did not bind to the Pdx-1 promoter in the control group, and interference of basal Ets-1 gene transcription did not affect PDX-1 expression (Fig. 5a,b), which indicated that Ets-1 only inhibits PDX-1 expression under certain pathological situations such as hyperglycaemia. Similarly, the FOXO1 transcription factor demonstrated almost no binding to the Pdx-1 gene promoter, which was consistent with our previous results [41]. Thus, neither Ets-1 nor FOXO1 regulated PDX-1 expression by binding to the Pdx-1 promoter under basal situation.
The decrease of PDX-1 expression and DNA binding activities was implicated previously in chronic hyperglycaemia-induced beta cell dysfunction [17]. However, the potential mechanisms were unclear. Here, we established a connection between high glucose, Ets-1 elevation and reduction of PDX-1 expression. We described in detail the cause-and-effect relationship between the increased level of Ets-1 and decreased levels of PDX-1 in vivo and in vitro. High glucose increased the activity of Ets-1 binding to the Pdx-1 promoter (Fig. 6b). On the basis of the existing investigations, we speculated that Ets-1 was responsible for the reduction in PDX-1 expression under the hyperglycaemia condition.
In conclusion, our study demonstrates that chronic hyperglycaemia contributes to upregulation of Ets-1 expression by mediating hyperacetylation of histone H3 and H4 at the Ets-1 gene promoter, leading to reduction of PDX-1 expression, and finally to beta cell dysfunction. This process may be one of the molecular mechanisms responsible for glucotoxicity in beta cells.
Abbreviations
- Ad-Ets-1 :
-
Ets-1 overexpression adenovirus
- BW:
-
Body weight
- ChIP:
-
Chromatin immunoprecipitation
- ChIP-qPCR:
-
Chromatin immunoprecipitation and quantitative real-time PCR
- Ets-1:
-
V-ets avian erythroblastosis virus E26 oncogene homologue 1
- FOX:
-
Forkhead box
- GCK:
-
Glucokinase
- GSI:
-
Glucose-stimulated index
- GSIS:
-
Glucose-stimulated insulin secretion
- IP:
-
Immunoprecipitation
- Neuro D1:
-
Neurogenic differentiation 1
- PDX1:
-
Pancreatic and duodenal homeobox 1
- PH2:
-
PDX-1 homology region 2
- si-Ets-1 :
-
Small interfering (RNA)-Ets-1 adenovirus
- TSA:
-
Trichostatin A
References
Henquin JC (2000) Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49:1751–1760
Neutzsky-Wulff AV, Andreassen KV, Hjuler ST et al (2012) Future detection and monitoring of diabetes may entail analysis of both beta-cell function and volume: how markers of beta-cell loss may assist. J Transl Med 10:214
Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H (1998) Beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev 12:1763–1768
Jonas JC, Sharma A, Hasenkamp W et al (1999) Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J Biol Chem 274:14112–14121
Tokuyama Y, Sturis J, DePaoli AM et al (1995) Evolution of beta-cell dysfunction in the male Zucker diabetic fatty rat. Diabetes 44:1447–1457
Brunner Y, Schvartz D, Priego-Capote F, Coute Y, Sanchez JC (2009) Glucotoxicity and pancreatic proteomics. J Proteomics 71:576–591
Poitout V, Amyot J, Semache M, Zarrouki B, Hagman D, Fontes G (2010) Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys Acta 1801:289–298
Melloul D (2004) Transcription factors in islet development and physiology: role of PDX-1 in beta-cell function. Ann N Y Acad Sci 1014:28–37
Glick E, Leshkowitz D, Walker MD (2000) Transcription factor BETA2 acts cooperatively with E2A and PDX1 to activate the insulin gene promoter. J Biol Chem 275:2199–2204
Waeber G, Thompson N, Nicod P, Bonny C (1996) Transcriptional activation of the GLUT2 gene by the IPF-1/STF-1/IDX-1 homeobox factor. Mol Endocrinol 10:1327–1334
Watada H, Kajimoto Y, Umayahara Y et al (1996) The human glucokinase gene beta-cell-type promoter: an essential role of insulin promoter factor 1/PDX-1 in its activation in HIT-T15 cells. Diabetes 45:1478–1488
Carty MD, Lillquist JS, Peshavaria M, Stein R, Soeller WC (1997) Identification of cis- and trans-active factors regulating human islet amyloid polypeptide gene expression in pancreatic beta-cells. J Biol Chem 272:11986–11993
Marshak S, Benshushan E, Shoshkes M, Havin L, Cerasi E, Melloul D (2000) Functional conservation of regulatory elements in the pdx-1 gene: PDX-1 and hepatocyte nuclear factor 3beta transcription factors mediate beta-cell-specific expression. Mol Cell Biol 20:7583–7590
Liberzon A, Ridner G, Walker MD (2004) Role of intrinsic DNA binding specificity in defining target genes of the mammalian transcription factor PDX1. Nucleic Acids Res 32:54–64
Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF (1997) Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet 15:106–110
Stoffers DA, Ferrer J, Clarke WL, Habener JF (1997) Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet 17:138–139
Kawamori D, Kajimoto Y, Kaneto H et al (2003) Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun NH(2)-terminal kinase. Diabetes 52:2896–2904
Kitamura T, Nakae J, Kitamura Y et al (2002) The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest 110:1839–1847
Issad T, Kuo M (2008) O-GlcNAc modification of transcription factors, glucose sensing and glucotoxicity. Trends Endocrinol Metab 19:380–389
Nakae J, Biggs WH 3rd, Kitamura T et al (2002) Regulation of insulin action and pancreatic beta-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet 32:245–253
Kitamura T, Ido Kitamura Y (2007) Role of FoxO proteins in pancreatic beta cells. Endocr J 54:507–515
Asada S, Daitoku H, Matsuzaki H et al (2007) Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell Signal 19:519–527
Mosley AL, Corbett JA, Ozcan S (2004) Glucose regulation of insulin gene expression requires the recruitment of p300 by the beta-cell-specific transcription factor Pdx-1. Mol Endocrinol 18:2279–2290
Pham TX, Lee J (2012) Dietary regulation of histone acetylases and deacetylases for the prevention of metabolic diseases. Nutrients 4:1868–1886
Seeger FH, Chen L, Spyridopoulos I, Altschmied J, Aicher A, Haendeler J (2009) Downregulation of ETS rescues diabetes-induced reduction of endothelial progenitor cells. PLoS One 4:e4529
Zhang X, Zhang J, Yang X, Han X (2007) Several transcription factors regulate COX-2 gene expression in pancreatic beta-cells. Mol Biol Rep 34:199–206
Han X, Sun Y, Scott S, Bleich D (2001) Tissue inhibitor of metalloproteinase-1 prevents cytokine-mediated dysfunction and cytotoxicity in pancreatic islets and beta-cells. Diabetes 50:1047–1055
He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A 95:2509–2514
Meng ZX, Nie J, Ling JJ et al (2009) Activation of liver X receptors inhibits pancreatic islet beta cell proliferation through cell cycle arrest. Diabetologia 52:125–135
Zhang X, Yong W, Lv J et al (2009) Inhibition of forkhead box O1 protects pancreatic beta-cells against dexamethasone-induced dysfunction. Endocrinology 150:4065–4073
Lin HY, Yin Y, Zhang JX et al (2012) Identification of direct forkhead box O1 targets involved in palmitate-induced apoptosis in clonal insulin-secreting cells using chromatin immunoprecipitation coupled to DNA selection and ligation. Diabetologia 55:2703–2712
Yamasaki S, Yagishita N, Sasaki T et al (2007) Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. EMBO J 26:113–122
Laybutt DR, Weir GC, Kaneto H et al (2002) Overexpression of c-Myc in beta-cells of transgenic mice causes proliferation and apoptosis, downregulation of insulin gene expression, and diabetes. Diabetes 51:1793–1804
Oshima H, Taketo MM, Oshima M (2006) Destruction of pancreatic beta-cells by transgenic induction of prostaglandin E2 in the islets. J Biol Chem 281:29330–29336
Kaneko K, Ueki K, Takahashi N et al (2010) Class IA phosphatidylinositol 3-kinase in pancreatic beta cells controls insulin secretion by multiple mechanisms. Cell Metab 12:619–632
Kobberup S, Nyeng P, Juhl K, Hutton J, Jensen J (2007) ETS-family genes in pancreatic development. Dev Dyn 236:3100–3110
Picarella DE, Kratz A, Li CB, Ruddle NH, Flavell RA (1992) Insulitis in transgenic mice expressing tumor necrosis factor beta (lymphotoxin) in the pancreas. Proc Natl Acad Sci U S A 89:10036–10040
Mosley AL, Ozcan S (2003) Glucose regulates insulin gene transcription by hyperacetylation of histone h4. J Biol Chem 278:19660–19666
Saleh MC, Fatehi-Hassanabad Z, Wang R (2008) Mutated ATP synthase induces oxidative stress and impaired insulin secretion in beta-cells of female BHE/cdb rats. Diabetes Metab Res Rev 24:392–403
Kajimoto Y, Matsuoka T, Kaneto H, Watada H, Fujitani Y, Kishimoto M, Sakamoto K, Matsuhisa M, Kawamori R, Yamasaki Y, Hori M (1999) Induction of glycation suppresses glucokinase gene expression in HIT-T15 cells. Diabetologia 42:1417–1424
Chen F, Zhu Y, Tang X (2011) Dynamic regulation of PDX-1 and FoxO1 expression by FoxA2 in dexamethasone-induced pancreatic beta-cells dysfunction. Endocrinology 152:1779–1788
Acknowledgements
The authors gratefully acknowledge the generous support of the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine of Jiangsu Province (Nanjing, China).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by grants from: (1) the Special Funds for Major State Basic Research Program of China (973 Program, 2011CB504003) and the State Key Program of National Natural Science of China (81130013) to XH; (2) the National Natural Science Foundation of China (81200558) to FC.
Contribution statement
All authors took part in the conception and design of the study, as well as either drafting or critically revising the manuscript. All authors have approved the final version of the manuscript. MS, YW, TW, WS, JL and WZ collected the data and carried out the data analysis. XH is responsible for the integrity of the work as a whole.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 106 kb)
ESM Fig. 1
(PDF 141 kb)
ESM Fig. 2
(PDF 780 kb)
ESM Fig. 3
(PDF 162 kb)
ESM Table 1
(PDF 90 kb)
Rights and permissions
About this article

Cite this article
Chen, F., Sha, M., Wang, Y. et al. Transcription factor Ets-1 links glucotoxicity to pancreatic beta cell dysfunction through inhibiting PDX-1 expression in rodent models. Diabetologia 59, 316–324 (2016). https://doi.org/10.1007/s00125-015-3805-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3805-3