
Abstract
Aims/hypothesis
Normal glucose metabolism depends on pancreatic secretion of insulin and glucagon. The bihormonal hypothesis states that while lack of insulin leads to glucose underutilisation, glucagon excess is the principal factor in diabetic glucose overproduction. A recent study reported that streptozotocin-treated glucagon receptor knockout mice have normal glucose tolerance. We investigated the impact of acute disruption of glucagon secretin or action in a mouse model of severe diabetes by three different approaches: (1) alpha cell elimination; (2) glucagon immunoneutralisation; and (3) glucagon receptor antagonism, in order to evaluate the effect of these on glucose tolerance.
Methods
Severe diabetes was induced in transgenic and wild-type mice by streptozotocin. Glucose metabolism was investigated using OGTT in transgenic mice with the human diphtheria toxin receptor expressed in proglucagon producing cells allowing for diphtheria toxin (DT)-induced alpha cell ablation and in mice treated with either a specific high affinity glucagon antibody or a specific glucagon receptor antagonist.
Results
Near-total alpha cell elimination was induced in transgenic mice upon DT administration and resulted in a massive decrease in pancreatic glucagon content. Oral glucose tolerance in diabetic mice was neither affected by glucagon immunoneutralisation, glucagon receptor antagonism, nor alpha cell removal, but did not deteriorate further compared with mice with intact alpha cell mass.
Conclusions/interpretation
Disruption of glucagon action/secretion did not improve glucose tolerance in diabetic mice. Near-total alpha cell elimination may have prevented further deterioration. Our findings support insulin lack as the major factor underlying hyperglycaemia in beta cell-deficient diabetes.
Similar content being viewed by others

Introduction
Glucose homeostasis is mainly controlled by insulin and glucagon. Insulin, secreted in response to hyperglycaemia, decreases hepatic glucose output and promotes glucose disposal and storage in insulin-sensitive tissues. Glucagon is released during hypoglycaemia and increases hepatic glucose output to re-establish normal blood glucose levels. Originally, lack of insulin was thought to explain all diabetic abnormalities [1]; however, the bihormonal hypothesis stated that while lack of insulin will lead to glucose underutilisation, it is an inappropriate excess of glucagon that is the principal factor in diabetic glucose overproduction [2]. Glucagon receptor knockout mice (Gcgr –/–) [3] have lower (but not hypoglycaemic) blood glucose levels during the day and normal body weight, food intake and energy expenditure, but marked alpha cell hyperplasia and hypersecretion. The beta-cytotoxic compound streptozotocin (STZ) was used by Lee et al [4] to ablate beta cells, in Gcgr –/– as well as Gcgr +/+ mice. STZ treatment rendered Gcgr +/+ mice severely hyperglycaemic while Gcgr –/– mice showed fed and fasted glucose levels within normal range. To further support their theory, Lee et al transiently restored glucagon receptor expression in STZ-treated Gcgr –/– mice and found a marked increase in blood glucose levels [5]. According to these observations, totally pancreatectomised individuals should not show hyperglycaemia but have a well-regulated glucose metabolism. However, total pancreatectomy always renders patients severely diabetic.
Thorel et al reported that a 98% alpha cell ablation in mice did not induce changes in fed or fasted blood glucose levels and the animals were not resistant to STZ-induced diabetes [6]. We developed another transgenic mouse model (GCG.DTR) [7] in which the diphtheria toxin (DT) receptor is expressed on all proglucagon producing cells. Upon DT administration alpha cells as well as L-cells in the gastrointestinal tract are eliminated. The L-cells reappear after 7 days. This mouse showed a 90% decrease in pancreatic glucagon content and revealed normal fasting blood glucose levels and a normal glucose tolerance. To elucidate further on the effect of glucagon and the alpha cell contribution to the diabetic phenotype in mice with massive beta cell loss, we now studied the effects of massive alpha cell loss, antagonism of the glucagon receptor and immunoneutralisation with glucagon antibodies on oral glucose tolerance in an STZ-induced mouse model of diabetes.
Methods
Animals
The GCG.DTR mouse was developed as previously described [7]. GCG.DTR and wild-type (WT) mice were bred at our own breeding facilities, Department of Experimental Medicine, Panum Institute, University of Copenhagen, Denmark. Male C57BL/6J mice were purchased from Taconic, Lille Skensved, Denmark. Animals were housed individually in individually ventilated cages with constant temperature under a light/dark cycle of 12 h (lights on 6 am to 6 pm) with ad libitum access to normal chow and water. All experiments were conducted in accordance with Institutional Guidelines and approved by the local ethics committee as well as the Animal Experiments Inspectorate, Ministry of Food, Agriculture and Fisheries, Danish Veterinary and Food Administration in Denmark.
Studies were neither randomised nor blinded. No animals were excluded in these studies.
Destruction of pancreatic beta and alpha cells
Male GCG.DTR and WT litter mates (7–8 weeks old) were grouped based on body weight on day 0 (n = 10–14/group). Beta cells were destroyed with two s.c. injections of STZ (150 mg/kg body weight followed after 7 days by an additional 150 mg/kg body weight) (Sigma Aldrich, St Louis, MO, USA). Body weight, blood glucose, ketone and lactate levels were measured twice weekly for 5 weeks. On day 28, animals were injected i.p. with either DT (100 μg/kg body weight) or vehicle. Animals were killed on day 37 by cervical dislocation and tissues and blood were removed for further analysis.
OGTT
OGTTs were performed on days 21 and 35. Glucose (2 mg/g body weight) was delivered by oral gavage after a 16–18 h fast and blood glucose levels were measured at 0, 20, 40, 60, 90, 120, 150 and 180 min. A blood sample was taken from v. facialis at time 0 min.
Acute elimination of glucagon action in diabetic C57BL/6 animals
Beta cell destruction was induced by STZ in male C57BL/6J mice (25–29 g) as described above and OGTT performed 20 days after. Glucagon action was blocked using neutralising antibodies [8] or an antagonist [9] (gifts from Novo Nordisk, Måløv, Denmark). Glu-001, a monoclonal murine anti-glucagon antibody, or the inactive monoclonal antibody, anti-2, 4, 6-trinitrophenyl (designated control antibody), were administered i.p. at 4 mg/kg body weight at time −120 min [8]. The glucagon receptor antagonist, 25–2648, was administered by oral gavage as a suspension in a dose of 100 mg/kg body weight at time −180 min. The 25-2648 was dissolved in 5% ethanol, 20% propyleneglycol, 10% 2-hydroxypropyl-β-cyclodextrin (vol./vol.) and phosphate buffer at pH 7.5–8.0 at a concentration of 10 mg/ml. Diluent was used as vehicle in control experiments. C57BL/6J mice were allocated to one of the four groups (n = 10/group) based on body weight on the day of OGTT.
Plasma measurements
Blood glucose levels were measured with glucometers (Accu-chek compact plus, F. Hoffmann-La Roche AG, Basel, Switzerland or Alphatrak 2, Animed Direct, Suffolk, UK) and blood ketone and lactate levels with a ketone (Freestyle Precision, Abbott Laboratories, Green Oaks, IL, USA) and a lactate apparatus (Lactate Plus, Nova Biomedical, Waltham, MA, USA). Plasma insulin levels were measured using the Ultra-Sensitive Mouse Insulin ELISA kit (Chrystal Chem, Downers Grove, IL, USA). Plasma glucose-dependent insulinotropic polypeptide (GIP) levels were measured using the rat/mouse GIP (total) ELISA kit (Millipore, St Charles, MO, USA) and IGF-1 with the Mouse/Rat IGF-1 ELISA kit (Mediagnost, Reutlingen, Germany). Plasma triacylglycerol levels were measured using the Serum Triglyceride Determination Kit (Sigma Aldrich). Plasma glucagon levels were measured using the Glucagon ELISA kit (Mercodia, Uppsala, Sweden). For all assays, included quality controls were within required limits. Overall, GIP was measured as single determination, insulin and leptin were measured as duplicates, and triacylglycerol was measured in triplicate. The remaining assays were evaluated by running samples as possible and depending on the amount of plasma available.
Pancreatic protein extraction and analysis
Tissue from GCG.DTR and WT animals was homogenised in 1% trifluoroacetic acid (vol./vol.). Homogenates were purified using Sep-Pak tc18 cartridges (Waters, Midford, MA, USA), eluted with 70% ethanol + 0.1% trifluoroacetic acid, evaporated and resuspended in assay buffer. Glucagon was determined using the side-viewing 4304 anti-serum (in-house) [10]. Glucagon-like peptide-1 (GLP-1) was determined using the in-house GLP-1 390 antibody [11]. Insulin was determined using the Ultra-Sensitive Mouse Insulin ELISA kit (Chrystal Chem).
Immunohistochemistry
Pancreases were fixed in cold 4% paraformaldehyde (wt/vol.) in 0.1 mol/l phosphate buffer pH 7.4. Samples were embedded in paraffin and cut into 5 μm sections. Sections were incubated in 2% BSA (wt/vol.) followed by incubation with either anti-insulin or anti-glucagon serum (in-house guinea pig anti-insulin, serum no. 2006 [12] diluted 1:12.800, and in-house rabbit anti-glucagon, serum no. 4304 [10], diluted 1:4.800, respectively) and subsequent incubation with secondary antibodies, biotinylated goat anti-guinea pig immunoglobulins (Vector labs, Burlingame, CA, USA, diluted 1:200) or goat anti-rabbit immunoglobulins (Vector labs; diluted 1:200), respectively. StreptABComplex/horseradish peroxidase (DakoCytomation, Carpinteria, CA, USA; diluted 1:200) was the third layer followed by staining with 3,3-diaminobenzidine, counterstained with haematoxylin. Images of identical islets stained for glucagon and insulin on neighbouring sections were captured by means of a Leica Ortoplan microscope, fitted with a Leica DFC 420 C camera.
Glycogen content
Glycogen was extracted from the biopsy by boiling (100°C) the samples for 10 min in 0.4 mol/l KOH. The pH of the extract was adjusted to 5.0 by 1 mol/l citric acid. Amyloglucosidase was added (5.2 mg/ml), and the extract incubated for 2 h at 40°C. On this extract, glucose was assayed enzymatically, as described [13].
Statistics
Data are presented as means ± SEM. Graphs were constructed using GraphPad Prism software (version 5.04, GraphPad Software, San Diego, CA, USA) and statistical analyses were performed with the same program either using two-way ANOVA for repeated measurements followed by Bonferroni post hoc test or Student’s t test.
Results
Development in body weight, fed blood glucose levels and oral glucose tolerance
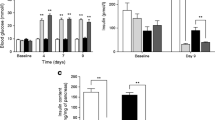
Throughout the study STZ-treated animals were in a deteriorating condition and did not recover after DT treatment. Weight loss was seen in GCG.DTR and WT mice upon STZ treatment and no difference was registered between DT and non-DT-treated groups (Fig. 1a–d). Fed blood glucose levels instantly rose following STZ administration in all groups (Fig. 1e–h) and the alpha cell destroying effect of DT administration to GCG.DTR mice offered no decline (Fig. 1e). Surprisingly, a fall in blood glucose levels was seen after DT treatment of WT animals (Fig. 1f). Glucose tolerance tests performed after STZ treatment revealed high levels of fasting blood glucose as well as a poor glucose clearance in all groups (Fig. 2a–f). A minor difference in glucose tolerance was seen in DT-treated GCG.DTR animals compared with non-DT-treated animals at the second test (Fig. 2c). However, comparison with the first OGTT revealed that there was no change in the DT group but impairment in the control group. No difference was seen between DT and non-DT-treated WT animals (Fig. 2d).

Development in body weight and blood glucose levels. (a) Body weight in DT- (black circles, n = 15) and non-DT-treated (grey circles, n = 14) GCG.DTR mice. (b) Body weight in DT- (black circles, n = 14) and non-DT-treated (grey circles, n = 13) WT mice. (c) Body weight in antagonist- (black triangles, n = 10) and vehicle-treated (grey triangles, n = 10) C57BL/6 mice. (d) Body weight in antibody-treated (white triangles with black border, n = 10) and control (white triangles with grey border, n = 10) C57BL/6 mice. (e) Blood glucose levels in DT- (black circles) and non-DT-treated (grey circles) GCG.DTR mice. (f) Blood glucose concentrations in DT- (white circles with black border) and non-DT-treated (white circles with grey border) WT mice. *p < 0.05. (g) Two hour fasted blood glucose concentrations in C57BL/6 mice used for the antagonist studies. (h) Two hour fasted blood glucose concentrations in C57BL/6 mice used for the immunoneutralisation studies. BW, body weight

Glucose concentration curves during OGTT before alpha cell elimination (a, b), after alpha cell elimination (c, d), with or without glucagon receptor antagonist (e), and with and without glucagon neutralising antibody (f). Inserts: AUC of glucose concentration curves. (a) Diabetic GCG.DTR mice before DT (black circles, n = 13) or vehicle (grey circles, n = 11) treatment. (b) Diabetic WT mice before DT (white circles with black border, n = 14) or vehicle (white circles with grey border, n = 13) treatment. (c) DT- (black squares, n = 13) and non-DT-treated (grey squares, n = 11) diabetic GCG.DTR mice. (d) DT- (white squares with black border, n = 13) and non-DT-treated (white squares with grey border, n = 12) diabetic WT mice. (e) Diabetic C57BL/6 mice receiving the glucagon antagonist 2648 (black triangles, n = 10) or matching vehicle (grey triangles, n = 10) at time −180 min before an OGTT. (f) Diabetic C57BL/6 mice receiving anti-glucagon antibody Glu-001 (white triangles with black border, n = 10) or control antibody (white triangles with grey border, n = 10) at time −120 min before an OGTT. *p < 0.05; ****p < 0.0001; † p < 0.05, as shown
Effects of STZ and DT on pancreatic insulin, glucagon, and GLP-1 content
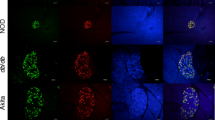
There were no differences in insulin content between DT and non-DT-treated GCG.DTR or WT mice (Fig. 3b, c, i, j). Glucagon and total GLP-1 were markedly reduced only in DT-treated GCG.DTR mice (Fig. 3f, g, m, n). Histological analysis revealed similar results, showing no or only a few glucagon-positive cells in DT-treated GCG.DTR animals (Fig. 3a). Non-DT-treated GCG.DTR mice as well as DT and non-DT-treated WT mice had an excessive amount of glucagon-positive cells compared with that usually seen in the alpha cell mantel (Fig. 3d, h, k).

Pancreatic islets in diabetic WT and diabetic GCG.DTR mice. Histological images of representative islets from DT-treated GCG.DTR (a, b), non-DT-treated GCG.DTR (d, e), DT-treated WT (h, i) and non-DT-treated WT (k, l) mice with regard to glucagon-positive cells (a, d, h, k) and insulin-positive cells (b, e, i, l). Scale bars, 50 μm. Islet content of insulin (c, j), glucagon (f, m) and GLP-1 (g, n) in diabetic GCG.DTR mice (c, f, g) treated with DT (black bars, n = 13) and vehicle (white bars, n = 11), and in diabetic WT mice (j, m, n) treated with DT (grey bars, n = 11) or vehicle (white bars, n = 13). ***p < 0.001; ****p < 0.0001
Effects of STZ and DT on plasma hormone and triacylglycerol levels
Fasting blood samples were taken just before the two OGTTs. No difference was seen between DT and non-DT-treated GCG.DTR animals on the respective days with regard to insulin levels (Fig. 4a, b), IGF-1 or GIP levels (data not shown). However, treatment with control antibodies significantly increased plasma insulin concentration during the OGTT (Fig. 4d). Fasting plasma glucagon levels were low and similar before and after DT treatment (data not shown). Fed plasma glucagon levels were very low in DT-treated GCG.DTR mice at day 37 (Fig. 4e, f). Blood levels of ketone and lactate were measured twice a week throughout the experiment. No difference was found between DT and non-DT-treated groups (data not shown). Triacylglycerol levels were measured throughout the study at fed and fasted states, but there were no differences between DT and non-DT-treated GCG.DTR or WT mice (data not shown).

Fasting plasma insulin levels before and after DT (black/grey bars) or vehicle (white bars) administration in GCG.DTR mice (a) and WT mice (b). Plasma insulin concentration during OGTT in C57BL/6 mice administered glucagon antagonist (grey bars, c), glucagon-neutralising antibodies (grey bars, d) or respective vehicles (white bars) (c, d). Plasma glucagon levels measured at the end of study in GCG.DTR mice (e) and WT mice (f) administered DT (black/grey bars) or vehicle (white bars). *p < 0.05; ***p < 0.001. ND, not detected
Glycogen content
Hepatic glucose production was evaluated indirectly by measurement of glycogen content in liver. No differences were found when comparing DT and non-DT-treated animals (data not shown).
Discussion
It is often assumed that the secretion of glucagon is abnormal in type 1 diabetes and responsible for the inappropriate increase in hepatic glucose output (which is otherwise well explained by lack of insulin). Lee et al [4] reported unaffected OGTTs comparing STZ-treated and non-STZ-treated Gcgr –/– mice. Apparently, presence or absence of beta cells had no effect on glucose tolerance in mice lacking glucagon action. Lee et al postulated that insulin action during glucose absorption is largely directed towards overcoming the hepatic actions of glucagon during the pre-glucose fast. If glucagon is absent, the liver is in storage mode, and insulin is irrelevant. We evaluated the metabolic effects in diabetic mice of disruption of glucagon action at three different stages in glucagon signalling: (1) acute alpha cell depletion; (2) immunoneutralisation; and (3) antagonism of the glucagon receptor.
No differences were found in the development of body weight and fed blood glucose levels between any of the groups. The effect of DT treatment was confirmed by measurements of the pancreatic content of insulin and glucagon. As expected, only a few glucagon-positive cells remained after DT treatment in GCG.DTR mice, and glucagon content was markedly reduced. Before alpha cell elimination all groups had similar glucose tolerance, but after, DT-treated GCG.DTR mice exhibited a lower blood glucose concentration at late timepoints during glucose tolerance compared with non-DT-treated mice; this could indicate a positive effect of alpha cell elimination on glucose clearance. However, blood glucose levels of DT-treated mice were identical with the levels before DT treatment, while levels in non-DT-treated mice were slightly elevated, indicating that the difference is due to further deterioration in glucose tolerance in non-DT-treated mice rather than an improvement in DT-treated mice. Similarly to the elimination experiments, we did not detect any significant change of oral glucose tolerance following immunoneutralisation of plasma glucagon or antagonism at the glucagon receptor. Fasting plasma insulin levels before were similar before and after DT treatment and plasma insulin concentrations were not affected by antagonism of glucagon receptors. However, control antibody administration in the immunoneutralisation study was associated with an unexpected increase of plasma insulin during the OGTT, which we cannot explain.
Injection with DT is generally considered harmless to mice. However, DT-treated WT mice showed significantly lower but still severely diabetic blood glucose concentrations at the end of the study. This probably reflects the deterioration of the STZ-induced diabetes.
Lee et al investigated plasma levels of leptin and IGF-1 to determine whether the insulin-like responses found in their study were caused by an increase in a hormone with insulin-mimetic activity. None of the hormones rose in response to a glucose challenge, but IGF-1 levels were markedly reduced in Gcgr –/– mice compared with Gcgr +/+ controls. We also looked for an IGF-1-lowering effect of lack of glucagon action but found no difference in IGF-1 levels between DT and non-DT-treated animals. GIP is a hormone with important effects on insulin and glucagon secretion [14, 15] but we found no difference between the groups. In addition, the hepatic glycogen content was similar between DT and non-DT-treated animals and, furthermore, we evaluated the expression levels of PEPCK, G6Pase and glucokinase in the liver by quantitative PCR but found no difference between DT and non-DT animals (not shown). These results further support the notion that presence or absence of glucagon did not matter for the hepatic glucose production during insulinopenia. This is in contrast to insulin-independent diabetes, where both glucagon antagonism and immunoneutralisation lowers blood glucose [8, 16, 17].
Gcgr –/– mice have a compensatory increase in glucagon gene transcription and hence a substantial increase in circulating GLP-1 levels [18]. The contribution of increased GLP-1 concentrations to the altered glucose tolerance in Gcgr –/– has been investigated in STZ-treated Gcgr –/– mice, and inhibition of GLP-1 action had no effect on glucose tolerance but eliminated the improvement found in the STZ-treated Gcgr –/– mice [19]. The double knockout mouse, Gcgr –/– Glp1r –/– [20] has normal random fed blood glucose levels and a normal i.p. glucose tolerance. Hence, an insulin increasing effect of GLP-1 is not responsible for the improved glucose tolerance in Gcgr –/– mice. In our study the amount of total GLP-1 in the pancreas was markedly reduced in DT-treated GCG.DTR mice. This could support the notion that unknown effects of elevated GLP-1 in the STZ-treated Gcgr –/– mice are responsible for their improved glucose tolerance. Lee et al suggest that when totally depancreatised individuals are deprived of insulin, there is an increased production of extrapancreatic glucagon, but this cannot explain the lack of effect of glucagon immunoneutralisation and antagonism in our study.
Thus, hyperglycaemia following STZ-induced beta cell destruction was neither corrected by acute near-total alpha cell elimination, glucagon immunoneutralisation, nor antagonism of the glucagon receptors. In contrast, near-normal blood glucose levels were observed in the STZ-treated Gcgr –/– mouse and the animals did not develop the metabolic characteristics and lethal phenotype normally associated with STZ-induced diabetes [4]. Congenital alterations, metabolic adaptations and abnormal pancreatic islets should be considered when comparing studies. The GCG.DTR mouse had normal islets before DT treatment whereas the Gcgr –/– mice have marked alpha cell hyperplasia. A disadvantage of the GCG.DTR model is that glucagon removal is not complete. Thorel et al reported an even higher degree of glucagon removal and also did not observe any improvement in glucose tolerance and hence concluded that the few preserved alpha cells produced sufficient glucagon for maintaining glucagon action [6]. We had few alpha cells left, and the plasma levels as well as the pancreatic content of glucagon were very low. Therefore, it remains a theoretical possibility that a very small amount of glucagon is sufficient to maintain its normal functions. In a study by Brand et al [17], huge amounts of a high affinity glucagon antibody were used to remove all glucagon effect. Similarly, in that study, there was no effect of what was considered complete glucagon neutralisation in severely STZ-diabetic animals. In contrast, Wang et al [21] recently showed that weekly administration of a monoclonal glucagon receptor-targeting antibody caused normalisation of blood glucose in severely diabetic mice. Perhaps chronic blockade of the glucagon receptor elicits changes in the pancreas similar to GCGR-deletion or antagonist administration [16]. In the present study, all of the three methods were supposed to cause immediate and near complete glucagon deficiency. In agreement with results reported by Brand et al neither treatment had an effect on the diabetic hyperglycaemia [17]. We therefore must conclude that none of the applied attempts to eliminate the metabolic potential of glucagon had any influence on the diabetogenic effect of extensive beta cell destruction. Our conclusions are in strong contrast with those presented by Lee et al [4, 5]. How do we reconcile these findings? We propose that the explanation must be found in the congenital alterations resulting from deletion of the glucagon receptor, namely the dramatic alpha cell hyperplasia, and also hyperplasia of gut endocrine cells, with ensuing hypersecretion of GLP-1 and oxyntomodulin (which has glucagon action!) [22]. It needs to be explained how hypersecretion of GLP-1 can influence glucose metabolism in animals without insulin and glucagon action, but the answer may lie in the recent descriptions of GLP-1 actions on hepatic glucose production [23].
Abbreviations
- DT:
-
Diphtheria toxin
- GCG.DTR :
-
Strain name: A glucagon promoter-human diphtheria toxin receptor mouse
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GLP-1:
-
Glucagon-like peptide-1
- STZ:
-
Streptozotocin
- WT:
-
Wild-type
References
Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA (1922) Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J 12:141–146
Unger RH, Orci L (1975) The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet 1:14–16
Gelling RW, Du XQ, Dichmann DS et al (2003) Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci U S A 100:1438–1443
Lee Y, Wang MY, Du XQ, Charron MJ, Unger RH (2011) Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes 60:391–397
Lee Y, Berglund ED, Wang MY et al (2012) Metabolic manifestations of insulin deficiency do not occur without glucagon action. Proc Natl Acad Sci U S A 109:14972–14976
Thorel F, Damond N, Chera S et al (2011) Normal glucagon signaling and β-cell function after near-total α-cell ablation in adult mice. Diabetes 60:2872–2882
Pedersen J, Ugleholdt RK, Jorgensen SM et al (2013) Glucose metabolism is altered after loss of L cells and alpha-cells but not influenced by loss of K cells. Am J Physiol Endocrinol Metab 304:E60–E73
Sorensen H, Brand CL, Neschen S et al (2006) Immunoneutralization of endogenous glucagon reduces hepatic glucose output and improves long-term glycemic control in diabetic ob/ob mice. Diabetes 55:2843–2848
Kodra JT, Jorgensen AS, Andersen B et al (2008) Novel glucagon receptor antagonists with improved selectivity over the glucose-dependent insulinotropic polypeptide receptor. J Med Chem 51:5387–5396
Holst JJ (1980) Evidence that glicentin contains the entire sequence of glucagon. Biochem J 187:337–343
Orskov C, Rabenhoj L, Wettergren A, Kofod H, Holst JJ (1994) Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes 43:535–539
Brand CL, Jorgensen PN, Knigge U et al (1995) Role of glucagon in maintenance of euglycemia in fed and fasted rats. Am J Physiol 269:E469–E477
Katz J, Golden S, Wals PA (1976) Stimulation of hepatic glycogen synthesis by amino acids. Proc Natl Acad Sci U S A 73:3433–3437
Holst JJ (2007) The physiology of glucagon-like peptide 1. Physiol Rev 87:1409–1439
Baggio LL, Drucker DJ (2007) Biology of incretins: GLP-1 and GIP. Gastroenterology 132:2131–2157
Kelly RP, Garhyan P, Raddad E et al (2015) Short-term administration of the glucagon receptor antagonist LY2409021 lowers blood glucose in healthy people and in those with type 2 diabetes. Diabetes Obes Metab 17:414–422
Brand CL, Rolin B, Jorgensen PN, Svendsen I, Kristensen JS, Holst JJ (1994) Immunoneutralization of endogenous glucagon with monoclonal glucagon antibody normalizes hyperglycaemia in moderately streptozotocin-diabetic rats. Diabetologia 37:985–993
Hayashi Y (2011) Metabolic impact of glucagon deficiency. Diabetes Obes Metab 13(Suppl 1):151–157
Omar BA, Andersen B, Hald J, Raun K, Nishimura E, Ahren B (2014) Fibroblast growth factor 21 (FGF21) and glucagon-like peptide 1 contribute to diabetes resistance in glucagon receptor-deficient mice. Diabetes 63:101–110
Ali S, Lamont BJ, Charron MJ, Drucker DJ (2011) Dual elimination of the glucagon and GLP-1 receptors in mice reveals plasticity in the incretin axis. J Clin Invest 121:1917–1929
Wang MY, Yan H, Shi Z et al (2015) Glucagon receptor antibody completely suppresses type 1 diabetes phenotype without insulin by disrupting a novel diabetogenic pathway. Proc Natl Acad Sci U S A 112:2503–2508
Baldissera FG, Holst JJ, Knuhtsen S, Hilsted L, Nielsen OV (1988) Oxyntomodulin (glicentin-(33-69)): pharmacokinetics, binding to liver cell membranes, effects on isolated perfused pig pancreas, and secretion from isolated perfused lower small intestine of pigs. Regul Pept 21:151–166
Seghieri M, Rebelos E, Gastaldelli A et al (2013) Direct effect of GLP-1 infusion on endogenous glucose production in humans. Diabetologia 56:156–161
Acknowledgements
We thank our technical personnel for assistance: S. Pilgaard, for assistance with RIA, H. M. Paulsen for assistance with histology, and P. J. Suku and S. L. Jepsen for assistance with genotyping, all from The University of Copenhagen, Novo Nordisk Foundation Center for Basic Metabolic Research, Translational Metabolic Physiology, Copenhagen, Denmark.
Funding
This work was supported by a grant from Augustinus Fonden (13-3752).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
JP and JJH designed the study. VRS and SMJ acquired and analysed the data, and drafted the article. SSP, ANM, BH, BQ and JAW analysed data. All authors revised the manuscript critically. All authors gave final approval for the current version to be published. JJH is responsible for the integrity of the work as a whole.
Author information
Authors and Affiliations
Corresponding author
Additional information
Vivi R. Steenberg and Signe M. Jensen contributed equally to this work.
Rights and permissions
About this article

Cite this article
Steenberg, V.R., Jensen, S.M., Pedersen, J. et al. Acute disruption of glucagon secretion or action does not improve glucose tolerance in an insulin-deficient mouse model of diabetes. Diabetologia 59, 363–370 (2016). https://doi.org/10.1007/s00125-015-3794-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3794-2