
Abstract
Aims/hypothesis
Studies have shown that dipeptidyl peptidase-4 (DPP4) inhibitors stimulate insulin secretion and increase beta cell mass in rodents. However, in these models hyperglycaemia has been induced early on in life and the treatment periods have been short. To explore the long-term effects of DPP4 inhibition on insulin secretion and beta cell mass, we have generated a high-fat diet (HFD)-induced-obesity model in mice of advanced age (10 months old).
Methods
After 1 month of HFD alone, the mice were given the DPP4 inhibitor vildagliptin for a further 11 months. At multiple time points throughout the study, OGTTs were performed and beta cell area and long-term survival were evaluated.
Results
Beta cell function and glucose tolerance were significantly improved by vildagliptin with both diets. In contrast, in spite of the long treatment period, beta cell area was not significantly different between vildagliptin-treated mice and controls. Mice of advanced age chronically fed an HFD displayed clear and extensive pancreatic inflammation and peri-insulitis, mainly formed by CD3-positive T cells, which were completely prevented by vildagliptin treatment. Chronic vildagliptin treatment also improved survival rates for HFD-fed mice.
Conclusions/interpretation
In a unique advanced-aged HFD-induced-obesity mouse model, insulin secretion was improved and the extensive peri-insulitis prevented by chronic DPP4 inhibition. The improved survival rates for obese mice chronically treated with vildagliptin suggest that chronic DPP4 inhibition potentially results in additional quality-adjusted life-years for individuals with type 2 diabetes, which is the primary goal of any diabetes therapy.
Similar content being viewed by others

Introduction
Type 2 diabetes is characterised by both insulin resistance and insulin insufficiency, resulting in elevated fasting and postprandial blood glucose levels. Type 2 diabetes is also associated with a reduced beta cell mass [1]. The recently introduced incretin-based treatments for type 2 diabetes, which target the incretin hormones, have resulted in improvement in islet function. Thus, both glucagon-like peptide 1 (GLP-1) receptor agonists and dipeptidyl peptidase-4 (DPP4) inhibitors, which increase the amount of active GLP-1 and glucose-dependent insulinotropic polypeptide (GIP) in the circulation, improve insulin secretion and lower blood glucose levels in both type 2 diabetic patients and experimental animal models of diabetes [2].
In rodent models of diabetes, it has also been demonstrated that DPP4 inhibitors can preserve beta cell mass [3], mainly through increasing beta cell proliferation and protecting beta cells from apoptosis [4, 5]. The models used to show improved beta cell function and increased beta cell mass by incretin-based therapy have been genetically deficient rodents, rodents given an exogenous beta cell toxin, and animals with diet-induced obesity (DIO) subsequent to dietary changes introduced at a young age. For example, in many of the studies reporting changes in beta cell mass and function in rodents in response to DPP4 inhibitor treatment, the rodents were either newborn or less than 7 weeks old at the start of the studies and several were treated with the beta cell toxin streptozotocin prior to DPP4 treatment [3, 6–8].
The shortness of therapy and the use of young animals are important limitations of previous rodent studies on effects of DPP4 inhibition on islet function and beta cell mass, given the differences in phenotype seen between these models and human type 2 diabetes [9]. Many studies using DIO models are done on animals only a few weeks old at the start of the study [5, 7], whereas postprandial hyperglycaemia in humans occurs most often in middle age [10]. The duration of treatment has also been limited in most studies. For example, studies of chronic dosing of DIO mice with DPP4 inhibitors report treatment only up to 11 weeks [3, 7, 8], which is a relatively short time in relation to the lifespan of most mouse strains. Another phenotypic difference between mouse models and human type 2 diabetes is the change in beta cell mass with insulin resistance. We have previously shown that feeding young mice an HFD for up to 1 year results in fourfold increases in beta cell area with no beta cell loss seen at 1 year despite significant hyperglycaemia [11]. In human obesity, beta cell area increases by 50%, but significant loss of beta cell area occurs by the onset of diabetes [12]. A potential explanation for the difference in capacity to increase beta cell area in response to insulin resistance lies in the young age of rodents at the start of DIO studies. Whether this capacity is retained in more age-appropriate DIO models, and whether chronic DPP4 inhibition can affect beta cell area in such a model, is not known. In this study we therefore studied the effects of extensive long-term therapy of the DPP4 inhibitor vildagliptin on beta cell function and beta cell area in a DIO model in which the diet was given at middle age, and not young age. We thus generated a DIO model in advanced-aged mice to mimic the time at which hyperglycaemia debuts in humans. These animals were then treated with vildagliptin for the remainder of their healthy life. This resulted in a treatment period covering 46 weeks (∼11 months). The effect of vildagliptin treatment on glycaemia and insulin secretion was determined at several time points throughout the lifespan of the mice and at the termination of the study the effect of vildagliptin on pancreatic beta cell area was determined. The unique study design also allowed for an evaluation of survival and longevity as treatment was given up until the end of healthy life for the mice or the end of the 1 year long study.
Methods
Animals and study design
Female C57BL/6JBomTac mice were from Taconic (Skensved, Denmark). The mice were 10 months old on arrival. After 1 week of acclimatisation the mice were divided into two groups, one receiving a standard rodent diet with 10% of energy from fat (Research Diets D12450B) and another receiving an HFD with 60% energy from fat (Research Diets D12492). After 1 month of feeding with the respective diets, a subgroup of mice on each diet was given the DPP4 inhibitor vildagliptin (LAF-237, Novartis, Cambridge, MA, USA; 3 μmol/day) in the drinking water for 46 weeks (∼11 months); this dose was predicted to result in continuous blocking of GLP-1 inactivation by DPP4 [4], which is the same inhibition profile that is achieved in man [13]. Food intake and body weight was measured once weekly. Body composition was determined by dual-energy x-ray absorptiometry (DEXA) using a PIXImus imager (GE Lunar, Madison, WI, USA) on anaesthetised mice. The principles of animal laboratory care were followed. All animal studies were approved by the regional ethics committee in Lund, Sweden.
OGTT
Mice were fasted for 5 h and d-glucose was administered at 75 mg/mouse by oral gavage. Blood was sampled from the intraorbital–retrobulbar sinus at 0, 15, 30, 60 and 120 min. Plasma samples were stored at −20°C until being assayed for glucose and insulin. OGTT was performed after 1 month with HFD, prior to the start of the treatment with the DPP4 inhibitor and then after 2, 5, 8 and 11 months with vildagliptin.
Beta cell and alpha cell area determination
Pancreases were removed and fixed in neutral-buffered formalin and embedded in paraffin. Sections, 5 μm thick, were mounted onto Superfrost Plus (Thermo scientific, Rockford, IL, USA) glass slides. Briefly, after deparaffinisation, sections were blocked in PBS containing 3% H2O2 (vol./vol.) and 10% methanol (vol./vol.) and again in 10% (vol./vol.) normal rabbit serum prior to overnight incubation with a polyclonal guinea pig anti-insulin/proinsulin antibody (EuroProxima, Arnhem, the Netherlands) or a monoclonal mouse anti-glucagon antibody (Abcam, Cambridge, UK). Sections were washed in PBS and incubated with horseradish-peroxidase-conjugated rabbit anti-guinea pig (DAKO, Glostrup, Denmark) or sheep anti-mouse (GE Healthcare, Amersham, UK) secondary antibodies, washed again and developed with 3,3′-diaminobenzidine chromogen reagent (DAKO, Glostrup Denmark). Sections were cross-stained with haematoxylin, dehydrated and mounted. Slides were scanned in a ScanScope digital slide scanner (Aperio, Vista, CA, USA) and the insulin- or glucagon-positive and total pancreas areas of each scanned slide were determined using the cellSens Dimensions software package (Olympus, Tokyo, Japan).
Pancreatic insulin content
Tissue samples were taken from both the head and tail regions of the pancreas, weighed and homogenised in 0.5 ml ice-cold acid-ethanol prior to sonication. Homogenates were centrifuged and the supernatant fractions were transferred to fresh tubes. Insulin was determined by ELISA (Mercodia, Uppsala, Sweden) and protein concentrations were determined by the Bradford protein assay. Insulin content is expressed as ng insulin/mg total protein.
Islet isolation and glucose-stimulated insulin secretion
Pancreatic islets of Langerhans were isolated by collagenase digestion and handpicked using an inverted microscope. Batches of freshly isolated islets were preincubated in HEPES balanced salt solution containing 125 mmol/l MgCl2, 25 mmol/l HEPES (pH 7.4), 5.6 mmol/l glucose and 0.1% fatty-acid-free BSA (Boehringer Mannheim, Germany) for 60 min. Islets were then incubated in groups of three in 200 μl of the buffer described above with varying glucose concentrations.
Islet immunohistochemistry
Slides that had been stained for insulin were evaluated for the presence of peri-insulitis. Peri-insulitis was determined as the percentage of islets showing the presence of mononuclear cell masses within or immediately adjacent to the islet. The percentage of islets exhibiting peri-insulitis was determined in at least three slides per mouse, with each slide representing a different region of the pancreas. Double immunofluorescent staining was performed on a subset of sections with primary antibodies specific for insulin, CD3 and ionised calcium-binding adaptor molecule 1 (IBA-1). Briefly, after deparaffinisation sections were washed in PBS buffer containing 0.25% Triton X-100 and incubated overnight at 4°C with antiserum raised in guinea pig against human proinsulin (Euro-Diagnostica, Malmö, Sweden; 9003, 1:5,000), antiserum raised in goat against mouse CD3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; sc-1127, 1:1,000) or IBA-1. Sections were washed and incubated 1 h at room temperature with DyLight488- and DyLight594-conjugated secondary antibodies (Jackson Immunoresearch, PA, USA; 1:1,000). The specificity of the antisera was tested by pre-absorbing with synthetic peptide, proinsulin [14] and CD3 (Santa Cruz Biotechnology; sc-1127 P).
Statistical analysis
All data are presented as mean ± SEM unless otherwise indicated. The AUC was calculated using the trapezoidal method. The insulinogenic index (IGI) was calculated as the incremental AUC (iAUC) for insulin (iAUCins) from time 0 to 60 min divided by the iAUC for glucose (iAUCglu) during the same time period. Oral disposition index was calculated as IGI × HOMA-insulin resistance (IR)−1. Statistical analysis was determined using one-way ANOVA with Tukey’s post test. Survival was determined with the Kaplan–Meier analysis using GraphPad Prism 5 software (GraphPad, San Diego, CA, USA). Differences were considered statistically significant when p < 0.05.
Results
The advanced-aged DIO mouse model
In order to generate a mouse model with obesity and hyperglycaemia at a time that mimics the debut of hyperglycaemia in humans, female C57BL/6J mice that were 10 months old at the start of the study were subjected to an HFD for 12 months. After 1 month of HFD feeding, vildagliptin was added to the drinking water in a subset of mice that continued consuming their respective diets for the remainder of the study. As seen in Fig. 1a, there was a rapid gain in weight as a result of consumption of the HFD, with significantly higher bodyweights seen after 1 month of HFD feeding. Vildagliptin had no effect on bodyweight development in either the HFD- or the normal-diet-fed groups. Total fat mass and lean body mass were similar between vildagliptin-treated and untreated mice (Fig. 1b). There were no effects of vildagliptin on food consumption (data not shown).

Effect of chronic vildagliptin treatment on weight gain in advanced-aged DIO mice. (a) 10-month-old mice were fed a normal diet (black circles) or HFD (black squares) for 1 month and continued thereafter with the diet either alone or supplemented with vildagliptin (LAF-237, 3 μmol/day in the drinking water) (white circles, normal diet + vildagliptin; white squares, HFD + vildagliptin) for 11 months. n = 30–45 per group; *** p < 0.001 when comparing each HFD group with its normal-diet control group. (b) Body composition as determined by DEXA after 8 months of treatment. Black bars, normal diet; white bars, normal diet + vildagliptin; dark grey bars, HFD; light grey bars, HFD + vildagliptin. n = 8–10 per group
Beta cell function is improved by chronic vildagliptin treatment
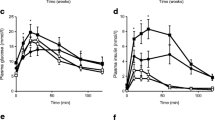
OGTTs were performed frequently during the study to evaluate the changes in glucose tolerance and beta cell function in response to HFD in the advanced-aged model as well as to evaluate the effect of chronic DPP4 inhibition with vildagliptin. Insulin secretion in response to oral glucose was significantly higher in the vildagliptin-treated animals fed either the normal diet or the HFD at all time points (Fig. 2d–f), resulting in lower glucose excursions compared with their respective controls (Fig. 2a–c). In normal-diet-fed mice, vildagliptin treatment improved glucose tolerance, determined by AUC for glucose, at each time point measured (Fig. 3a). Glucose tolerance was significantly impaired after 1 month of HFD feeding (Fig. 3b). HFD-fed mice receiving normal water had no change in glucose tolerance after 3 months, whereas HFD-fed mice treated with vildagliptin showed improvements in glucose tolerance that were maintained for the duration of the study (Fig. 3b).

Effect of chronic vildagliptin treatment on oral glucose tolerance in advanced-aged DIO mice. Mice were subjected to OGTTs at 2 (a,d), 5 (b,e) and 11 (c,f) months after the start of vildagliptin treatment. Mice were given a single oral bolus of d-glucose (75 mg/mouse) and plasma glucose (a–c) and insulin (d–f) were determined for each of the time points indicated. Black circles, normal diet; black squares, HFD; white circles, normal diet + vildagliptin; white squares, HFD + vildagliptin. n = 9–10 per group. The 8 month OGTT was omitted for clarity

Effect of chronic vildagliptin treatment on oral glucose tolerance and insulin secretion. OGTTs were performed in normal-diet- and HFD-fed mice treated with (white bars) or without (black bars) vildagliptin (3 μmol/day) for the months of treatment indicated. (a,b) Glucose tolerance, as the AUC for glucose (0–120 min), (c,d) IGI, as iAUCins/iAUCglu, and (e,f) oral disposition index, IGI × HOMA-IR−1, were determined from the OGTT data for the time period 0–60 min (a,c,e, normal diet; b,d,f, HFD). n = 9–10 per group; * p < 0.05, ** p < 0.01 and *** p < 0.001, when comparing treated and non-treated groups at the indicated time
In both diet groups, treatment with vildagliptin significantly increased IGI at all time points (Fig. 3c, d and electronic supplementary material [ESM] Table 1). The IGI is a measure of beta cell function that does not account for the underlying insulin sensitivity in the individual mice. To address this, the IGI was normalised to a measure of insulin sensitivity (HOMA-IR−1) similar to the oral disposition index. As seen in Fig. 3e, f, the oral disposition index was improved in vildagliptin-treated mice in both diet groups, and the improvements increased incrementally during the course of the study.
Beta cell area is not significantly altered by chronic vildagliptin treatment in the advanced-aged DIO mouse
As changes in beta cell mass in rodents are often reported using young animals, we determined whether beta cell area increases as a result of chronic HFD feeding, chronic DPP4 inhibition or both in advanced-aged animals. After 12 months of HFD feeding, there were no differences in beta cell area compared with the normal-diet group (Fig. 4a). Furthermore, chronic DPP4 inhibition with vildagliptin for 11 months did not significantly increase beta cell area in advanced-aged mice in either diet group (Fig. 4a). There was a trend towards decreased alpha cell area in the HFD mice treated with vildagliptin but this was not statistically significant (p = 0.10) (ESM Fig. 1). In addition, pancreatic insulin content was not different between groups, although there was a non-significant trend towards an increase in HFD mice treated with vildagliptin (Fig. 4b). After the completion of the in vivo study, an in vitro washout experiment was performed. Islets were isolated from the mice, washed and incubated in the absence of vildagliptin for 24 h prior to evaluation of glucose-stimulated insulin secretion. After the in vitro washout, glucose-stimulated insulin secretion was approximately equal between all groups, regardless of diet and prior treatment (Fig. 4c).

Effect of chronic vildagliptin treatment on beta cell and alpha cell area in advanced-aged DIO mice. (a) Beta cell area determined in pancreas sections immunostained for insulin. The insulin-positive area per total pancreas area in each group is shown. n = 3–4 per group. (b) Pancreatic insulin content after 11 months of treatment. n = 4–7 per group. (c) Glucose-stimulated insulin secretion in isolated islets. Isolated islets of Langerhans were incubated with the indicated concentrations of glucose for 1 h. Black bars, normal diet; white bars, normal diet + vildagliptin; dark grey bars, HFD; light grey bars, HFD + vildagliptin. n = 4–8 per group. ND, normal diet; VIL, vildagliptin treated
Advanced-aged mice fed chronically with an HFD display peri-insulitis, which is prevented by vildagliptin treatment
During the quantification of beta cell mass, islets with altered morphology were observed. When these islets were observed under a higher magnification, the presence of mononuclear cells adjacent to, or at times infiltrating, the islet was observed (Fig. 5a). The cellular phenotype was reminiscent of the peri-insulitis seen in NOD mice, albeit less severe [15]. When the peri-insulitis was quantified it was clear that chronic feeding of an HFD in advanced-aged mice resulted in a significant increase in the percentage of islets with peri-insulitis and that chronic treatment of HFD-fed mice with vildagliptin completely prevented this increase (Fig. 5b). Using double immunofluorescent staining we were able to determine that a major portion of the infiltrating cell mass was comprised of CD3+ T cells (Fig. 5c). In addition, the infiltrating cell mass contained appreciable amounts of phagocytic macrophages (ESM Fig. 2).

Peri-islet inflammation in advanced-aged DIO mice. (a) Pancreatic sections from HFD-fed mice were stained with haematoxylin and eosin or insulin (brown) and haematoxylin for evaluation of peri-insulitis. Scale bar represents 100 μm. (b) Peri-insulitis in advanced-aged DIO mice in percentage of total islets per section. n = 4; * p < 0.05. (c) Sections from the same animals were double immunofluorescent stained for insulin (red) and CD3 (green). Scale bar represents 50 μm. ND, normal diet; VIL, vildagliptin treated
Advanced-aged DIO mice treated with vildagliptin had improved survival rates
The specific time of death, and thereby length of life, for each mouse used in the study was recorded. According to the ethical guidelines where the study was carried out, animals in poor health were to be killed for humane reasons. Both these cases and spontaneous deaths were used for survival calculation. Survival was higher than 50% in all of the experimental groups (Fig. 6). The mice chronically fed an HFD had a tendency towards lower survival than normal-diet-fed mice (p = 0.08). Treating HDF-fed mice with vildagliptin throughout the course of the study resulted in higher survival rates compared with untreated HFD-fed mice (p = 0.05) (Fig. 6). There was also a trend towards higher survival in normal-diet-fed mice treated with vildagliptin compared with untreated normal-diet-fed mice. Spontaneous deaths were more common in the untreated HFD group than the vildagliptin-treated group, but this observation was not statistically significant because of the low number of observations.

Percentage of mice surviving after the 1 year long study. Kaplan–Meier curves shown for normal-diet- or HFD-fed mice, vildagliptin-treated and untreated mice. The x-axis shows the time since the initiation of the respective diets. p = 0.05 for the difference between treated and untreated HFD-fed mice at the end of the study. Grey lines, normal diet; black lines, HFD; solid lines, untreated; dashed lines, vildagliptin treated
Discussion
It is currently well known that beta cell function and mass deteriorate during the lifetime of individuals with type 2 diabetes [16, 17]. While changes in beta cell function can be measured over the lifetime of an individual, it is not currently possible to measure changes in beta cell mass. Therefore, experimental models have been used to describe the effects of DPP4 inhibition on beta cell mass and function in rodents. The majority of these studies were carried out using young rodents (<10 weeks old at the start of the study) or in rodents treated for a short period (<20 weeks) in relation to the lifespan of mice [18]. It is with this knowledge that we determined the long-term effects of chronic treatment with DPP4 inhibition on beta cell function and area using the DPP4 inhibitor vildagliptin in advanced-aged DIO mice (10 months old at study start), in order to more closely mimic the age of onset of hyperglycaemia in individuals with type 2 diabetes.
We and others have shown that DPP4 inhibition increases insulin secretion in experimental models using rats [19, 20] and mice [6, 21]. In this study we demonstrate that chronic treatment with the DPP4 inhibitor vildagliptin in an advanced-aged DIO mouse model improved insulin secretion and beta cell function throughout the course of the long study period of 11 months. We also report the novel findings that, in spite of this long-term therapy, beta cell area was not significantly affected by vildagliptin treatment in advanced-aged mice, despite improvements in beta cell function. Furthermore, in advanced-aged DIO mice, multiple instances of peri-insulitis were observed and these were nearly completely ablated by vildagliptin treatment. This may have contributed to the increased survival of vildagliptin-treated DIO mice.
It is well established that DPP4 inhibition lowers blood glucose and improves insulin secretion in humans and animal models [22]. Human studies have established that, acutely, DPP4 inhibitors improve the glucose sensitivity of the beta cells and insulin secretory capacity [13]. These effects in humans depend on increased levels of GLP-1 (and possibly GIP) but are reversible as soon as the levels of these incretins return to pre-meal levels [13]. The small increases in insulin secretory capacity were lost after a 3 month washout from vildagliptin, suggesting that this increased capacity was not due to increased beta cell mass. In the current study, 1 year of treatment with vildagliptin greatly increased insulin secretion in vivo where there was constant vildagliptin exposure; however, the effect of vildagliptin was lost after a 24 h washout in vitro. This suggests that in mice, as in man, the effects of chronic vildagliptin treatment on insulin secretion depend on in vivo exposure to the drug.
There is currently no evidence in man that incretin-based therapy, including DPP4 inhibition, leads to increased beta cell mass [23]. In contrast, many studies have shown that beta cell mass is improved by treatment with DPP4 inhibitors in rodents. However, all of these studies have been done in relatively young rodents. Several studies have shown that the proliferative capacity of beta cells is temporally limited in both aged rodents and humans [24–26], whereas in younger rodents, the proliferative capacity of the beta cells is high [27]. Consequently, studies in aged mice have shown that beta cell mass does not change significantly as a result of high-fat feeding [26]. In addition, treatment with the GLP-1 receptor agonist exendin-4 had no effect on beta cell mass in these mice.
Another study showed that the regenerative capacity of mouse beta cells in response to partial pancreatectomy is nearly ablated after 12 months of age [25]. Furthermore, a lack of a proliferative effect of exendin-4 in 14-month-old mice was reported [25]. The mice in our study were 10 months of age at the start of HFD feeding and 11 months of age at the start of vildagliptin treatment. In agreement with previous findings, we saw no increases in beta cell area in response to vildagliptin treatment. Furthermore, in some of the previously used models, the turnover of beta cells has been maximised by the model itself, like the neonatal streptozotocin model in rats [4] or the combination of HFD and streptozotocin in mice [3]. In contrast, in our model with HFD as the sole metabolic challenge, the beta cell turnover is expected to be lower and therefore not as easily affected. The conclusion is thus that when introducing DPP4 inhibition in an aged-mouse model, no significant increase in beta cell area is seen. The current study was carried out in female mice while many studies of DIO use male mice. A recent study of similar design, although involving only 4 months of treatment vs 1 year in our study, with sitagliptin in male mice in an advanced-aged DIO model showed a similar lack of effect on beta cell mass despite improving glucose tolerance [28].
During the course of the beta cell area determination we observed the presence of masses of T cells adjacent to, and at times infiltrating, the islets of advanced-aged HFD-fed mice. To our knowledge this has not been previously reported for DIO mouse models on the C57BL/6 background. We have previously given young mice an HFD for 1 year but did not see any similar pathology in that study [11]. The development of insulitis and subsequent beta cell destruction in the NOD mouse is well characterised [15, 29]. The peri-insulitis observed in our study was not as severe as that seen in NOD mice nor was it as prevalent. It was, however, robustly suppressed by vildagliptin treatment in advanced-aged HFD-fed mice. The suppression of islet inflammation by DPP4 inhibitors in mouse and rat models has been reported previously [30–33]. Dobrian et al reported a reduction of the mRNA expression of a number of inflammatory cytokines including monocyte chemotactic protein 1 (MCP-1) and IL-6 in the islets of DIO mice treated with sitagliptin for 12 weeks [30]. There were, however, no reports of the presence of an inflammatory cell infiltrate in the pancreas of this DIO model in any of the experimental groups. Two additional reports in NOD mice and another in Zucker diabetic fatty (ZDF) rats have shown a reduced lymphocytic infiltrate in the islets of animals treated with DPP4 inhibitors [31–33]. The studies in NOD mice offered some insight into potential mechanisms whereby DPP4 inhibition could reduce inflammation and immune cell infiltration into pancreatic islets. DPP4 is present on the outer cell surface of both resting and activated T cells [34]. An elegant study in NOD mice recently showed that treatment with the DPP4 inhibitor sitagliptin reduced diabetes incidence [32]. Furthermore, CD4+ T cells isolated from the spleen and lymph nodes of diabetic NOD mice treated with sitagliptin ex vivo showed reduced cell migration. Splenic T cells injected into NOD mice after in vitro treatment with GLP-1 and GIP were subsequently less abundant in the pancreatic infiltrate than untreated cells [32]. These results suggest that DPP4 inhibition and the resulting increase in circulating active GLP-1 and GIP could inhibit T cell migration and by this mechanism could reduce the amount of T cells in the pancreatic infiltrate.
In the unique advanced-aged DIO mouse model, survival and longevity appeared to be compromised similar to the profile of humans with type 2 diabetes. The natural process of ageing is associated with cellular oxidative stress [35]. Type 2 diabetes is characterised by an increased glucose-induced oxidative stress in multiple cell types including pancreatic beta cells [36]. Recently, vildagliptin has been shown to potently reduce markers of oxidative stress in type 2 diabetic humans by minimising the large daily fluctuations in blood glucose [37]. Reduced daily glucose fluctuations were probably occurring in the advanced-aged DIO mouse model. This was demonstrated by the lower glucose excursions in the OGTTs performed at different stages of treatment. As the treatment was for nearly half of the natural life of the mice, the reduced daily fluctuation of glucose over a long period would correspond to less cellular stress and increased longevity. In humans, this would correspond to an increased number of quality-adjusted life-years for a type 2 diabetic individual, which is the most relevant measure of a treatment’s efficacy.
In conclusion, DIO initiated in advanced-aged mice results in a unique and clinically relevant model with glucose intolerance and pancreatic immune cell infiltration, both of which are prevented by chronic treatment with vildagliptin. Chronic treatment with vildagliptin in this model does not, however, affect beta cell mass. Our results also present the novel finding that DPP4 inhibition increases survival and longevity in insulin-resistant/glucose-intolerant states. These findings offer insight into the effectiveness of DPP4 inhibitors in treating multiple pathophysiological defects in type 2 diabetes.
Abbreviations
- DEXA:
-
Dual-energy x-ray absorptiometry
- DIO:
-
Diet-induced obesity
- DPP4:
-
Dipeptidyl peptidase-4
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GLP-1:
-
Glucagon-like peptide 1
- HFD:
-
High-fat diet
- HOMA-IR:
-
HOMA-insulin resistance
- iAUC:
-
Incremental AUC
- iAUCglu :
-
Incremental AUC for glucose
- iAUCins :
-
Incremental AUC for insulin
- IBA-1:
-
Ionised calcium-binding adaptor molecule 1
- IGI:
-
Insulinogenic index
References
Ritzel RA, Butler AE, Rizza RA, Veldhuis JD, Butler PC (2006) Relationship between beta cell mass and fasting blood glucose concentration in humans. Diabetes Care 29:717–718
Ahren B (2011) The future of incretin-based therapy: novel avenues—novel targets. Diabetes Obes Metab 13(Suppl 1):158–166
Mu J, Woods J, Zhou YP et al (2006) Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta cell mass and function in a rodent model of type 2 diabetes. Diabetes 55:1695–1704
Duttaroy A, Voelker F, Merriam K et al (2011) The DPP-4 inhibitor vildagliptin increases pancreatic beta cell mass in neonatal rats. Eur J Pharmacol 650:703–707
Takeda Y, Fujita Y, Honjo J et al (2012) Reduction of both beta cell death and alpha cell proliferation by dipeptidyl peptidase-4 inhibition in a streptozotocin-induced model of diabetes in mice. Diabetologia 55:404–412
Furuta Y, Horiguchi M, Sugaru E et al (2010) Chronic administration of DSP-7238, a novel, potent, specific and substrate-selective DPP IV inhibitor, improves glycaemic control and beta cell damage in diabetic mice. Diabetes Obes Metab 12:421–430
Mu J, Petrov A, Eiermann GJ et al (2009) Inhibition of DPP-4 with sitagliptin improves glycemic control and restores islet cell mass and function in a rodent model of type 2 diabetes. Eur J Pharmacol 623:148–154
Zhang X, Wang Z, Huang Y, Wang J (2011) Effects of chronic administration of alogliptin on the development of diabetes and beta cell function in high fat diet/streptozotocin diabetic mice. Diabetes Obes Metab 13:337–347
Muhlhausler BS (2009) Nutritional models of type 2 diabetes mellitus. Meth Mol Biol 560:19–36
Koopman RJ, Mainous AG 3rd, Diaz VA, Geesey ME (2005) Changes in age at diagnosis of type 2 diabetes mellitus in the United States, 1988 to 2000. Ann Fam Med 3:60–63
Ahren J, Ahren B, Wierup N (2010) Increased beta cell volume in mice fed a high-fat diet: a dynamic study over 12 months. Islets 2:353–356
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta cell deficit and increased beta cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
Ahren B, Schweizer A, Dejager S, Villhauer EB, Dunning BE, Foley JE (2011) Mechanisms of action of the dipeptidyl peptidase-4 inhibitor vildagliptin in humans. Diabetes Obes Metab 13:775–783
Myrsen U, Keymeulen B, Pipeleers DG, Sundler F (1996) Beta cells are important for islet innervation: evidence from purified rat islet-cell grafts. Diabetologia 39:54–59
Aoki CA, Borchers AT, Ridgway WM, Keen CL, Ansari AA, Gershwin ME (2005) NOD mice and autoimmunity. Autoimmun Rev 4:373–379
Henry RR (1998) Type 2 diabetes care: the role of insulin-sensitizing agents and practical implications for cardiovascular disease prevention. Am J Med 105:20S–26S
Meigs JB, Muller DC, Nathan DM, Blake DR, Andres R (2003) The natural history of progression from normal glucose tolerance to type 2 diabetes in the Baltimore Longitudinal Study of Aging. Diabetes 52:1475–1484
Blackwell BN, Bucci TJ, Hart RW, Turturro A (1995) Longevity, body weight, and neoplasia in ad libitum-fed and diet-restricted C57BL6 mice fed NIH-31 open formula diet. Toxicol Pathol 23:570–582
Miura K, Kitahara Y, Yamagishi S (2010) Combination therapy with nateglinide and vildagliptin improves postprandial metabolic derangements in Zucker fatty rats. Horm Metab Res 42:731–735
Inaba W, Mizukami H, Kamata K, Takahashi K, Tsuboi K, Yagihashi S (2012) Effects of long-term treatment with the dipeptidyl peptidase-4 inhibitor vildagliptin on islet endocrine cells in non-obese type 2 diabetic Goto-Kakizaki rats. Eur J Pharmacol 691:297–306
Reimer MK, Holst JJ, Ahren B (2002) Long-term inhibition of dipeptidyl peptidase IV improves glucose tolerance and preserves islet function in mice. Eur J Endocrinol 146:717–727
Ahren B, Foley JE (2008) The islet enhancer vildagliptin: mechanisms of improved glucose metabolism. Int J Clin Pract Suppl (159):8–14
Foley JE, Bunck MC, Moller-Goede DL et al (2011) Beta cell function following 1 year vildagliptin or placebo treatment and after 12 week washout in drug-naive patients with type 2 diabetes and mild hyperglycaemia: a randomised controlled trial. Diabetologia 54:1985–1991
Cnop M, Hughes SJ, Igoillo-Esteve M et al (2010) The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 53:321–330
Rankin MM, Kushner JA (2009) Adaptive beta cell proliferation is severely restricted with advanced age. Diabetes 58:1365–1372
Tschen SI, Dhawan S, Gurlo T, Bhushan A (2009) Age-dependent decline in beta cell proliferation restricts the capacity of beta cell regeneration in mice. Diabetes 58:1312–1320
Bonner-Weir S, Trent DF, Weir GC (1983) Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J Clin Invest 71:1544–1553
Rankin MM, King KB, Kim JJ, Tu JX, Li C, Kushner JA (2012) New B cells are not generated by prolonged administration of incretin-based therapies (exendin-4 or sitagliptin) in adult mouse pancreas. Diabetes 61:LB34
Giarratana N, Penna G, Adorini L (2007) Animal models of spontaneous autoimmune disease: type 1 diabetes in the nonobese diabetic mouse. Meth Mol Biol 380:285–311
Dobrian AD, Ma Q, Lindsay JW et al (2011) Dipeptidyl peptidase IV inhibitor sitagliptin reduces local inflammation in adipose tissue and in pancreatic islets of obese mice. Am J Physiol Endocrinol Metab 300:E410–E421
Ferreira L, Teixeira-de-Lemos E, Pinto F et al (2010) Effects of sitagliptin treatment on dysmetabolism, inflammation, and oxidative stress in an animal model of type 2 diabetes (ZDF rat). Mediat Inflamm 2010:592760
Kim SJ, Nian C, Doudet DJ, McIntosh CH (2009) Dipeptidyl peptidase IV inhibition with MK0431 improves islet graft survival in diabetic NOD mice partially via T cell modulation. Diabetes 58:641–651
Tian L, Gao J, Hao J et al (2010) Reversal of new-onset diabetes through modulating inflammation and stimulating beta cell replication in nonobese diabetic mice by a dipeptidyl peptidase IV inhibitor. Endocrinology 151:3049–3060
Morimoto C, Schlossman SF (1998) The structure and function of CD26 in the T cell immune response. Immunol Rev 161:55–70
Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408:239–247
Leibowitz G, Bachar E, Shaked M et al (2010) Glucose regulation of beta cell stress in type 2 diabetes. Diabetes Obes Metab 12(Suppl 2):66–75
Rizzo MR, Barbieri M, Marfella R, Paolisso G (2012) Reduction of oxidative stress and inflammation by blunting daily acute glucose fluctuations in patients with type 2 diabetes: role of dipeptidyl peptidase-IV inhibition. Diabetes Care 35:2076–2082
Acknowledgements
The authors would like to acknowledge K. Andersson, L. Kvist, L. Bengtsson and C. Blennow from Lund University, Lund, Sweden for their outstanding technical assistance and C. Boitard from Inserm U561 Paris, France for advice on interpretation of the histopathology.
Funding
This study was funded by research grants from the Swedish Research Council, Region Skåne, Lund University Medical Faculty and Novartis Pharmaceuticals.
Duality of interest
M. S. Winzell is an employee of AstraZeneca. J. E. Foley is an employee of and holds stock in Novartis Pharmaceuticals. B. Ahrén is on the scientific advisory board for GlaxoSmithKline, Merck and Sanofi-Aventis, is a member of the speakers’ bureaus for Novartis and Merck and holds stock in Novo Nordisk. All other authors declare that there is no duality of interest associated with their contribution to this manuscript
Author contribution
BAO, JV, MSW, UV and EE acquired data, and performed data analysis and interpretation. JV, MSW, UV and EE performed experiments and data analysis. JEF and BOA designed the study and participated in interpretation and discussion of the data. BAO and BA wrote the manuscript. JV, MSW, UV, EE and JEF carried out critical revision of the manuscript. All authors approved the final version of the manuscript. BA is the guarantor of this work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
Alpha cell area determined in pancreas sections immunostained for glucagon. The glucagon positive area per total pancreas area in each group is indicated. n = 3–4 per group (PDF 12 kb)
ESM Fig. 2
Characterization of immune cell infiltrate in an untreated advanced age DIO mouse at study termination. (a) insulin staining in an insulitic islet (b) staining with the pan-T-cell marker CD3 from the same islet. (c) Staining with the macrophage marker IBA-1 (in a different periinsulitic islet from the same animal). The white arrows indicate CD3 positive cells that have fully infiltrated the islet. Scale bar represents 20 μm. (PDF 179 kb)
Table S1
(PDF 51 kb)
Rights and permissions
About this article
Cite this article
Omar, B.A., Vikman, J., Winzell, M.S. et al. Enhanced beta cell function and anti-inflammatory effect after chronic treatment with the dipeptidyl peptidase-4 inhibitor vildagliptin in an advanced-aged diet-induced obesity mouse model. Diabetologia 56, 1752–1760 (2013). https://doi.org/10.1007/s00125-013-2927-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2927-8