
Abstract
Aims/hypothesis
Cardiotrophin 1 (CT-1) is a recently described cytokine originally isolated from the heart where it has been shown to play an important role in apoptotic protection of cardiomyocytes and heart hypertrophy. Its beneficial properties have also been described in other organs such as liver and neuromuscular tissue. In the present study, we investigated whether CT-1 can confer protection against pro-apoptotic stimuli in pancreatic beta cells, and its role in insulin secretion and diabetes development.
Methods
The effects of CT-1 on apoptosis and function were studied using MIN6B1 cells and freshly isolated murine pancreatic islets. The impact on the development of diabetes was evaluated in Ct1-null (Ct1 −/−) mice (the gene Ct1 is also known as Ctf1) using two streptozotocin (STZ)-induced models of diabetes.
Results
CT-1 has a protective effect in MIN6B1 cells and murine islets under the pro-apoptotic stimulus of serum deprivation, which correlates with the expression of B cell lymphoma-extra large, or following exposure to a mixture of cytokines. In addition, CT-1 enhances glucose-stimulated insulin secretion in MIN6B1 cells and this was repressed by inhibitors of phospholipase C. Furthermore, Ct1 −/− mice were more prone to develop diabetes, and their glucose tolerance test showed impaired plasma glucose clearance which correlated with decreased pancreatic insulin secretion.
Conclusions/interpretation
The results obtained from both in vitro and in vivo experiments show that CT-1 improves beta cell function and survival, and protects mice against STZ-induced diabetes.
Similar content being viewed by others


Introduction
Diabetes is a chronic disease, defined and diagnosed by chronic hyperglycaemia that requires a lifelong therapeutic self-care regimen to maintain normal blood glucose levels [1]. Despite improvements in therapy, the life expectancy of diabetic patients is still below that of the general population, and they experience an extraordinarily high incidence of heart attacks, stroke, kidney failure, blindness and other complications [1]. An absolute or relative decrease in beta cell mass and function underlies type 1 and type 2 diabetes, respectively, rooted in diverse mechanisms. Inter-organ communication may play an important role in the pathophysiology of diabetes, possibly contributing to decreased beta cell functional mass, especially in type 2 diabetes. For example, adipokines including leptin [2] and as yet to be identified myokines [3] can influence beta cell function, survival and proliferation.
Cardiotrophin-1 (CT-1) is a member of the IL-6 family of cytokines that was originally isolated from cardiac tissue and identified by its ability to induce hypertrophy in cardiomyocytes [4]. Its receptor consists of a heterodimer made of glycoprotein 130 (GP130) receptor and leukaemia inhibitory factor receptor (LIFR), together with a yet to be described third component [5]. It is also known that CT-1 has beneficial effects in several tissues, such as motoneurons, by protecting them from apoptosis or by participating in regeneration mechanisms [6]. Recent studies have reported the protective role of CT-1 in the liver; CT-1 is upregulated during liver regeneration and exerts anti-apoptotic effects on hepatocytes [7]. Consistent with these findings, the overexpression of CT-1 in the liver efficiently protects rats from fulminant hepatic failure after subtotal hepatectomy [8]. Furthermore, the CT-1-related cytokine IL-6 has been shown to act as a protective agent against apoptosis induced by pro-inflammatory cytokines in murine islets [9] while also impacting beta cell function indirectly through induction of glucagon-like peptide-1 (GLP-1) secretion from neighbouring alpha cells [10]. In this context, Il6-null mice showed altered carbohydrate and lipid metabolism as well as impairment in glucose homeostasis [11].
Interestingly, the liver and the pancreas are closely related organs with a common embryonic origin [12]. Both organs share important metabolic pathways, transcription factors and transmembrane receptors [13]. Based on these premises, the main goal of this study was to analyse whether the protective effect of CT-1 observed in liver tissue could be extended to pancreatic beta cells. Also, we investigated the role of CT-1 in glucose metabolism and the development of streptozotocin (STZ)-induced diabetes in Ct1-null (Ct1 −/−) mice (the gene Ct1 is also known as Ctf1).
Methods
Cell culture
MIN6B1 cells [14] were maintained in Dulbecco’s modified Eagle medium (Gibco, Paisley, UK) containing 15% fetal calf serum (FCS), 71 μmol/l β-mercaptoethanol, 2 mmol/l glutamine, 100 U/ml penicillin, 100 mg/l streptomycin and 110 mg/ml sodium pyruvate (Sigma-Aldrich, St Louis, MO, USA) on adherent 175 cm2 flasks.
Mice
C57Bl/6J wild-type (WT) and C57Bl/6J Ct1 knockout mice (Ct1 −/−) were bred and housed in pathogen-free animal facilities at the University of Navarra. Ct1 −/− mice were originally generated by Oppenheim et al [15]. All the mice had free access to water and food. Animal procedures were performed in accordance with approved protocols and following the criteria outlined in the Guide of the Care and Use of Laboratory Animals by the National Academy of Sciences.
Isolation and culture of islets of Langerhans
Murine islets were isolated from 6- to 9-week-old male C57/BL6J mice using collagenase digestion, followed by Ficoll gradient purification, and manually harvested as described by Li et al [16]. The purified islets were collected using a glass pipette under a stereo microscope (Olympus SZ51, Bilbao, Spain) and maintained in Petri dishes. The isolated murine islets were seeded on 48- or 96-well plates pre-coated with poly l-lysine (Sigma-Aldrich). In order to obtain dispersed cells, the islets were digested by trypsin/EDTA (Biowhitaker, Verviers, Belgium) treatment (37°C, 3 min) followed by gentle dispersion with a pipette.
Gene expression evaluation by real-time PCR
Total RNA was isolated from 5 × 106 P19 and MIN6B1 cells and 200 freshly isolated pancreatic islets. cDNA was synthesised from 1 μg total RNA using the First Strand cDNA Synthesis Kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. Glyceraldehyde 3-phosphate dehydrogenase (Gapdh), Lifr, Gp130 and Ct1 expression were evaluated using a real-time PCR quantitative system (Applied Biosystems, Foster City, CA, USA) with the oligonucleotides presented in electronic supplementary material (ESM) Table 1. The relative amount of mRNA was normalised according to the levels of Gapdh as housekeeping gene and using the mouse embryonal carcinoma cell line P19 and total RNA (BD Biosciences, Palo Alto, CA, USA) as positive controls.
Apoptosis induction protocols
MIN6B1 cell line and murine islets were subjected to serum deprivation to stimulate apoptosis by incubating the cells with their corresponding medium without FCS for 48, 96 and 120 h. In addition, murine islets were treated for 24 h with a cocktail of pro-inflammatory cytokines (50 U/ml IL-1β; 1,000 U/ml TNF-α; 1,000 U/ml IFN-γ) (Cytomix; eBioscience, Frankfurt, Germany) to induce apoptosis. CT-1 (DRO Biosystems, San Sebastian, Spain) or IL-6 (AbD Serotec, Oxford, UK) was added to the media at 100 ng/ml. Apoptotic levels were determined by the luminescence assay kit Caspase-Glo 3/7 assay (Promega, Madrid, Spain), which measures caspase-3 and -7 activities in the cell lysates. For the MIN6B1 assays, the rate of cell death was analysed using TUNEL staining (Roche, Mannheim, Germany), which detects nuclei in cells undergoing apoptosis or necrosis by immunofluorescence. It has been shown previously that under these conditions of culture, cell death detected by TUNEL is due predominantly to apoptosis [17]. The percentage of apoptotic cells was quantified using ImageJ-based software (NIH, Bethesda, MD, USA), where a brightness threshold detection system quantifies apoptotic cells (TUNEL-positive cells) in relation to the total number of cells (stained with 4′,6-diamidino-2-phenylindole [DAPI]; Vector Labs, Peterborough, UK).
Analysis of STAT3/pSTAT3, ERK/pERK, BCL-xL and GLUT2 expression by western blot
Protein was extracted from 1–2 × 106 MIN6B1 cells and/or 200–300 murine islets after treatment with lysis buffer (150 μl, 15 min). Samples were spun at 21,000 g (20 min, 4°C) and the pellets were discarded. Protein concentrations of the supernatant fractions were assessed with a BCA kit (Pierce; Rockford, IL, USA). Fifteen micrograms of protein were resuspended in loading buffer, separated in a 12% polyacrylamide gel, and transferred onto a nitrocellulose membrane. Immunoprobing was performed by incubating the membranes with primary antibody (signal transducer and activator of transcription 3 [STAT3], pSTAT3, extracellular signal-regulated kinase [ERK] and pERK; Cell Signaling, Beverly, MA, USA), B cell lymphoma-extra large (BCL-xL; BD Biosciences) and GLUT2 (Millipore, Darmstadt, Germany) overnight at 4°C. Anti-rabbit IgG coupled to alkaline phosphatase was used as a secondary antibody (1 h, room temperature). Membranes were revealed to detect the presence of alkaline phosphatase activity using a Tropix revealing kit (Applied Biosystems). Relative quantities of STAT3, pSTAT3, ERK, pERK, BCL-xL and GLUT2 were determined using densitometer software (Bio-Rad, Madrid, Spain). β-Actin or β-tubulin (both purchased from Sigma-Aldrich) were used to normalise the samples in the same membrane.
Flow cytometry was used to measure the ability of fresh isolated WT and Ct1 −/− islets to take up the non-metabolisable glucose analogue 6-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-6-Deoxyglucose (6-NBDG; Invitrogen) (200 μmol/l, 37°C, 10 min).
Insulin secretion assay
MIN6B1 cells were seeded at 50,000 cells/cm2 on 24-well plates and incubated under standard conditions for 48 h. Subsequently, cells were preincubated for 2 h in Hanks’ balanced salt solution (HBSS) supplemented with 1% bovine serum albumin (BSA) and 2.8 mmol/l glucose at 37°C. After washing, cells were incubated for 1 h with the same solution to determine the basal levels of insulin secretion. Subsequently, they were incubated for 1 h with HBSS supplemented with 1% BSA and 16.7 mmol/l glucose in combination or not (controls) with CT-1 (100 ng/ml) and the phospholipase C (PLC) inhibitors: neomycin (10 μmol/l; Sigma-Aldrich) and U73122 (2 μmol/l; Sigma-Aldrich) or its structural inactive analogue U73343 (2 μmol/l; Sigma-Aldrich). Finally, the cells were treated with a mixture of ethanol/acetic acid (95:5) to release the intracellular content of insulin. The supernatant fractions were collected and stored at −20°C. Insulin was detected using a commercial ELISA kit (Mercodia, Uppsala, Sweden).
Animal models of diabetes
Diabetes was induced by intraperitoneal administration of STZ (Sigma-Aldrich) to 10- to 12-week-old C57Bl/6J WT and C57Bl/6J Ct1 −/− male mice according to two models: an ‘acute model’, performed by the injection of a single dose of STZ (150 mg/kg), and a ‘chronic model’, based on the administration of multiple low doses of STZ [18] in which mice were treated with STZ at 50 mg kg−1 day−1 for four consecutive days. Once mice were treated with STZ, blood samples were collected daily (09:00–11:00 hours) from the tail vein of non-fasted mice and glucose was measured with a blood glucose meter (Accu-Chek Aviva; Roche). Mice were considered diabetic when non-fasting blood glucose levels were >16.7 mmol/l for at least three consecutive days.
Intraperitoneal glucose tolerance test
C57Bl/6J WT and C57Bl/6J Ct1 −/− male mice were fasted overnight (16–18 h), weighed and injected with d-glucose (Sigma-Aldrich) at a dose of 2 g/kg body weight intraperitoneally. Blood samples (50 μl) were obtained from the tail vein before and at 5, 10, 15, 20, 25, 30, 45, 60 and 120 min after glucose administration. Blood glucose levels were determined using a glucose meter (Accu-Chek Aviva).
Histology and measurement of pancreatic insulin content
Once the animals were killed, pancreases were isolated and fixed in 10% formalin (6 h at room temperature). Tissue samples were embedded in paraffin, cut in 5 μm sections, deparaffinised, and analysed by immunohistochemistry. Immunolabelling was performed with a primary antibody specific to insulin (dilution 1:100; Dako, Glostrup, Denmark). Signal amplification was performed using either the Envision system (Dako) or chromogen diaminobenzidine (Dako). All sections were viewed under a Nikon Eclipse E600 microscope (L'Hospitalet de Llobregat, Barcelona, Spain).
For the determination of total pancreatic insulin content, pancreases obtained from normal mice and STZ mouse models of diabetes (WT and Ct1 −/−, acute and chronic models) were homogenised and insulin was extracted with a mixture of ethanol/acetic acid (95:5). The insulin present in the supernatant fractions was detected using a commercial ELISA kit (Mercodia) and was measured in reference to the weight of the total pancreas.
Statistical analysis
All data were expressed as mean ± SD. The Gaussian distribution was confirmed using the Kolmogorov–Smirnov test. In experiments where data differences between two groups were assessed, statistical significance was analysed using Student’s unpaired t test. For non-parametric data, the Kruskal–Wallis test and Mann–Whitney U test were used. All statistical analyses were performed using the GraphPad Instat software version 3.00 for Windows (GraphPad Software, San Diego, CA, USA). All p values <0.05 were considered statistically significant.
Results
Pancreatic beta cells express CT-1 and its receptor
To explore the role of CT-1 in beta cell function, we first analysed the expression of Ct1 and the two known components of its receptor, Gp130 and Lifr, in both freshly isolated murine islets and MIN6B1 cells. Using real-time quantitative PCR, we detected similar mRNA expression levels in both cell sources (Table 1). Previous studies performed in cardiomyocytes and hepatocytes have reported that CT-1 activates important signalling pathways, such as Janus kinase (JAK)/STAT3, ERK1/2 and AKT [19–21]. To investigate whether CT-1 activates these pathways in beta cells, fresh isolated murine islets were stimulated in vitro with CT-1 (100 ng/ml) for 15, 30 and 60 min. Our results show that CT-1 treatment significantly induces transient phosphorylation of STAT3 and ERK1/2, peaking at 30 and 15 min, respectively (Fig. 1a, b).

CT-1 activates STAT3 and ERK pathways in pancreatic beta cells. Representative western blot for STAT3 (a) and ERK (b) phosphorylation in MIN6B1 protein extracts at different time points (15, 30 and 60 min) after CT-1 (100 ng/ml) stimulation. Data (mean ± SD) from three independent experiments were normalised to β-actin. *p < 0.05; **p < 0.01
CT-1 induces an anti-apoptotic effect on beta cells
IL-6 has been shown to play an important role in the protection of neuroendocrine tumoral PC12 cells subjected to serum deprivation [22]. Also, IL-6 exerts an anti-apoptotic effect on MIN6 cells and pancreatic islets treated with pro-inflammatory cytokines [9, 23], while CT-1, a member of the IL-6 family of cytokines, has proven to be a survival factor for hepatocytes subjected to serum deprivation [7]. To analyse whether CT-1 also displayed cytoprotective effects on pancreatic beta cells, MIN6B1 cells were cultured for up to 120 h under serum deprivation conditions in the presence or absence of CT-1 (100 ng/ml). After 120 h of serum deprivation, there was a significant reduction (38%) in the number of TUNEL-positive apoptotic cells in MIN6B1 cells treated with CT-1 (Fig. 2a). There was furthermore a significant decrease in apoptosis (active caspase-3 and -7) in the presence of CT-1 at both 96 (24.8%) and 120 h (41.7%) of serum deprivation (Fig. 2b, c). We also observed a protective effect for IL-6 (100 ng/ml) at 120 h (Fig. 2c). Similar results were observed when islets obtained from mouse pancreas were subjected to Cytomix (eBioscience) containing pro-inflammatory cytokines IL-1β, TNF-α and IFN-γ (ESM Fig. 1).

CT-1 protects MIN6B1 cells from apoptosis induced by serum deprivation. (a) TUNEL assay for the detection of apoptotic nuclei of MIN6B1 cells after 120 h of serum deprivation. The graph shows the percentage of TUNEL-positive cells, and representative images of three independent experiments performed in triplicate are shown. (b, c) Detection of caspase-3 and -7 activity in MIN6B1 cells under serum deprivation and CT-1 or IL-6 treatment for 96 h (b) and 120 h (c). (d) BCL-xL detection by western blot in freshly isolated islets under 40 h of serum deprivation and CT-1 (100 ng/ml) treatment. (e) BCL-xL quantification by western blot in MIN6B1 cells under 48 and 96 h of serum deprivation and CT-1 or IL-6 (100 ng/ml) treatment. Data are expressed as mean ± SD and normalised to β-actin. For all experiments, MIN6B1 cells incubated with 10% FCS for 48, 96 and 120 h were used as control samples. Images shown are representative of three independent experiments. S.D., serum deprivation. *p < 0.05, **p < 0.01; ***p < 0.001
CT-1 treatment induces expression of BCL-xL
Since BCL-xL has been demonstrated to be involved in IL-6-mediated suppression of apoptosis in murine islets [9], we analysed the levels of BCL-xL in MIN6B1 cells treated with CT-1 (100 ng/ml) by western blot. CT-1 treatment of freshly isolated islets subjected to 40 h of serum deprivation significantly increased BCL-xL content (Fig. 2d). We also compared the levels of BCL-xL between MIN6B1 cells treated with CT-1 or IL-6. Our results show that CT-1 and IL-6 were able significantly to increase the levels of BCL-xL to a similar extent after 48 h and 96 h of serum deprivation (Fig. 2e).
CT-1 enhances glucose-stimulated insulin secretion from MIN6B1 cells through the activation of the PLC pathway
Recent studies have shown that co-family member IL-6 can induce insulin secretion in MIN6 cells [24, 25]. We now show that in the presence of high glucose, CT-1 treatment likewise stimulates insulin secretion from MIN6B1 cells (Fig. 3b). This effect was observed independently of the CT-1 concentration used (from 100 to 400 ng/ml) (data not shown).

CT-1 stimulates insulin secretion from MIN6B1 cells. (a) Experimental design of the insulin secretion assay in MIN6B1 cells. (b) Relative insulin secretion (fold) observed in MIN6B1 cells stimulated with glucose (16.7 mmol/l) alone or with the combination of glucose and CT-1 (100 ng/ml). (c) Relative insulin secretion (fold) in MIN6B1 cells in the presence of high glucose (16.7 mmol/l) in the combination of CT-1 (100 ng/ml) and the PLC inhibitor neomycin (10 μmol/l). Data are expressed as mean ± SD from three independent experiments in triplicate. *p < 0.05, **p < 0.01; ***p < 0.001
The activation of PLC has been implicated in the mechanism of calcium release and activation of other secondary effectors that mediate insulin secretion [26]. In addition, it has been demonstrated that IL-6 activates the PLC pathway in different cell types [22], including beta cells [25]. Concordant with these results, Suzuki et al [25] showed that different PLC inhibitors were able to block insulin secretion mediated by IL-6. Therefore, we tested the effect of treating MIN6B1 cells with the PLC inhibitor neomycin (10 μmol/l) (Fig. 3c) or U73122 (and its inactive structural analogue U73343) (ESM Fig. 2) in the presence or absence of CT-1 (100 ng/ml) and glucose (16.7 mmol/l). Our results demonstrate that both neomycin and U73122 were able to block significantly the insulin secretion stimulated by CT-1 observed in combination with high glucose (16.7 mmol/l) treatment (Fig. 3c and ESM Fig. 2), suggesting that the underlying mechanism involves the activation of the PLC pathway.
Endogenous CT-1 protects mice from STZ-induced diabetes
To determine whether CT-1 might be a critical endogenous factor in the protection of pancreatic beta cells against pro-apoptotic stimuli in vivo, we determined the sensitivity of Ct1 −/− and WT mice to the induction of diabetes by STZ based on two models of diabetes (acute and chronic). First, we evaluated the levels of the receptor (GLUT2) through which STZ is taken up by beta cells. Islets obtained from WT or Ct1 −/− mice showed similar levels of GLUT2 (ESM Fig. 3a). By flow cytometry, we also show that both WT and Ct1 −/− islets were equally able to take up the fluorescent glucose analogue 6-NBDG (ESM Fig. 3b).
Regarding the two diabetic models used, the acute model consisted of a single high dose of STZ (150 mg/kg body weight) administered intraperitoneally, which induced hyperglycaemia within the first 3 days, with 30% of the mice displaying glycaemia >16.7 mmol/l by the end of the experimental period (Fig. 4a). The second model based on multiple low-dose injections of STZ (50 mg kg−1 day−1 for 4 days) resulted in a more gradual acquisition of hyperglycaemia, with 44% of the mice displaying glycaemia >16.7 mmol/l by the end of the experimental period (Fig. 4c). Ct1 −/− mice were more susceptible to both acute and chronic induction of diabetes, with 90% and 91%, respectively, of the mice becoming diabetic by the end of the study (Fig. 4b, d). After monitoring the blood glucose levels for 1 month, the results showed a progressive acquisition of hyperglycaemia in the chronic model (Fig. 4c) compared with the acute model (Fig. 4a), with higher mean levels of glycaemia in Ct1 −/− mice from 15 and 10 days, respectively, following STZ administration. In addition, during the course of the induction of diabetes in the two models, Ct1 −/− mice showed a slightly higher body weight in comparison with WT mice (data not shown).

Ct1 −/− mice are more prone to develop diabetes, both in acute and chronic STZ-induced diabetic models. (a) Blood glucose levels measured in acute STZ-induced models in WT (black squares) and Ct1 −/− (white squares) mice (150 mg/kg STZ, single administration). (c) Blood glucose levels measured in chronic STZ-induced models in WT (black squares) and Ct1 −/− (white squares) mice (50 mg/kg STZ, four consecutive doses). For both models, glucose levels were determined daily during 1 month (a, c). The percentage of animals showing levels of glycaemia under 16.7 mmol/l (white bar, non-diabetic) and over 16.7 mmol/l (black bar, diabetic) at the end of the study in both mouse strains and both diabetic models are shown (b, d). Data are expressed as mean ± SD for ten mice per group. *p < 0.05
In order to evaluate the effect of CT-1 on beta cell mass in non-diabetic and diabetic conditions, we determined the total insulin content per pancreas in WT and Ct1 −/− mice. In non-diabetic conditions, there was no significant difference in total insulin content between WT and Ct1 −/− mice (Fig. 5a, b). After treatment with STZ, insulin content was lower in both WT and Ct1 −/− mice subjected to either acute (Fig. 5c, d) or chronic diabetes (Fig. 5e, f) compared with untreated mice (Fig. 5a, b). Regardless of whether acute or chronic diabetes was induced, Ct1 −/− mice presented a more important reduction in total insulin content (Fig. 5d, f; white bars) compared with WT mice (black bars). These results suggest that Ct1 −/− mice are more prone to develop diabetes induced by STZ as a consequence of decreased beta cell mass.

Histological analysis of islets of Langerhans from non-diabetic and STZ-induced acute and chronic diabetic models in WT and Ct1 −/− mice. (a) Representative images of sections of pancreases obtained from non-diabetic WT and Ct1 −/− mice (×40), stained for insulin. Quantification of pancreas insulin content (b) in non-diabetic WT (black bar) and Ct1 −/− (white bar) mice is also shown. (c, d, e, f) Representative images of insulin-positive cells detected in pancreases of acute diabetic (c, d) and chronic diabetic (e, f) WT and Ct1 −/− mice (×40). Quantification of pancreas insulin content in acute diabetic (d) and chronic diabetic (f) WT (black bars) and Ct1 −/− (white bars) mice are also represented. Data are expressed as mean ± SD for ten mice per group. ***p < 0.001
Impaired glucose tolerance test in Ct1 −/− mice
Finally, we investigated the impact of Ct1 knockout on glucose tolerance. While fasting glycaemia was unchanged, Ct1 −/− mice presented mild glucose intolerance, with slightly higher glucose levels from 20 to 40 min following glucose injection compared with WT mice (Fig. 6a), accompanied by decreased insulin secretion (Fig. 6b).

Ct1 −/− mice are glucose intolerant. (a) Blood glucose values determined following intraperitoneal glucose administration (2 g/kg body weight) in WT (black squares) and Ct1 −/− (white squares) mice. Data are presented as mean ± SD for four mice per group. (b) Relative insulin secretion (fold) measured in plasma of WT and Ct1 −/− mice (n = 4). Blood samples were obtained 10 min after intraperitoneal glucose administration (2 g/kg body weight). *p < 0.05; **p < 0.01
Discussion
The cytoprotective effect of CT-1 previously described in various cell types, such as cardiomyocytes and motoneurons [15, 21], has been extended to cultured hepatocytes according to in vivo models of acute severe liver damage [7, 8], suggesting that similar beneficial effects may also apply to other tissues. The present study shows that CT-1 is indeed able to protect pancreatic beta cells from apoptosis induced by serum deprivation and inflammatory cytokines; furthermore, it had a stimulatory effect on insulin secretion in the presence of glucose, which could be prevented via PLC inhibition. Consistent with these in vitro effects of CT-1 on beta cell survival and function, in vivo studies also demonstrate the role of CT-1 in normal glucose tolerance and in preventing the development of diabetes induced by STZ.
MIN6B1 is a subclone derived from the murine insulinoma cell line MIN6 with features, mainly in terms of adhesion and insulin secretion, that resemble those of primary beta cells [16, 27]. We show that both MIN6B1 and murine islets express Ct1 mRNA along with the components of its receptor, Gp130 and Lifr. All members of the IL-6 family, including CT-1, play important roles in diverse cellular processes such as proliferation, survival, adhesion and insulin secretion, through a common signalling pathway that involves the GP130 receptor protein [28]. Here, we show that CT-1 induces the activation of mitogen-activated protein kinase and JAK/STAT3 pathways through ERK1/2 and STAT3 phosphorylation, respectively. Moreover, CT-1 treatment promotes the protection of MIN6B1 cells from apoptotic stimuli such as serum deprivation through the upregulation of pro-survival protein BCL-xL, as has been shown for co-family member IL-6 in various cell types, including murine islets [29]. It is important to note that IL-6 has also been shown to improve beta cell function in mice mainly through an indirect route involving the induction of GLP-1 secretion from neighbouring alpha cells [10]. It will be interesting in future studies to investigate whether CT-1 also impacts alpha cell function.
The hallmark of beta cells is their capacity for glucose-stimulated insulin secretion. Our results suggest that CT-1 potentiates glucose-stimulated insulin secretion, indicating a possible synergistic effect between CT-1 and glucose which could be likened to the action of an incretin [30], albeit through different signalling pathways. Neomycin and U-73122 PLC inhibitors prevent this effect in agreement with the role of PLC activation in Ca2+ release from the endoplasmic reticulum and further in the release of insulin granules [31].
CT-1 appears to protect mice from STZ-induced diabetes in vivo, as Ct1 −/− mice show higher susceptibility to develop diabetes and greater loss in insulin content upon STZ-induced damage compared with WT mice based on both acute [32] and chronic [33] diabetic mouse models. Ct1 −/− mice also present mild glucose intolerance compared with WT mice, accompanied by reduced insulin secretion in response to the glucose load (measured 10 min after injection), although glycaemia was also normalised at later time points in Ct1 −/− mice. This suggests that impaired beta cell function may underlie the glucose intolerance of Ct1 −/− mice. However, there was a significant increase in the body weight of the Ct1 −/− mice (data not shown) that may also contribute to glucose intolerance, and it will be interesting in future studies to investigate insulin sensitivity in these animal models. Regardless, these data indicate an important role for CT-1 in glucose homeostasis.
Different authors have described the role of cytokines such as IL-6, ciliary neurotrophic factor and TNF-α in adipose tissue metabolism [34], with elevated levels in the blood associated with obesity [35]. CT-1 levels in plasma [35] and adipose tissue [36] have recently been correlated with the pathology of the metabolic syndrome, suggesting that CT-1 may be involved in a protective mechanism against the metabolic changes that occur during the initial stages of the disease [37]. Moreover, the presence of CT1 polymorphisms have also been associated with the development of hypertension [38] and cardiomyopathies [39]. Thus, a similar analysis of the CT1 polymorphisms may provide a genetic link between CT-1 protection and the risk of developing diabetes.
In summary, our data here show a novel role for CT-1 in beta cell function and survival both in vitro and in vivo. CT-1 could be a candidate to promote islet survival and function during islet transplantation and may also prove useful in the treatment of diabetes or to slow its progression.
Abbreviations
- BCL-xL:
-
B cell lymphoma-extra large
- BSA:
-
Bovine serum albumin
- CT-1:
-
Cardiotrophin 1
- Ct1 −/− :
-
Cardiotrophin 1-null mouse
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- ERK:
-
Extracellular signal-regulated kinase
- FCS:
-
Fetal calf serum
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GLP-1:
-
Glucagon-like peptide-1
- GP130:
-
Glycoprotein 130
- HBSS:
-
Hanks’ balanced salt solution
- JAK:
-
Janus kinase
- LIFR:
-
Leukaemia inhibitory factor receptor
- 6-NBDG:
-
6-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-6-Deoxyglucose
- PLC:
-
Phospholipase C
- STAT3:
-
Signal transducer and activator of transcription 3
- STZ:
-
Streptozotocin
- WT:
-
Wild-type
References
American Diabetes Association (2010) Diagnosis and classification of diabetes mellitus. Diabetes Care 33(Suppl 1):S62–S69
Maedler K, Sergeev P, Ehses JA et al (2004) Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc Natl Acad Sci U S A 101:8138–8143
Bouzakri K, Plomgaard P, Berney T, Donath MY, Pedersen BK, Halban PA (2011) Bimodal effect on pancreatic beta-cells of secretory products from normal or insulin-resistant human skeletal muscle. Diabetes 60:1111–1121
Pennica D, King KL, Shaw KJ et al (1995) Expression cloning of cardiotrophin 1, a cytokine that induces cardiac myocyte hypertrophy. Proc Natl Acad Sci U S A 92:1142–1146
Robledo O, Fourcin M, Chevalier S et al (1997) Signaling of the cardiotrophin-1 receptor. Evidence for a third receptor component. J Biol Chem 272:4855–4863
Mitsumoto H, Klinkosz B, Pioro EP et al (2001) Effects of cardiotrophin-1 (CT-1) in a mouse motor neuron disease. Muscle Nerve 24:769–777
Bustos M, Beraza N, Lasarte JJ et al (2003) Protection against liver damage by cardiotrophin-1: a hepatocyte survival factor up-regulated in the regenerating liver in rats. Gastroenterology 125:192–201
Iñíguez M, Berasain C, Martinez-Anso E et al (2006) Cardiotrophin-1 defends the liver against ischemia-reperfusion injury and mediates the protective effect of ischemic preconditioning. J Exp Med 203:2809–2815
Choi SE, Choi KM, Yoon IH et al (2004) IL-6 protects pancreatic islet beta cells from pro-inflammatory cytokines-induced cell death and functional impairment in vitro and in vivo. Transpl Immunol 13:43–53
Ellingsgaard H, Hauselmann I, Schuler B et al (2011) Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat Med 17:1481–1489
Wallenius V, Wallenius K, Ahrén B et al (2002) Interleukin-6-deficient mice develop mature-onset obesity. Nat Med 8:75–79
Deutsch G, Jung J, Zheng M, Lora J, Zaret KS (2001) A bipotential precursor population for pancreas and liver within the embryonic endoderm. Development 128:871–881
Zaret KS, Watts J, Xu J, Wandzioch E, Smale ST, Sekiya T (2008) Pioneer factors, genetic competence, and inductive signaling: programming liver and pancreas progenitors from the endoderm. Cold Spring Harb Symp Quant Biol 73:119–126
Lilla V, Webb G, Rickenbach K et al (2003) Differential gene expression in well-regulated and dysregulated pancreatic beta-cell (MIN6) sublines. Endocrinology 144:1368–1379
Oppenheim RW, Wiese S, Prevette D et al (2001) Cardiotrophin-1, a muscle-derived cytokine, is required for the survival of subpopulations of developing motoneurons. J Neurosci 21:1283–1291
Li DS, Yuan YH, Tu HJ, Liang QL, Dai LJ (2009) A protocol for islet isolation from mouse pancreas. Nat Protoc 4:1649–1652
Yermen B, Tomas A, Halban PA (2007) Pro-survival role of gelsolin in mouse beta-cells. Diabetes 56:80–87
Kannan Y, Tokunaga M, Moriyama M, Kinoshita H, Nakamura Y (2004) Beneficial effects of troglitazone on neutrophil dysfunction in multiple low-dose streptozotocin-induced diabetic mice. Clin Exp Immunol 137:263–271
Negoro S, Kunisada K, Tone E et al (2000) Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc Res 47:797–805
Railson JE, Liao Z, Brar BK et al (2002) Cardiotrophin-1 and urocortin cause protection by the same pathway and hypertrophy via distinct pathways in cardiac myocytes. Cytokine 17:243–253
Sheng Z, Knowlton K, Chen J, Hoshijima M, Brown JH, Chien KR (1997) Cardiotrophin 1 (CT-1) inhibition of cardiac myocyte apoptosis via a mitogen-activated protein kinase-dependent pathway. Divergence from downstream CT-1 signals for myocardial cell hypertrophy. J Biol Chem 272:5783–5791
Lee YH, Bae SS, Seo JK, Choi I, Ryu SH, Suh PG (2000) Interleukin-6-induced tyrosine phosphorylation of phospholipase C-gamma1 in PC12 cells. Mol Cells 10:469–474
Park KH, Na K, Jung SY, Kim SW, Cha KY, Chung HM (2005) Insulinoma cell line (MIN6) adhesion and spreading mediated by Arg-Gly-Asp (RGD) sequence conjugated in thermo-reversible gel. J Biosci Bioeng 99:598–602
Shimizu H, Ohtani K, Kato Y, Mori M (2000) Interleukin-6 increases insulin secretion and preproinsulin mRNA expression via Ca2+-dependent mechanism. J Endocrinol 166:121–126
Suzuki T, Imai J, Yamada T et al (2011) Interleukin-6 enhances glucose-stimulated insulin secretion from pancreatic beta-cells: potential involvement of the PLC-IP3-dependent pathway. Diabetes 60:537–547
Zawalich WS, Zawalich KC (1996) Regulation of insulin secretion by phospholipase C. Am J Physiol 271:E409–E416
Jaques F, Jousset H, Tomas A et al (2008) Dual effect of cell-cell contact disruption on cytosolic calcium and insulin secretion. Endocrinology 149:2494–2505
Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L (1998) Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334:297–314
Schwarze MM, Hawley RG (1995) Prevention of myeloma cell apoptosis by ectopic bcl-2 expression or interleukin 6-mediated up-regulation of bcl-xL. Cancer Res 55:2262–2265
Peters A (2010) Incretin-based therapies: review of current clinical trial data. Am J Med 123(3 Suppl):S28–S37
Yamazaki H, Zawalich KC, Zawalich WS (2010) Physiologic implications of phosphoinositides and phospholipase C in the regulation of insulin secretion. J Nutr Sci Vitaminol (Tokyo) 56:1–8
Bonal C, Avril I, Herrera PL (2008) Experimental models of beta-cell regeneration. Biochem Soc Trans 36:286–289
Amirshahrokhi K, Dehpour AR, Hadjati J, Sotoudeh M, Ghazi-Khansari M (2008) Methadone ameliorates multiple-low-dose streptozotocin-induced type 1 diabetes in mice. Toxicol Appl Pharmacol 232:119–124
Coppack SW (2001) Pro-inflammatory cytokines and adipose tissue. Proc Nutr Soc 60:349–356
Natal C, Fortuno MA, Restituto P et al (2008) Cardiotrophin-1 is expressed in adipose tissue and upregulated in the metabolic syndrome. Am J Physiol Endocrinol Metab 294:E52–E60
Zvonic S, Hogan JC, Arbour-Reily P, Mynatt RL, Stephens JM (2004) Effects of cardiotrophin on adipocytes. J Biol Chem 279:47572–47579
Moreno-Aliaga MJ, Perez-Echarri N, Marcos-Gomez B et al (2011) Cardiotrophin-1 is a key regulator of glucose and lipid metabolism. Cell Metab 14:242–253
Robador PA, Moreno MU, Beloqui O et al (2010) Protective effect of the 1742(C/G) polymorphism of human cardiotrophin-1 against left ventricular hypertrophy in essential hypertension. J Hypertens 28:2219–2226
Erdmann J, Hassfeld S, Kallisch H, Fleck E, Regitz-Zagrose V (2000) Genetic variants in the promoter (g983G>T) and coding region (A92T) of the human cardiotrophin-1 gene (CTF1) in patients with dilated cardiomyopathy. Hum Mutat 16:448
Funding
This work was supported by the Fundación Mutua Madrileña and Caja de Ahorros de Navarra, and grant number 31003A_127276 from the Swiss National Science Fund to PAH.
Contribution statement
All authors contributed to the concept and design of the study or to the acquisition and interpretation of data, and to drafting the article or revising it critically. All authors gave approval of the final version to be published.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
CT-1 protects mouse islets from apoptosis induced by CYTOMIX. Detection of caspase 3/7 activity in murine islets with CYTOMIX (50 U/ml IL-1β + 1000 U/ml TNF-α + 1000 U/ml IFN-γ) apoptosis induction, in the presence of CT-1 (100 ng/ml, 24 h). The data represent the percentage of apoptotic cells obtained from 3 independent experiments in triplicate. **p<0.01 (PDF 29 kb)
ESM Fig. 2
Inhibition of PLC in MIN6B1 cells blocks glucose-stimulated insulin secretion mediated by CT-1. Fold-increase in insulin secretion observed in MIN6B1 cells in the presence of high glucose (16.7 mmol/l) in combination of CT-1 (100 ng/ml) and PLC inhibitor U73122 (2 μmol/l) or its structural inactive analogue U73343 (2 μmol/l). Data are expressed as mean ± SD for 3 independent experiments in triplicate. *p<0.05; **p<0.01 (PDF 48 kb)
ESM Fig. 3
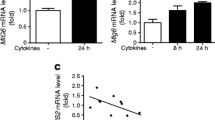
GLUT2 expression and uptake of glucose analogue 6-NBDG by WT and CT-1−/− mouse islets. (a) Detection of GLUT2 expression by western blot analysis. The expression of GLUT2 was determined in 500 murine islets obtained from WT and CT-1−/− pancreases and 250,000 MIN6B1 cells. GLUT2 expression is represented as mean ± SD normalized to β-tubulin and images shown are representative of 3 independent experiments. (b) The capability of WT (red line) and CT-1−/− (blue line) islets to uptake glucose was evaluated by incubating them with the fluorescent non-metabolizable glucose analogue 6-NBDG (200 μmol/l). Non-treated islets were used as a negative control (gray line). The measure of 6-NBDG uptake was determined by flow cytometry, showing a representative analysis of 3 independent experiments (PDF 82 kb)
ESM Table 1
(PDF 63 kb)
Rights and permissions
About this article
Cite this article
Jiménez-González, M., Jaques, F., Rodríguez, S. et al. Cardiotrophin 1 protects beta cells from apoptosis and prevents streptozotocin-induced diabetes in a mouse model. Diabetologia 56, 838–846 (2013). https://doi.org/10.1007/s00125-012-2822-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2822-8