
Abstract
Aims/hypothesis
Type 2 diabetes is closely associated with pathological lipid accumulation in the liver, which is suggested to actively contribute to the development of insulin resistance. We recently identified serine/threonine protein kinase 25 (STK25) as a regulator of liver steatosis, whole-body glucose tolerance and insulin sensitivity in a mouse model system. The aim of this study was to assess the role of STK25 in the control of lipid metabolism in human liver.
Methods
Intracellular fat deposition, lipid metabolism and insulin sensitivity were studied in immortalised human hepatocytes (IHHs) and HepG2 hepatocellular carcinoma cells in which STK25 was overexpressed or knocked down by small interfering RNA. The association between STK25 mRNA expression in human liver biopsies and hepatic fat content was analysed.
Results
Overexpression of STK25 in IHH and HepG2 cells enhanced lipid deposition by suppressing β-oxidation and triacylglycerol (TAG) secretion, while increasing lipid synthesis. Conversely, knockdown of STK25 attenuated lipid accumulation by stimulating β-oxidation and TAG secretion, while inhibiting lipid synthesis. Furthermore, TAG hydrolase activity was repressed in hepatocytes overexpressing STK25 and reciprocally increased in cells with STK25 knockdown. Insulin sensitivity was reduced in STK25-overexpressing cells and enhanced in STK25-deficient hepatocytes. We also found a statistically significant positive correlation between STK25 mRNA expression in human liver biopsies and hepatic fat content.
Conclusions/interpretation
Our data suggest that STK25 regulates lipid partitioning in human liver cells by controlling TAG synthesis as well as lipolytic activity and thereby NEFA release from lipid droplets for β-oxidation and TAG secretion. Our findings highlight STK25 as a potential drug target for the prevention and treatment of type 2 diabetes.
Similar content being viewed by others

Introduction
Type 2 diabetes is closely associated with ectopic lipid deposition within the liver, which actively contributes to the development of hepatic and systemic insulin resistance [1, 2]. A comprehensive understanding of the molecular mechanisms controlling intrahepatic lipid accumulation is therefore critically needed to support the development of new treatments for type 2 diabetes.
In the search for novel targets that contribute to the pathogenesis of type 2 diabetes, we identified serine/threonine protein kinase 25 (STK25 [also referred to as YSK1 or SOK1]), a member of the sterile 20 (STE20) kinase superfamily [3], as a critical regulator of ectopic lipid deposition, systemic glucose and insulin homeostasis [4–7]. We found that partial knockdown of STK25 in the rat myoblast cell line L6 by small interfering (si)RNA improves insulin-stimulated glucose uptake [4]. Furthermore, genetic disruption of STK25 in knockout mice provides protection from the detrimental metabolic consequences of high-fat-diet exposure on liver and skeletal muscle lipid accumulation, accompanied by better preserved systemic glucose tolerance, reduced hepatic glucose production and increased whole-body insulin sensitivity [7]. These findings are reciprocal to the metabolic phenotype of high-fat-fed transgenic mice overexpressing STK25, which develop marked liver steatosis combined with hyperinsulinaemia, impaired systemic glucose tolerance and insulin resistance compared with wild-type littermates [5, 6].
The function of STK25 in the regulation of lipid accumulation in human liver has not been studied. Furthermore, the global depletion and overexpression of STK25 in knockout and transgenic mice, respectively, do not allow investigators to address whether the impact of STK25 on hepatic lipid homeostasis is direct or secondary to the action of STK25 in tissues other than liver. The present study provides several lines of evidence to support the key cell-specific role of STK25 in the control of lipid deposition and insulin sensitivity in human liver cells.
Methods
Cell culture
Immortalised human hepatocytes (IHHs) (a gift from B. Staels, the Pasteur Institute of Lille, University of Lille Nord de France, Lille, France [8]) were maintained in Complete William’s E medium (Gibco, Paisley, UK) supplemented with bovine insulin (20 U/l; Sigma-Aldrich, St Louis, MO, USA) and dexamethasone (50 nmol/l; Sigma-Aldrich). HepG2 cells (hepatocellular carcinoma, human, American Type Culture Collection, Manassas, VA, USA) were maintained in DMEM (Lonza, Basel, Switzerland). Both culture media were supplemented with 10% (vol./vol.) FBS, l-glutamine (2 mmol/l) and 1% (vol./vol.) penicillin/streptomycin (Gibco). Cells were demonstrated to be free of mycoplasma infection by use of the MycoAlert Mycoplasma Detection kit (Lonza).
Transient overexpression
Cells were transfected with pFLAG-STK25 (GeneCopoeia, Rockville, MD, USA) or an empty control plasmid using Lipofectamine 2000 (Invitrogen, San Diego, CA, USA).
RNA interference
Cells were transfected with anti-STK25 siRNA (a mixture of seven sets of siRNA against human STK25; s20570; Ambion, Austin, TX, USA) or scrambled siRNA (AM4635; Ambion) using Lipofectamine RNAiMax (Invitrogen).
Western blot and immunofluorescence
Western blot was performed as previously described [5] using anti-STK25 primary antibody, working dilution 1:1,000 (anti-YSK1; sc-6865; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and horseradish-peroxidase-conjugated anti-goat IgG secondary antibody, working dilution 1:1,000 (sc-2020; Santa Cruz Biotechnology). For immunofluorescence, cells were probed with anti-STK25 antibody, working dilution 1:200, followed by incubation with cyanine-3-labelled anti-goat IgG, working dilution 1:500 (20333; Biotium, Hayward, CA, USA). The validation of anti-STK25 antibody has been provided by using Stk25-knockout mice [6].
Lipid content and mitochondrial function
Cells were stained with Nile Red or Oil Red O for lipids, and MitoTracker Red for mitochondria (see the electronic supplementary material [ESM] Methods). Lipids were extracted using the Folch method [9] and quantified using ultraperformance liquid chromatography/mass spectrometry and direct-infusion mass spectrometry [10]. Citrate synthase activity was measured in the isolated mitochondrial fraction by monitoring the rate of reduction of 5,5′-dithio-bis(2-nitrobenzoic acid) at 412 nm.
Assessment of lipid metabolism and insulin sensitivity
To measure β-oxidation, the cells were incubated in the presence of [9,10-3H(N)]-palmitic acid, and [3H]-labelled water was measured as the product of NEFA oxidation (see the ESM Methods). Triacylglycerol (TAG) secretion, oleic acid (OA) and palmitic acid uptake, and incorporation of [14C]OA and [14C]glucose into TAG, were measured as described in the ESM Methods. Glucose uptake and production in response to insulin (100 nmol/l; Actrapid Penfill; Novo Nordisk, Bagsværd, Denmark) and glucose production in response to 5-amino-4-imidazole-carboxamideriboside (AICAR; 1 and 2 mmol/l; Toronto Research Chemicals, North York, ON, Canada) were assessed as previously described [6].
TAG hydrolase activity
The activity of TAG hydrolase was determined in total cell lysates using [3H]triolein (PerkinElmer, Waltham, MA, USA) as the substrate [11].
Assessments in Stk25-knockout mice
For a description of the in vivo and ex vivo assays performed in Stk25 −/− mice (a gift from B. Howell, Department of Neuroscience and Physiology, State University of New York Upstate Medical University, Syracuse, NY, USA), see the ESM Methods. All animal experiments were performed after approval from the Ethics Committee for Animal Studies at the Administrative Court of Appeals in Gothenburg, Sweden, and followed appropriate guidelines.
Quantitative real-time PCR in liver biopsies of human participants
The expression of STK25 mRNA was measured in liver tissue samples obtained from 62 white individuals (men, n = 35; women, n = 27) who underwent open abdominal surgery for Roux-en-Y bypass, sleeve gastrectomy, explorative laparotomy or elective cholecystectomy. Liver biopsy donors fulfilled the following inclusion criteria: (1) men and women, age >18 years; (2) indication for elective laparoscopic or open abdominal surgery; (3) BMI between 18 and 50 kg/m2; (4) abdominal MRI feasible; and (5) signed written informed consent. The exclusion criteria were: (1) significant acute or chronic inflammatory disease or clinical signs of infection; (2) C-reactive protein (CrP) >952.4 nmol/l; (3) type 1 diabetes and/or antibodies against GAD and islet cell antibodies (ICA); (4) systolic blood pressure >140 mmHg and diastolic blood pressure >95 mmHg; (5) clinical evidence of either cardiovascular or peripheral artery disease; (6) thyroid dysfunction; (7) alcohol or drug abuse; and (8) pregnancy. All participants gave their written informed consent before taking part in the study. All investigations were approved by the Ethics Committee of the University of Leipzig, Germany (363-10-13122010 and 017-12-230112) and carried out in accordance with the Declaration of Helsinki. For details on the measurement of total body and liver fat, see Kannt et al and Hussain et al [12, 13]. For participant characteristics and details on quantitative real-time (qRT)-PCR, see ESM Table 1 and the ESM Methods, respectively.
Statistical analysis
Statistical significance between the groups was calculated with an unpaired two-tailed Student’s t test or by two-way ANOVA, followed by Tukey’s post hoc test, with a value of p < 0.05 considered statistically significant. Correlation between STK25 expression in human liver and hepatic fat content was assessed by Spearman’s rank correlation analysis after the Kolmogorov–Smirnov test was performed to assess normality of data. All statistical analyses were performed using SPSS statistics (v22) (IBM Corporation, Armonk, NY, USA).
The nonclinical experiments were not blinded. No outlier data or samples have been excluded from analysis and no results were omitted from the reporting.
Results
Overexpression of STK25 induces lipid accumulation in IHHs and HepG2 cells as a consequence of reduced β-oxidation and TAG secretion combined with increased lipid synthesis
Our previous studies show that high-fat-fed Stk25 transgenic mice display a dramatic increase in liver steatosis compared with wild-type littermates because of reduced β-oxidation and VLDL-TAG secretion [6]. Here, we examined lipid metabolism in liver cells of human origin overexpressing STK25. The IHHs and HepG2 were transiently transfected with human STK25 expression plasmid or the empty control plasmid. Cells transfected with STK25 expression plasmid had substantially higher STK25 mRNA and protein abundance (99.2 ± 7.2-fold and 11.3 ± 0.5-fold increase in mRNA and 6.7 ± 0.6-fold and 10.8 ± 0.6-fold increase in protein in IHHs and HepG2 cells, respectively; ESM Fig. 1a, Fig. 1a). Immunofluorescence analysis identified STK25 staining inside or on the lipid droplets (LDs) visualised with the lipophilic dye Nile Red in IHHs and HepG2 cells transfected with the vector control (Fig. 1b); this cellular localisation of STK25 was not altered when hepatocytes were incubated with insulin and/or OA (ESM Fig. 2). Similarly to the endogenous protein, overexpressed STK25 was targeted to the LDs (Fig. 1b).

Overexpression of STK25 induces lipid accumulation in human hepatocytes. IHHs and HepG2 cells were transfected with STK25 expression plasmid or vector control (mock). (a) Representative western blot with anti-STK25 antibodies; actin was used as a loading control (endogenous STK25 48 kDa, FLAG-tagged STK25 51 kDa). (b) Representative immunofluorescence images of cells double-stained with antibodies for STK25 (red) and Nile Red (green); nuclei stained with DAPI (blue). Scale bars, 10 μm. (c) Oil Red O staining under basal conditions and after OA supplementation. Representative cell images stained with Oil Red O and counterstained with haematoxylin. Scale bars, 100 μm. (d) Spectrophotometric measurement of eluted Oil Red O. (e, f) LD number (e) and size distribution (f) measured under basal conditions. (g) Lipidomics analysis performed in IHHs. For (d) and (g), results are means ± SEM from 6–8 wells. The data shown in (a) and (b) are representative of at least two independent transfection experiments with similar results. The data shown in (c, d), (e, f) and (g) originate from three independent transfection experiments. * p < 0.05 and ** p < 0.01 comparing cells transfected with STK25 expression plasmid vs vector control. † p < 0.05 and †† p < 0.01, and ‡ p < 0.05 and ‡‡ p < 0.01 comparing basal conditions vs OA supplementation in cells transfected with vector control and STK25 expression plasmid, respectively. White bars, cells transfected with vector control (mock); black bars, cells transfected with STK25 expression plasmid
To analyse lipid deposition, cells were stained with Oil Red O, which detects neutral lipids. STK25 overexpression increased lipid accumulation approximately two- to threefold compared with the vector control in both cell lines based on colorimetric quantification of Oil Red O staining (Fig. 1c, d). Morphometric analysis further confirmed a marked increase in the total number of LDs and a shift in the overall LD size distribution toward larger LDs in STK25-overexpressing cells (Fig. 1e, f).
In parallel to the assessment of lipid accumulation under basal culture conditions, we exposed the cells to OA, known to efficiently induce steatosis in vitro [14–17]. As expected, OA supplementation increased Oil Red O staining in all cells; however, lipid deposition remained substantially higher in STK25-overexpressing cells compared with vector control (Fig. 1c, d). Of note, Oil Red O staining in cells overexpressing STK25 under basal conditions was similar to that observed in cells transfected with vector control when challenged by OA (Fig. 1c, d).
Lipidomics analysis, performed only in IHHs, confirmed enhanced accumulation of cholesteryl esters, TAG, lysophosphatidylcholines, sphingomyelins and ceramides in cells overexpressing STK25 both under basal conditions and after OA challenge, while increases in phosphatidylcholines and phosphatidylethanolamines were observed only under basal conditions (Fig. 1g, ESM Table 2).
Furthermore, we investigated the mechanisms underlying the increased lipid accumulation by STK25. In both IHHs and HepG2 cells, STK25 overexpression resulted in a significant reduction in β-oxidation compared with vector control (Fig. 2a). Consistently, staining with MitoTracker Red, a fluorescent dye that specifically accumulates within respiring mitochondria, was lower in both cell lines overexpressing STK25 (ESM Fig. 3a, Fig. 2b). Of note, the activity of the mitochondrial matrix enzyme citrate synthase was significantly reduced by STK25 overexpression (ESM Fig. 3c). The secretion of TAG into the media was significantly suppressed in STK25-overexpressing cells (Fig. 2c). Interestingly, despite the repressed fatty acid uptake in cells overexpressing STK25 (Fig. 2d, ESM Fig. 4a), the incorporation of media-derived [14C]-labelled OA and [14C]-labelled glucose into intracellular TAG was significantly increased (Fig. 2e, f). Of note, we found that TAG hydrolase activity was lower in IHHs and HepG2 cells transfected with STK25 expression plasmid (Fig. 2g).

Overexpression of STK25 in human hepatocytes reduces β-oxidation, TAG secretion and fatty acid uptake and increases lipid synthesis. IHHs and HepG2 cells were transfected with STK25 expression plasmid or vector control (mock); the assessments were performed under basal cell culture conditions. (a) Oxidation of radiolabelled palmitate. (b) Assessment of relative mitochondrial area by MitoTracker Red staining. (c) Secretion of [3H]TAG into the media. (d) Uptake of [3H]-labelled OA. (e, f) TAG synthesis from [14C]-labelled OA (e) and [14C]-labelled glucose (f). (g) TAG hydrolase activity. For (a) and (c–g), results are means ± SEM from 4–10 wells. The data shown in (a) and (c, d) are representative of two independent transfection experiments with similar results; a trend for repressed TAG hydrolase activity in HepG2 cells (g) was observed in an independent experiment (p = 0.08). The data shown in each figure part originate from an independent transfection experiment. In bar diagrams, * p < 0.05 and ** p < 0.01 comparing cells transfected with STK25 expression plasmid vs vector control. In line diagrams † p < 0.05, †† p < 0.01 and ‡ p < 0.05, and ‡‡ p < 0.01 comparing cells transfected with STK25 expression plasmid vs vector control in IHHs and HepG2 cells, respectively. White bars, cells transfected with vector control (mock); black bars, cells transfected with STK25 expression plasmid. White triangles and circles, IHH and HepG2 cells, respectively, transfected with vector control (mock); black triangles and circles, IHH and HepG2 cells, respectively, transfected with STK25 expression plasmid
Knockdown of STK25 reduces lipid deposition in IHHs and HepG2 cells via increased β-oxidation and TAG secretion combined with repressed lipid synthesis
Our recent studies demonstrate that mice with genetic disruption of STK25 are protected from diet-induced liver steatosis [7]. To investigate the impact of STK25 knockdown on lipid metabolism in human hepatocytes, we transfected IHHs and HepG2 cells with STK25-specific siRNA or with a non-targeting control (NTC) siRNA. In both cell lines transfected with anti-STK25 siRNA, the STK25 mRNA expression was repressed by approximately 80% (ESM Fig. 1b), whereas the protein level of STK25 was below the detection limit of western blot (Fig. 3a, b).

Knockdown of STK25 suppresses lipid accumulation in human hepatocytes supplemented with OA. IHHs and HepG2 cells were transfected with anti-STK25 or NTC siRNA. (a) Representative western blot with anti-STK25 antibodies; actin was used as a loading control. (b) Representative immunofluorescence images of cells double-stained with antibodies for STK25 (red) and Nile Red (green); nuclei stained with DAPI (blue). Scale bars, 10 μm. (c) Oil Red O staining under basal conditions and after OA supplementation. Representative cell images stained with Oil Red O and counterstained with haematoxylin. Scale bars, 100 μm. (d) Spectrophotometric measurement of eluted Oil Red O. (e, f) LD number (e) and size distribution (f) measured after OA supplementation. (g) Lipidomics analysis performed in IHHs. For (d) and (g), results are means ± SEM from 6–8 wells. The data shown in (a) and (b) are representative of at least two independent transfection experiments with similar results. The data shown in (c, d), (e, f) and (g) originate from three independent transfection experiments. * p < 0.05 and ** p < 0.01 comparing cells transfected with anti-STK25 vs NTC siRNA. † p ≤ 0.05 and †† p < 0.01 comparing basal conditions vs OA supplementation in cells transfected with NTC siRNA. White bars, cells transfected with NTC siRNA; grey bars, cells transfected with anti-STK25 siRNA
Knockdown of STK25 did not significantly alter lipid accumulation under basal culture conditions (Fig. 3c, d). However, when the cells were exposed to OA, the Oil Red O signal remained approximately 1.5- to threefold lower in both cell lines transfected with anti-STK25 siRNA compared with NTC siRNA (Fig. 3c, d). In fact, no significant increase in Oil Red O staining was observed in response to OA supplementation in STK25-deficient cells compared with basal conditions (Fig. 3c, d). Morphometric analysis further revealed that STK25 knockdown decreased the total number of LDs and caused a shift in the LD size distribution towards smaller droplets in cells exposed to OA (Fig. 3e, f). The observation that STK25 depletion only repressed lipid accumulation in hepatocytes after challenge with OA suggests compensation for the loss-of-gene function in cells transfected with anti-STK25 siRNA in basal but not in challenged conditions.
Lipidomics analysis, performed only in IHHs, confirmed that OA supplementation enhanced the levels of cholesteryl esters, TAG, lysophosphatidylcholines, sphingomyelins, and ceramides in cells transfected with NTC siRNA, but no increase was seen in cells transfected with anti-STK25 siRNA (Fig. 3g; ESM Table 3).
Silencing of STK25 mediated by siRNA resulted in a marked increase in β-oxidation in both IHHs and HepG2 cells (Fig. 4a). Consistently, the mitochondrial area was significantly augmented in STK25-deficient cells (ESM Fig. 3b, Fig. 4b). Of note, the activity of citrate synthase in the isolated mitochondrial fraction was not altered by STK25 depletion (ESM Fig. 3d). The concentration of de novo synthesised TAG secreted into the media was markedly higher in cells transfected with anti-STK25 siRNA (Fig. 4c). No significant change in fatty acid influx was observed in STK25-deficient cells; a tendency for increased fatty acid uptake was, nevertheless, seen in IHHs (p < 0.1; Fig. 4d, ESM Fig. 4b). The incorporation of media-derived [14C]-labelled OA and [14C]-labelled glucose into intracellular TAG was significantly reduced in hepatocytes in which STK25 was depleted (Fig. 4e, f). Interestingly, TAG hydrolase activity was significantly higher in both cells lines transfected with anti-STK25 siRNA (Fig. 4g).

Knockdown of STK25 in human hepatocytes supplemented with OA increases β-oxidation and TAG secretion and suppresses lipid synthesis. IHHs and HepG2 cells were transfected with anti-STK25 or NTC siRNA; the assessments were performed after 24 h incubation with OA. (a) Oxidation of radiolabelled palmitate. (b) Assessment of relative mitochondrial area by MitoTracker Red staining. (c) Secretion of [3H]TAG into the media. (d) Uptake of [3H]-labelled OA. (e, f) TAG synthesis from [14C]-labelled OA (e) and [14C]-labelled glucose (f). (g) TAG hydrolase activity. For (a) and (c–g), results are means ± SEM from 4–6 wells. The data shown in each figure part originate from an independent transfection experiment. In bar diagrams, § p < 0.1, * p < 0.05 and ** p < 0.01 comparing cells transfected with anti-STK25 vs NTC siRNA. In line diagrams, † p < 0.05 and ‡ p < 0.05 comparing cells transfected with anti-STK25 vs NTC siRNA in IHHs and HepG2 cells, respectively. White bars, cells transfected with NTC siRNA; grey bars, cells transfected with anti-STK25 siRNA. White triangles and circles, IHH and HepG2 cells, respectively, transfected with NTC siRNA; grey triangles and circles, IHH and HepG2 cells, respectively, transfected with anti-STK25 siRNA
Overexpression of STK25 induces insulin and AICAR resistance whereas STK25 knockdown improves insulin and AICAR sensitivity in IHHs and HepG2
Consistent with markedly increased lipid accumulation, insulin failed to regulate the glucose production and uptake in IHHs and HepG2 overexpressing STK25, whereas cells transfected with the vector control displayed the expected statistically significant suppression of glucose production and increase of glucose uptake by insulin treatment (Fig. 5a, b). Reciprocally, marked enhancement in response to insulin in terms of repression of glucose production and increase of glucose uptake was observed in cells transfected with anti-STK25 siRNA compared with NTC siRNA (Fig. 5d, e).

Overexpression of STK25 in human hepatocytes induces insulin resistance whereas knockdown of STK25 improves insulin sensitivity. (a–c) IHHs and HepG2 cells were transfected with STK25 expression plasmid or vector control (mock); the assessments were performed under basal cell culture conditions. (d–f) IHHs and HepG2 cells were transfected with anti-STK25 or NTC siRNA; the assessments were performed after 24 h incubation with OA. Glucose production (a, d) and uptake (b, e) in response to insulin. Glucose production in response to AICAR (c, f). Results are means ± SEM from 5–6 wells. The data shown in (a) and (c) are representative of two independent transfection experiments with similar results. The data shown in each figure part originate from an independent transfection experiment. * p < 0.05 and ** p < 0.01 comparing cells transfected with STK25 expression plasmid vs vector control (a–c) or anti-STK25 vs NTC siRNA (d–f). † p < 0.05 and †† p < 0.01 comparing basal vs insulin-stimulated conditions in control cells. ‡ p < 0.05 and ‡‡ p < 0.01 comparing basal vs insulin-stimulated conditions in cells transfected with STK25 expression plasmid (c) or anti-STK25 siRNA (d–f). White bars, cells transfected with vector control (mock; a–c) or NTC siRNA (d–f); black bars, cells transfected with STK25 expression plasmid; grey bars, cells transfected with anti-STK25 siRNA
Cross-talk between AMP-activated protein kinase (AMPK) and STK25 signalling pathways has been suggested based on interaction of STK25 with the upstream activator complex of AMPK [18, 19]. To investigate this interaction, we treated cells with the AMPK agonist AICAR [20]. Interestingly, the suppression of glucose production by AICAR was blunted in STK25-overexpressing hepatocytes and augmented in STK25-deficient hepatocytes compared with the respective control-transfected cells (Fig. 5c, f).
Depletion of STK25 stimulates hepatic VLDL-TAG secretion and β-oxidation in a mouse model
To extend the findings of STK25 knockdown in IHHs and HepG2 cells to in vivo settings, we assessed liver lipid metabolism in high-fat-fed Stk25 −/− mice and wild-type littermates. To measure hepatic VLDL-TAG secretion in vivo, the mice were injected with Triton WR-1339; under these conditions, circulating TAG is mainly derived from hepatic VLDL secretion [21]. We observed markedly higher levels of TAG and VLDL in the plasma of Stk25 −/− mice (Fig. 6a, b). To assess hepatic lipid uptake, the mice were injected with an intravenous bolus dose of Intralipid, thus bypassing intestinal absorption [22]. No difference in liver clearance of lipids was observed between the genotypes in this assay (Fig. 6c). Primary hepatocytes isolated from Stk25 −/− mice had a higher oxidative capacity than wild-type hepatocytes, while fatty acid influx was similar (Fig. 6d, e). These observations are reciprocal to our earlier findings in high-fat-fed transgenic mice overexpressing STK25, which display lower hepatic VLDL-TAG secretion in vivo without any alteration in liver clearance of lipids; hepatocytes of Stk25 transgenic mice had a lower oxidative capacity compared with wild-type hepatocytes, while fatty acid influx was not changed [6].

Depletion of STK25 in knockout mice increases hepatic VLDL-TAG secretion and β-oxidation. (a, b) TAG content in plasma and secretion rate of TAG after an intraperitoneal injection of Triton WR-1339 (a); lipoprotein profiling of plasma 4 h after the injection (b). (c) Plasma clearance of TAG measured after an intravenous injection of Intralipid. (d, e) β-oxidation (d) and OA uptake (e) in isolated primary hepatocytes. Results are means ± SEM from 7–11 mice/genotype. *p < 0.05 comparing wild-type vs knockout mice. White circles and bars, high-fat-fed wild-type mice; black circles and bars, high-fat-fed knockout mice. HFD, high-fat diet; WT, wild-type
Expression of STK25 mRNA is significantly and positively correlated with fat content in human liver
We examined the expression of STK25 mRNA in relation to the hepatic fat content in liver biopsy material collected from 62 individuals with a wide range of BMI (22.7–45.6 kg/m2), body fat (19.5–57.9%) and liver fat content (1.1–50.0%). Hepatic STK25 mRNA correlated significantly and positively with liver fat (Fig. 7a). Furthermore, we showed that STK25 expression was 2.3 ± 0.4-fold higher in the liver of individuals with high intrahepatic TAG (>6%) compared with liver from those with low intrahepatic TAG (<6%) (Fig. 7b). There was no correlation between hepatic STK25 mRNA and the BMI, body fat content or WHR of the participants (ESM Fig. 5), indicating that the increase in liver STK25 expression was not a consequence of obesity.

Expression of STK25 mRNA is significantly and positively correlated with fat content in human liver. (a) Correlation between hepatic fat content and STK25 expression in human liver biopsies (n = 62); r = 0.47, p = 0.00011. The variables were log10 transformed. (b) STK25 expression in livers of individuals with low (n = 17) vs high (n = 45) intrahepatic TAG content. For (b), results are means ± SEM. * p < 0.05. IHTAG, intrahepatic TAG; RQ, relative quantification
Discussion
In this study we provide several lines of evidence to support a key role for the protein kinase STK25 in regulating liver lipid partitioning. First, we found that overexpression of STK25 in the human hepatocyte cell lines IHH and HepG2 promoted lipid deposition by suppressing β-oxidation and TAG secretion and enhancing TAG synthesis. This is in agreement with our previous observation of increased lipid storage via reduced β-oxidation and repressed VLDL-TAG export in the liver of high-fat-fed Stk25 transgenic mice [6]. Conversely, we found that siRNA knockdown of STK25 in IHHs and HepG2 cells attenuated lipid accumulation by stimulating β-oxidation and TAG secretion and inhibiting TAG synthesis. Consistent with these results, we observed augmented hepatic β-oxidation and VLDL-TAG export in Stk25-knockout mice fed a high-fat diet, extending the results from our earlier studies showing that Stk25 −/− mice are protected against diet-induced liver steatosis [7]. Moreover, we found a statistically significant positive association between STK25 mRNA expression and fat content in human liver. Notably, we have only been able to measure mRNA and not protein abundance of STK25 in human liver biopsies, and it is currently not known whether any physiological situations exist in which the STK25 protein level is increased to an extent similar to that of the overexpressed protein in human hepatocytes transfected with the STK25 expression plasmid in this study or in the transgenic mice used in our previous experiments [5, 6], which is a limitation of the models used.
It is generally accepted that mitochondrial β-oxidation plays an important role in liver steatosis and hepatic insulin resistance, although the nature of this role is still under debate. Increased hepatic mitochondrial oxidation has been observed in patients and rodents with fatty liver [23, 24], which likely reflects a metabolic adaptation to elevated lipid burden to limit further fat accumulation. Indeed, the development of fatty liver and hepatic insulin resistance in response to high-fat feeding in rats can be prevented by increasing mitochondrial β-oxidation [25–28]. Furthermore, a primary defect in mitochondrial β-oxidation capacity in mice has been shown to result in liver steatosis and hepatic insulin resistance [29]. Of note, in this study we observed enhanced β-oxidation in human hepatocytes transfected with anti-STK25 siRNA compared with scrambled siRNA even under basal culture conditions when intrahepatocellular lipid storage was similar (ESM Fig. 6), suggesting that the stimulation of β-oxidation by STK25 depletion is likely a primary event, rather than a compensatory mechanism related to higher lipid content.
The liver exports TAG to extrahepatic tissues through VLDL secretion, and the formation of mature VLDL particles is highly dependent on the availability of cytosolic TAG [30, 31]. This might appear to be contradictory to our observation that STK25 overexpression reduced TAG secretion in human hepatocytes despite a dramatic increase in lipid accumulation, and the reciprocal effect was seen with STK25 knockdown. However, hepatic VLDL-TAG secretion is a highly regulated process and increased cytosolic TAG accumulation alone does not necessarily result in enhanced VLDL export. For example, accelerating hepatic TAG storage by liver-specific overexpression of the key TAG synthetic enzymes diacylglycerol O-acyltransferase (DGAT)1 or DGAT2 is not sufficient to affect VLDL secretion in vivo [32, 33]. Similarly, VLDL export is either unchanged [34] or even decreased [35] in leptin-deficient ob/ob mice, despite drastically increased hepatic TAG deposition. Moreover, recent studies show that TM6SF2, a gene with hitherto unknown function, has opposing effects on the hepatic lipid secretion and liver fat content. Knockdown of Tm6sf2 in mice decreased hepatic VLDL secretion by 50% and increased liver TAG content threefold [36]. Consistently, functional studies in human hepatoma Huh7 and HepG2 cells showed that TM6SF2 siRNA inhibition was associated with reduced secretion of lipids and increased cellular TAG storage [37]. These results are in line with data from population studies demonstrating associations between the region on chromosome 19 (19p13) flanking TM6SF2 and plasma TAG concentration and hepatic steatosis [38–45].
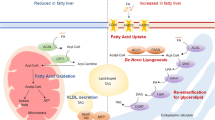
We found that STK25 coats LDs in human hepatocytes, in agreement with our earlier observation in mouse liver [6]. Hepatic LDs, once thought to be only inert energy storage depots, are increasingly recognised as organelles that play a key role in the regulation of liver lipid metabolism [46]. Intrahepatocellular LDs are the major source of TAG substrate for the biogenesis of VLDL via a process involving lipolysis [47, 48]. Alternatively, the NEFA released from liver LDs by lipase activity can be used for mitochondrial β-oxidation [49]. Because of its subcellular localisation, we hypothesised that STK25 regulates hepatic lipid catabolism by controlling release of NEFA from LDs. Indeed, we found that TAG hydrolase activity was significantly repressed in human hepatocytes overexpressing STK25 and increased in hepatocytes where STK25 was knocked down, compared with their respective control-transfected cells. These observations are consistent with our previous findings showing that STK25 overexpression in transgenic mice reduces the association of adipose triacylglycerol lipase (ATGL)/patatin-like phospholipase domain containing 2 (PNPLA2) with hepatic LDs [6]. A major hepatic lipase, ATGL normally remains constitutively associated with LDs and catalyses the initial step in TAG hydrolysis [11]. Reduced hepatic activity of ATGL has been reported in patients with liver steatosis and in obese mice [50, 51]. Hepatic depletion of ATGL in mice leads to severe liver steatosis and reduced β-oxidation, while hepatic overexpression of ATGL reduces hepatic steatosis, increases β-oxidation and improves insulin signal transduction [11, 50, 52]. Furthermore, liver-specific ablation of comparative gene identification-58 (CGI-58), a coactivator of ATGL, leads to marked liver steatosis through reduction in TAG hydrolase activity [53]. We therefore propose that STK25 regulates liver lipid catabolism by controlling lipolytic activity and thereby NEFA release from the LDs for VLDL-TAG secretion and β-oxidation (Fig. 8), possibly mediated by displacement of ATGL.

Putative model for STK25 function in regulating lipid accumulation in human hepatocytes (IHH and HepG2 cells). Overexpression of STK25 represses LD catabolism through suppressed β-oxidation and VLDL-TAG secretion, and promotes LD anabolism through enhanced TAG synthesis. Knockdown of STK25, conversely, increases β-oxidation and VLDL-TAG secretion and reduces TAG synthesis
We observed an increase in the incorporation of media-derived fatty acids and glucose into intracellular TAG in human hepatocytes overexpressing STK25. Reciprocally, knockdown of STK25 suppressed TAG synthesis in human hepatocytes. According to the prominent model, LDs are formed within the lipid bilayer of the endoplasmic reticulum (ER) and are subsequently budded; LDs then grow because of the action of various TAG and phospholipid synthetic enzymes present on the LD surface [46, 54]. Based on the subcellular localisation of STK25, we speculate that STK25 regulates TAG synthesis by directly controlling the activity of LD-associated enzymes that synthesise TAG. However, it is also possible that the effect on TAG synthesis observed in our experimental setup in fact reflects an altered rate of hydrolysis of the newly synthesised lipids.
Major contributions of the liver to systemic glucose homeostasis involve the regulation of glucose uptake and production by insulin. Ectopic lipid storage in the liver is known to contribute to the pathogenesis of insulin resistance and type 2 diabetes [1, 2]. We previously showed impaired systemic glucose and insulin homeostasis in Stk25 transgenic mice fed a high-fat diet [5] and the reciprocal phenotype in Stk25 −/− mice [7]. Consistent with these earlier findings, we observed that the effect of insulin on glucose uptake and production was lost in STK25-overexpressing human hepatocytes and significantly enhanced in STK25-deficient hepatocytes challenged by OA, which is consistent with the changes in lipid deposition pattern in these cells. Of note, we observed no enhancement in response to insulin in terms of repression of glucose production or increase of glucose uptake in hepatocytes transfected with anti-STK25 siRNA compared with scrambled siRNA under basal culture conditions when intrahepatocellular lipid storage was similar (ESM Fig. 7), supporting STK25 regulation of insulin sensitivity through changes in lipid content. Interestingly, we also found that suppression of glucose production by the pharmacological AMPK agonist AICAR was blunted in human hepatocytes overexpressing STK25 and enhanced in cells where STK25 was depleted, which suggests possible crosstalk between AMPK and STK25 signalling pathways.
Taken together, our studies using the transient overexpression and acute knockdown of STK25 in human hepatocytes in vitro, evaluation of Stk25-transgenic and -knockout mice in vivo, and finally expression analysis in human liver biopsies all provide consistent evidence for a cell-specific role of STK25 in the regulation of metabolic balance of hepatic lipid use vs lipid storage. Our findings provide a basis for further studies to increase our insight into the mechanisms of lipid-mediated liver injury while highlighting STK25 as a potential drug target for the prevention and treatment of type 2 diabetes and related metabolic complications.
Abbreviations
- AICAR:
-
5-Amino-4-imidazole-carboxamideriboside
- ATGL:
-
Adipose triacylglycerol lipase
- IHH:
-
Immortalised human hepatocyte
- LD:
-
Lipid droplet
- NTC:
-
Non-targeting control
- OA:
-
Oleic acid
- siRNA:
-
Small interfering RNA
- STK25:
-
Serine/threonine protein kinase 25
- TAG:
-
Triacylglycerol
References
Anstee QM, Targher G, Day CP (2013) Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol 10:330–344
Perry RJ, Samuel VT, Petersen KF, Shulman GI (2014) The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510:84–91
Sugden PH, McGuffin LJ, Clerk A (2013) SOcK, MiSTs, MASK and STicKs: the GCKIII (germinal centre kinase III) kinases and their heterologous protein-protein interactions. Biochem J 454:13–30
Nerstedt A, Cansby E, Andersson CX et al (2012) Serine/threonine protein kinase 25 (STK25): a novel negative regulator of lipid and glucose metabolism in rodent and human skeletal muscle. Diabetologia 55:1797–1807
Cansby E, Amrutkar M, Manneras Holm L et al (2013) Increased expression of STK25 leads to impaired glucose utilization and insulin sensitivity in mice challenged with a high-fat diet. FASEB J 27:3660–3671
Amrutkar M, Cansby E, Nunez-Duran E et al (2015) Protein kinase STK25 regulates hepatic lipid partitioning and progression of liver steatosis and NASH. FASEB J 29:1564–1576
Amrutkar M, Cansby E, Chursa U et al (2015) Genetic disruption of protein kinase STK25 ameliorates metabolic defects in a diet-induced type 2 diabetes model. Diabetes 64:2791–2804
Samanez CH, Caron S, Briand O et al (2012) The human hepatocyte cell lines IHH and HepaRG: models to study glucose, lipid and lipoprotein metabolism. Arch Physiol Biochem 118:102–111
Folch J, Lees M, Sloane Stanley GH (1957) A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226:497–509
Stahlman M, Fagerberg B, Adiels M et al (2013) Dyslipidemia, but not hyperglycemia and insulin resistance, is associated with marked alterations in the HDL lipidome in type 2 diabetic subjects in the DIWA cohort: impact on small HDL particles. Biochim Biophys Acta 1831:1609–1617
Reid BN, Ables GP, Otlivanchik OA et al (2008) Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J Biol Chem 283:13087–13099
Kannt A, Pfenninger A, Teichert L et al (2015) Association of nicotinamide-N-methyltransferase mRNA expression in human adipose tissue and the plasma concentration of its product, 1-methylnicotinamide, with insulin resistance. Diabetologia 58:799–808
Hussain HK, Chenevert TL, Londy FJ et al (2005) Hepatic fat fraction: MR imaging for quantitative measurement and display—early experience. Radiology 237:1048–1055
De Gottardi A, Spahr L, Ravier-Dall’Antonia F, Hadengue A (2010) Cannabinoid receptor 1 and 2 agonists increase lipid accumulation in hepatocytes. Liver Int 30:1482–1489
Ricchi M, Odoardi MR, Carulli L et al (2009) Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J Gastroenterol Hepatol 24:830–840
Pang J, Cui J, Gong H, Xi C, Zhang TM (2015) Effect of NAD on PARP-mediated insulin sensitivity in oleic acid treated hepatocytes. J Cell Physiol 230:1607–1613
Liu HY, Collins QF, Xiong Y et al (2007) Prolonged treatment of primary hepatocytes with oleate induces insulin resistance through p38 mitogen-activated protein kinase. J Biol Chem 282:14205–14212
Hao Q, Feng M, Shi Z et al (2014) Structural insights into regulatory mechanisms of MO25-mediated kinase activation. J Struct Biol 186:224–233
Matsuki T, Matthews RT, Cooper JA et al (2010) Reelin and stk25 have opposing roles in neuronal polarization and dendritic Golgi deployment. Cell 143:826–836
Hardie DG (2015) AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr Opin Cell Biol 33:1–7
Ye J, Li JZ, Liu Y et al (2009) Cideb, an ER- and lipid droplet-associated protein, mediates VLDL lipidation and maturation by interacting with apolipoprotein B. Cell Metab 9:177–190
Qi K, Al-Haideri M, Seo T, Carpentier YA, Deckelbaum RJ (2003) Effects of particle size on blood clearance and tissue uptake of lipid emulsions with different triglyceride compositions. JPEN J Parenter Enteral Nutr 27:58–64
Sunny NE, Parks EJ, Browning JD, Burgess SC (2011) Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab 14:804–810
Satapati S, Sunny NE, Kucejova B et al (2012) Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J Lipid Res 53:1080–1092
Samuel VT, Liu ZX, Qu X et al (2004) Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 279:32345–32353
Stefanovic-Racic M, Perdomo G, Mantell BS, Sipula IJ, Brown NF, O’Doherty RM (2008) A moderate increase in carnitine palmitoyltransferase 1a activity is sufficient to substantially reduce hepatic triglyceride levels. Am J Physiol Endocrinol Metab 294:E969–977
Flamment M, Gueguen N, Wetterwald C, Simard G, Malthiery Y, Ducluzeau PH (2009) Effects of the cannabinoid CB1 antagonist rimonabant on hepatic mitochondrial function in rats fed a high-fat diet. Am J Physiol Endocrinol Metab 297:E1162–1170
Valdecantos MP, Perez-Matute P, Gonzalez-Muniesa P, Prieto-Hontoria PL, Moreno-Aliaga MJ, Martinez JA (2012) Lipoic acid administration prevents nonalcoholic steatosis linked to long-term high-fat feeding by modulating mitochondrial function. J Nutr Biochem 23:1676–1684
Zhang D, Liu ZX, Choi CS et al (2007) Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci U S A 104:17075–17080
Adiels M, Taskinen MR, Packard C et al (2006) Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 49:755–765
Chan DC, Watts GF, Gan S, Wong AT, Ooi EM, Barrett PH (2010) Nonalcoholic fatty liver disease as the transducer of hepatic oversecretion of very-low-density lipoprotein-apolipoprotein B-100 in obesity. Arterioscler Thromb Vasc Biol 30:1043–1050
Monetti M, Levin MC, Watt MJ et al (2007) Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 6:69–78
Millar JS, Stone SJ, Tietge UJ et al (2006) Short-term overexpression of DGAT1 or DGAT2 increases hepatic triglyceride but not VLDL triglyceride or apoB production. J Lipid Res 47:2297–2305
Chen Z, Newberry EP, Norris JY et al (2008) ApoB100 is required for increased VLDL-triglyceride secretion by microsomal triglyceride transfer protein in ob/ob mice. J Lipid Res 49:2013–2022
Li X, Grundy SM, Patel SB (1997) Obesity in db and ob animals leads to impaired hepatic very low density lipoprotein secretion and differential secretion of apolipoprotein B-48 and B-100. J Lipid Res 38:1277–1288
Kozlitina J, Smagris E, Stender S et al (2014) Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 46:352–356
Mahdessian H, Taxiarchis A, Popov S et al (2014) TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A 111:8913–8918
Willer CJ, Sanna S, Jackson AU et al (2008) Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 40:161–169
Global Lipids Genetics C, Willer CJ, Schmidt EM et al (2013) Discovery and refinement of loci associated with lipid levels. Nat Genet 45:1274–1283
Aulchenko YS, Ripatti S, Lindqvist I et al (2009) Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet 41:47–55
Kathiresan S, Willer CJ, Peloso GM et al (2009) Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet 41:56–65
Teslovich TM, Musunuru K, Smith AV et al (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466:707–713
Speliotes EK, Yerges-Armstrong LM, Wu J et al (2011) Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 7, e1001324
Gorden A, Yang R, Yerges-Armstrong LM et al (2013) Genetic variation at NCAN locus is associated with inflammation and fibrosis in non-alcoholic fatty liver disease in morbid obesity. Hum Hered 75:34–43
Kathiresan S, Melander O, Guiducci C et al (2008) Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet 40:189–197
Mashek DG, Khan SA, Sathyanarayan A, Ploeger JM, Franklin MP (2015) Hepatic lipid droplet biology: getting to the root of fatty liver. Hepatology 62:964–967
Boren J, Taskinen MR, Olofsson SO, Levin M (2013) Ectopic lipid storage and insulin resistance: a harmful relationship. J Intern Med 274:25–40
Gibbons GF, Wiggins D (1995) Intracellular triacylglycerol lipase: its role in the assembly of hepatic very-low-density lipoprotein (VLDL). Adv Enzym Regul 35:179–198
Begriche K, Massart J, Robin MA, Bonnet F, Fromenty B (2013) Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 58:1497–1507
Turpin SM, Hoy AJ, Brown RD et al (2011) Adipose triacylglycerol lipase is a major regulator of hepatic lipid metabolism but not insulin sensitivity in mice. Diabetologia 54:146–156
Kato M, Higuchi N, Enjoji M (2008) Reduced hepatic expression of adipose tissue triglyceride lipase and CGI-58 may contribute to the development of non-alcoholic fatty liver disease in patients with insulin resistance. Scand J Gastroenterol 43:1018–1019
Sapiro JM, Mashek MT, Greenberg AS, Mashek DG (2009) Hepatic triacylglycerol hydrolysis regulates peroxisome proliferator-activated receptor alpha activity. J Lipid Res 50:1621–1629
Guo F, Ma Y, Kadegowda AK et al (2013) Deficiency of liver Comparative Gene Identification-58 causes steatohepatitis and fibrosis in mice. J Lipid Res 54:2109–2120
Wilfling F, Wang H, Haas JT et al (2013) Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell 24:384–399
Acknowledgements
The authors acknowledge the editorial assistance of R. Perkins, Wallenberg Laboratory, Department of Molecular and Clinical Medicine, University of Gothenburg, Sweden.
Funding
This work was supported by grants from the Swedish Research Council, the European Foundation for the Study of Diabetes/Lilly research grant, the Novo Nordisk Foundation, the Swedish Heart and Lung Foundation, the Diabetes Wellness Network Sweden, the Swedish Diabetes Foundation, the P. and A. Hedlunds Foundation, the Å. Wiberg Foundation, the Adlerbert Research Foundation, the I. Hultman Foundation, the S. and E. Goljes Foundation, the West Sweden ALF program and the F. Neubergh Foundation.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript
Author contributions
MA generated the bulk of the results. MK, EN-D, MS, EC, UC and ES contributed to the research data. JB and MB substantially contributed to the design and interpretation of data. MM directed the project, designed the study, interpreted the results and wrote the manuscript. All the authors revised the article critically for important intellectual content and approved the final version of the article to be published. MM is the guarantor of this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 133 kb)
ESM Fig. 1
(PDF 81 kb)
ESM Fig. 2
(PDF 378 kb)
ESM Fig. 3
(PDF 873 kb)
ESM Fig. 4
(PDF 118 kb)
ESM Fig. 5
(PDF 91 kb)
ESM Fig. 6
(PDF 77 kb)
ESM Fig. 7
(PDF 79 kb)
ESM Table 1
(PDF 69 kb)
ESM Table 2
(PDF 174 kb)
ESM Table 3
(PDF 174 kb)
Rights and permissions
About this article

Cite this article
Amrutkar, M., Kern, M., Nuñez-Durán, E. et al. Protein kinase STK25 controls lipid partitioning in hepatocytes and correlates with liver fat content in humans. Diabetologia 59, 341–353 (2016). https://doi.org/10.1007/s00125-015-3801-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3801-7