
Abstract
Aims/hypothesis
Downregulation of levels of endothelial progenitor cells (EPCs) during in-vitro short-term exposure to high glucose concentrations relates to reduced activity of silent information regulator 1 (SIRT1) and increased synthesis of platelet-activating factor (PAF). We investigated the possible relationship between PAF and SIRT1 pathways in EPCs during altered glucose homeostasis.
Methods
SIRT1 and PAF receptor (PAF-R) levels were determined by western blot, RT-PCR and confocal laser-scanning microscopy. In-vivo experiments were performed on 48 type 2 diabetic patients (25 with poor glycaemic control and 23 with good glycaemic control) and 20 control individuals. In-vitro experiments with the PAF-R antagonist CV3988 were performed on EPCs isolated from leucocyte-rich buffy coat of healthy human donors.
Results
Decreased SIRT1 protein levels were observed in EPCs from type 2 diabetic patients compared with control individuals (p < 0.01). Notably, the SIRT1 level was consistently lower in patients with poor glycaemic control than in those with good glycaemic control (p < 0.01). Diabetic patients also showed an upregulation of PAF-Rs; this response occurred to a greater extent in individuals with poor glycaemic control than in those with good glycaemic control. In-vitro experiments confirmed that EPCs respond to PAF stimulation with decreased SIRT1 protein and SIRT1 mRNA levels. Moreover, reduction of SIRT1 levels and activity were abolished by CV3988.
Conclusions/interpretation
These findings unveil a link between PAF and SIRT1 pathways in EPCs that contributes to the deleterious effect of hyperglycaemia on the functional properties of EPCs, crucial in diabetes and peripheral vascular complications.
Similar content being viewed by others


Introduction
Endothelial progenitor cells (EPCs), which comprise diverse subsets of cells, control neovascularisation and vascular repair [1, 2]. The therapeutic application of these cells improves the recovery of blood flow after ischaemia and atherosclerosis, suggesting that EPCs are active players in maintaining a healthy cardiovascular system [1, 2]. EPC reduction is considered a route through which risk factors negatively affect cardiovascular function. The loss of the endothelium modulation is a critical initiating factor in the development of diabetic vascular disease [3]. In-vitro and in-vivo studies have demonstrated that glucose toxicity in hyperglycaemia is associated with impaired EPC number and functional capacity [3]; as a result, a reduction in the endothelial repairing ability of the EPC ensues [3]. Indeed, the number of circulating EPCs, in association with an impaired proliferation and mobilisation, decreases in patients with type 1 and type 2 diabetes [4–7] and in patients with severe peripheral vascular complications of type 2 diabetes [8]. In addition, hyperglycaemia enhances senescence and reduces the tube-forming ability of early and late EPCs from adult peripheral blood [9]. An additional cause of the development of vascular disease in diabetes can be ascribed to an impaired secretion by late EPCs of vasoactive substances such as nitric oxide (NO), plasminogen activator inhibitor-1 (PAI-1), tissue plasminogen activator (t-PA), prostaglandin I2 (PGI2), and vascular endothelial growth factor (VEGF) [10]. It is also likely that either the low miR-126 level that occurs as a result of hyperglycaemia [11] or the increased production of advanced glycation end-products (AGEs) impairs the functional properties of EPCs through inhibition of the PI3K/Akt/eNOS pathway [12–14].
EPCs release paracrine factors promoting angiogenesis but, at the same time, also release a combination of detrimental factors that can affect the proinflammatory environment in the vasculature [15, 16]. Indeed, the paracrine release of proinflammatory mediators and the expression of proinflammatory adhesion molecules are augmented by TNF-α [15]. EPCs respond to the prolonged stimulation by TNF-α with a marked reduction in their number [17] and an increased cellular senescence [16], both regulated by a common mechanism mediated by the p38 MAPK pathway [16–18]. Early outgrown EPCs, which release platelet-activating factor (1-O-alkyl-2-acetyl-sn-glycero-3-phosphocholine, PAF) upon stimulation with TNF-α or high glucose concentration, respond to PAF signalling via a transient increase in cytoplasmic Ca2+ concentration since they express PAF-receptors (PAF-Rs) [19]. Thus, early outgrown EPCs might be directly involved in the pathological inflammatory processes as proinflammatory stimuli, such as TNF-α or high glucose, activate the PAF biosynthetic route [20, 21] and reduce the EPC’s ability to hydrolyse PAF [20, 21].
Another critical modulator of EPC dysfunction during alteration in glucose metabolism is the silent information regulator 1 (SIRT1), known to mediate many of the effects of calorie restriction on metabolic pathways of organisms and lifespan [22–24]. In mammals, SIRT1 deacetylates many key transcription factors and cofactors, such as the tumour suppressor p53, forkhead box class O (FOXO) proteins, peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) and nuclear factor-κB (NF-κB) [25]. Downregulation of EPC levels during short-term exposure to a high concentration of glucose relates to the reduced SIRT1 activity with a concomitant increase in acetyl-FoxO1 protein levels [26]. The link between SIRT1 and glucose homeostasis is substantiated by the SIRT1 downregulation observed in peripheral blood mononuclear cells (PBMCs) of individuals with impaired glucose tolerance, compared with individuals in whom glucose regulation is normal [27]. The regulation exerted by SIRT1 over the NF-κB system, which belongs to a pleiotropic transcription factor family with a well known role in inflammation, plays a pivotal role in suppressing inflammation in many tissues [28–30].
However, the underlying molecular mechanisms by which SIRT1 controls inflammatory processes have not been completely clarified. Despite this considerable amount of data, no information is available on the relationships between PAF and SIRT1 signalling in human EPCs in the presence of hyperglycaemia. Specifically, there is no direct demonstration that activation of PAF-Rs by PAF produced during exposure of EPCs to high glucose levels can alter SIRT1 signalling.
Therefore, this study was undertaken to test the hypothesis of a possible relationship between PAF and SIRT1 signalling pathways in EPCs during altered glucose homeostasis. To this end, we assessed the phenotype and number of circulating early outgrown EPCs (CD34+/KDR+ and CD34+/CD133+) by flow cytometry analysis in individuals with type 2 diabetes (one group with poor glycaemic control and another group with good glycaemic control) and determined SIRT1 and PAF-R protein levels. Furthermore, a set of in-vitro experiments on early outgrown EPCs isolated from PBMCs of human healthy volunteers was devised to directly test the response of EPCs to PAF stimulation.
Methods
Participants
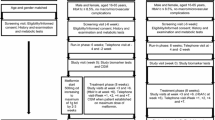
The study was approved by the ethics committee and is in accordance with the Helsinki Declaration of 1975. All participants in the study had given written informed consent before taking part. The study enrolled 48 patients with type 2 diabetes (23 men, 25 women) who attended the diabetic clinic during the period from March to January 2011. Their ages ranged from 41 to 76 years (mean ± SD, 60 ± 10 years) and the duration of diabetes varied from 1 to 15 years (mean ± SD, 10 ± 4.5 years). Clinical history and medications were evaluated together with fasting blood glucose (FBG) and HbA1c. Patients were divided into two groups according to criteria established by the ADA [31]; the first group had good glycaemic control (FBG ≤ 7 mmol/l and HbA1c ≤ 7.0% [53 mmol/mol]) and the second group had poor glycaemic control (FBG > 7 mmol/l and HbA1c > 7.0% [53 mmol/mol]). Patients with coronary artery disease, cerebrovascular disease, peripheral vascular disease, chronic inflammation and malignant disease were excluded. There were no significant differences in age, BMI, duration of diabetes, blood pressure or lipid profiles between diabetic patients with good and poor glycaemic control. Twenty-four healthy medical personnel served as controls (9 men, 15 women). Individuals in the control group were selected within the same age range as diabetic patients to avoid any age-related effect on EPC number. The ages ranged from 40 to 77 years (mean ± SD, 59 ± 10 years). None of the control individuals had a clinical history of diabetes or hyperlipidaemia and all had a normal FBG of <7 mmol/l. They had normal physical examination and had not received any medication.
Isolation and culture of EPCs
Fifteen millilitres of heparinised blood from type 2 diabetic patients and control individuals was used to isolate early outgrown EPCs, which are highly angiogenic and express myeloid lineage [17, 19, 26].To this end, PBMCs were isolated by density-gradient centrifugation on Histopaque-1077 (Sigma, St Louis, MO, USA) (400 g for 40 min at 4°C) according to the manufacturer’s protocol. After centrifugation, the interface cells were carefully removed and transferred to a new conical tube. Cells were washed twice with PBS, centrifuged at 300 g for 10 min at 4°C and then suspended in 9 ml of H2O and 3 ml KCl 0.6 mol/l to a final volume of 50 ml of PBS (1×). To isolate early EPCs, PBMCs were plated on fibronectin-coated culture dishes (5 × 106 cells/ml medium) and maintained in complete endothelial basal medium-2 (EBM-2) (Lonza Cologne, Walkersville, MD, USA). The early EPCs, obtained from the short-term culture (3–7 days), were prepared after 3 days of culture at 37°C with 5% CO2 in a humidified atmosphere. To this end, after 3 days’ culture in complete EBM-2, non-adherent cells were removed by washing with HBSS–10 mmol/l HEPES and adherent early EPC cells were used for the experiments on day 4 [17, 19, 26].
For in-vitro studies, early EPCs were isolated by density-gradient centrifugation of 15 ml of leucocyte-rich buffy coat on Histopaque-1077 (400 g for 40 min at 4°C), as previously described [17, 19, 26].
EPC phenotypic characterisation
Early EPCs from peripheral blood were quantified as described in detail in the electronic supplementary material (ESM) Methods section. Briefly, flow cytometry analysis was carried out on whole blood without any enrichment procedure to avoid enrichment artefacts. Blood (100 μl) was transferred into polypropylene test tubes and incubated with 20 μl FcR-blocking reagent (Miltenyi Biotech, Bologna, Italy) for 20 min at room temperature to avoid non-specific antibody binding. All staining procedures were performed on ice. Samples were incubated with 20 μl of CD34-FITC (Miltenyi Biotech) and 10 μl of KDR-PE (FAB 357P; R&D System, Minneapolis, MN, USA) or 20 μl of CD34-FITC and 20 μl of CD133-PE (Miltenyi Biotech) in the dark for 30 min. After erythrocyte lysis, the pellet was resuspended in 1 ml PBS for immediate FACS analysis. CD34+/KDR+ and CD34+/CD133+ cells isolated from leucocyte-rich buffy coat of healthy human donors were quantified by FACS analysis as previously described [19, 26]. Early EPCs were also characterised and counted after 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine-labelled acetylated LDL (Dil-Ac-LDL) (Biomedical Technologies, Stoughton, MA, USA) and lectin (Ulex europaeus) staining (Sigma), as previously described [17, 19, 26].
In-vitro EPC treatment
Isolated PBMCs (5 × 106 cells/ml medium) were incubated with different concentrations of PAF (up to 50 ng/ml) (Cayman Chemical Company, Ann Arbor, MI, USA) for 6 h at 37°C in serum-free media. Cells were then washed twice with HBSS–10 mmol/l HEPES and cultured for 3 days in complete EBM-2 media [19, 26]. When a PAF-R antagonist was used, cells were pre-treated with CV3988 (10 μmol/l) (Sigma) for 2 h at 37°C. After 2 h the PAF-R antagonist was removed and cells were washed twice with HBSS–10 mmol/l HEPES before treatment with PAF for 6 h in serum-free media. Control cells were cultured for 6 h in serum-free media and in basal conditions up to 3 days. Short-term exposure to high glucose concentration (15 mmol/l) was performed as previously described [19, 26]. Briefly, EPCs from total PBMCs were incubated with glucose (15 mmol/l) for 3 days. When treated with SIRT1 modulators, EPC were pre-treated with resveratrol (29 nmol/l) and nicotinamide (500 μmol/l) before successive treatments.
Confocal laser-scanning microscopy
Confocal microscope analysis of early EPCs was performed on cells cultured for 3 days in 24-well plate containing a fibronectin-coated microscope microglass, as described [19, 26] and reported in detail in the ESM Methods section.
Western blot analysis
Nuclear and cytosolic extracts were made as previously described [19, 26] and reported in detail in the ESM Methods section. Nitrocellulose membranes were incubated overnight at 4°C with antibody against SIRT1 (Abcam, Cambridge, UK) or PAF-R (Cayman Chemical Company) [19, 26].
RNA extraction and RT-PCR
Total RNAs were extracted from cultured EPCs using Trizol solution (Invitrogen Life Technologies Italia, Monza, Italy), according to the manufacturer’s instructions. Primer sequences and corresponding PCR conditions are shown in ESM Table 1 and ESM Methods section.
SIRT1 activity assay
Deacetylation activity of SIRT1 was measured using the Fluor de Lys kit (AK-555; Biomol, Enzo Life Sciences, Lausen, Switzerland) following the manufacturer’s protocol. Fluorescence was measured by excitation at 360 nm and emission at 460 nm and enzymatic activity was expressed in relative fluorescence units as previously described [26].
Statistical analysis
Data are given as mean ± SEM. Differences were assessed by t test and p < 0.05 was considered to be significant. For comparison, unpaired Student’s t tests or ANOVA tests were used. The relationship was calculated using Spearman’s correlation coefficient.
Results
Patient characteristics and levels of circulating CD34 + /KDR + progenitor cells
The characteristics of type 2 diabetic patients are given in Table 1. Anthropometric and clinical data were not different between the two study groups. Basal gluco-metabolic data (HbA1c, fasting glucose and postprandial glucose) were significantly higher in the group of patients with poor glycaemic control. Circulating EPCs, enumerated using FACS analysis, were identified as cells with a low side scatter (gated population) expressing CD34+KDR+ (Fig. 1a, b). EPC counts were expressed as percentage of CD34+KDR+ cells in the gated population. In agreement with previous studies [32], the level of CD34+KDR+ cells was significantly reduced in type 2 diabetic patients compared with healthy controls (p < 0.01). Notably, the percentage of circulating EPCs in diabetic patients with good glycaemic control was significantly higher than in patients with poor glycaemic control (0.23 ± 0.013 vs 0.15 ± 0.01, p < 0.05), but still significantly lower than in healthy control individuals (0.23 ± 0.013 vs 0.36 ± 0.01, p < 0.01) (Fig. 1a, b). FACS counting of the CD34+/CD133+ cells in the gated population (Fig. 1c) and the microscope counting of the merge DiLDL/lectin-positive cells (Fig. 1d) showed the same results. In diabetic patients, Spearman’s correlation analysis revealed that the number of circulating EPCs was inversely correlated with the concentrations of both FBG (r = 0.36, p < 0.05) and HbA1c (r = 0.41, p < 0.05). Correlation analyses using continuous data showed that the EPC number (CD34+/KDR+) was inversely correlated with the FBG (r = 0.35) and HbA1c (r = 0.41) (p < 0.05) (Fig. 2a, b).

FACS computed and microscope counting of EPCs in patients with type 2 diabetes with poor (n = 25) and good glycaemic control (n = 23). (a) Characterisation and counting of circulating EPCs by FACS analysis of the co-production of CD34/KDR (VEGFR2). (b) Representative FACS plots and gating profile set on the basis of the isotype control sample as used for the quantification of CD34+KDR+ cells. (c) FACS characterisation and counting of CD34+/CD133+ progenitor cell population. (d) EPC characterisation by microscope analysis for double positivity to DiLDL and lectin staining. Data are means ± SEM. **p < 0.01 vs control (n = 20); † p < 0.05 vs good glycaemic group (n = 23)

Correlation analyses of EPC levels in patients with type 2 diabetes. EPC number (CD34+/KDR+) correlation with continuous FBG (r = 0.3572) (a) and HbA1c values (r = 0.4082) (b). To convert values for HbA1c in % into mmol/mol, subtract 2.15 and multiply by 10.929
SIRT1 and PAF-R levels in early EPCs from patients with type 2 diabetes
We then looked at the differences in SIRT1 and PAF-R levels by western blot analysis and confocal laser-scanning microscopy. Results indicated that both groups of patients with type 2 diabetes (good glycaemic control and poor glycaemic control) showed a downregulation of SIRT1 levels compared with control individuals (p < 0.01) (Fig. 3a–c). However, interestingly, the fluorescence intensity of SIRT1 in diabetic patients with poor glycaemic control was significantly lower than that in those with good glycaemic control (29 ± 3.05 arbitrary fluorescence units [AFU] vs 66 ± 3.21, p < 0.01 vs control and p < 0.05 vs good glycaemic control) (Fig. 3a, b). Similarly, the protein levels detected by western blot analysis were 49 ± 8.3% of control arbitrary units (AU) in poor glycaemic control (p < 0.01 vs control and p < 0.05 vs good glycaemic control) and 80 ± 4.2% of AU in good glycaemic control (p < 0.01 vs control) (Fig. 3c). In contrast to SIRT1, an upregulation of PAF-R protein levels in early EPCs was observed in patients with type 2 diabetes (Fig. 4). Patients with poor glycaemic control showed a higher PAF-R fluorescence intensity (45 ± 3.01 AFU vs 21.8 ± 3.2 in control individuals, p < 0.01) than that observed in patients with good glycaemic control (34 ± 4.5 AFU vs 21.8 ± 3.2 in control individuals, p < 0.01) (Fig. 4a, b). Analysis of PAF-R protein levels by western blot showed a similar result (Fig. 4c).

SIRT1 levels and activity in EPCs from patients with type 2 diabetes. (a) Representative confocal images of EPCs incubated with specific antibodies against vimentin (green) and SIRT1 (red). Secondary antibodies were Alexa Fluor 488 and Alexa Fluor 633. (b) Immunofluorescence analysis of SIRT1 and vimentin protein levels. The fluorescence intensity was calculated with ImageJ software analysis and expressed as AFU. The mean (n = 6) of the unspecific fluorescence of the secondary antibodies was calculated with ImageJ software and subtracted. (c) Bar graph and representative image of the western blot analysis of SIRT1 protein (lane 1, control individual; lane 2, diabetic patient with good glycaemic control; lane 3, diabetic patient with poor glycaemic control). Data are means ± SEM. *p < 0.05 vs control (n = 20); **p < 0.01 vs control; † p < 0.05 vs good glycaemic group (n = 23)

PAF-R levels in EPCs from patients with type 2 diabetes. (a) Representative confocal images were performed using a Zeiss LSM 510 confocal microscope. (b) Immunofluorescence analysis of PAF-R (red) and vimentin (green) proteins. Bar graph shows AFU for PAF-R calculated with ImageJ software. (c) Bar graph and representative image of the western blot analysis of PAF-R levels (lane 1, control individuals; lane 2, diabetic patient with good glycaemic control; lane 3, diabetic patient with poor glycaemic control). Data are means ± SEM. **p < 0.01 vs control (n = 20); † p < 0.05 vs good glycaemic group (n = 23)
SIRT1 levels and activity in PAF-treated EPCs
Considering the in-vivo observations, we hypothesised that PAF might be involved in the cellular regulation of SIRT1. Therefore, we investigated the in-vitro effects of PAF on SIRT1 levels and activity in early EPCs from leucocyte-rich buffy coat of human healthy donors. To this end, PBMCs were pre-incubated for 6 h with PAF in serum-free media and then, after complete PAF removal, cultured for 3 days in complete culture media [19]. Results indicated that SIRT1 levels and activity are modulated by PAF in a time-dependent manner (Fig. 5a–d). SIRT1 protein and SIRT1 mRNA levels decreased in a dose-dependent manner upon PAF stimulation with a significant reduction at a PAF concentration of 50 ng/ml (p < 0.01 vs control) (Fig. 5a, b). Specifically, western blot analysis revealed that SIRT1 levels detected in EPC treated with PAF 50 ng/ml was 47% of the control AU (Fig. 5a). An even more consistent decrease in SIRT1 mRNA level was observed upon treatment with PAF 50 ng/ml (21% of the control) (Fig. 5b) (p < 0.01). Similarly, SIRT1 activity decreased from 750 ± 35 (AFU/μg protein) in control cells to 420 ± 34 (AFU/μg protein) in PAF (50 ng/ml)-treated cells (p < 0.01) (Fig. 5c). When nicotinamide and resveratrol were used as negative and positive controls [26], respectively, results indicated that SIRT1 activity of control untreated EPCs (750 ± 35 AFU/μg protein) was inhibited by in-vitro incubation with nicotinamide (185 ± 12 AFU/μg protein) (p < 0.05 vs control cells) and stimulated by resveratrol (1,925 ± 162 AFU/μg protein) (p < 0.01 vs control cells). Moreover, when treated with PAF 50 ng/ml, the fluorescence intensity of SIRT1 (red), assessed by immunocytochemistry against vimentin (green), was about 2.2-fold lower than untreated cells (31.2 ± 1.9 AFU vs 68.3 ± 4.5 AFU control cells) (p < 0.01 vs control) (Fig. 5d, e). The subtracted fluorescence intensity of the negative controls for SIRT1 secondary antibody (Alexa 488) was 6.2 ± 0.015 AFU.

Effect of PAF on SIRT1 levels and activity in early EPCs. Isolated PBMCs (5 × 106 cells/ml medium) were incubated with different concentrations of PAF (0–50 ng/ml) for 6 h at 37°C in serum-free media. Washed cells were then cultured for 3 days in complete media. (a) SIRT1 protein levels determined by western blot analysis after treatment with different concentration of PAF. Inset shows a representative image of western blot analysis (lane 1, absence of PAF; lane 2, PAF 5 ng/ml; lane 3, PAF 10 ng/ml; lane 4, PAF 50 ng/ml). (b) SIRT1 mRNA levels determined by RT-PCR (lane 1, absence of PAF; lane 2, PAF 5 ng/ml; lane 3, PAF 10 ng/ml; lane 4, PAF 50 ng/ml). (c) SIRT1 activity determined by Fluor de Lys enzyme assay as described under Methods section. (d) Bar graph of the SIRT1 levels determined by confocal laser-scanning microscopy. (e) Representative confocal images of SIRT1 (red) and vimentin (green). Data are means ± SEM (n = 6); **p < 0.01 vs control (absence of PAF)
Effect of high glucose concentration and PAF-R inhibitor on SIRT1
Short-term exposure of early EPCs to high glucose concentration induces downregulation of SIRT1 and activates PAF synthesis via the remodelling biosynthetic route [19, 26]. Furthermore, both high-glucose exposure and in-vitro treatment with PAF at a concentration of 50 ng/ml exert a detrimental effect on early EPC number [19, 26]. Based on these previous data, to evaluate whether the detrimental effect of high glucose concentration on SIRT1 and EPC number occurs through PAF signalling, we investigated the in-vitro effects of high glucose concentration in the presence and absence of CV3988, a PAF-receptor inhibitor. As shown in Fig. 6a, the inhibition of SIRT1 activity by treatment with a high concentration of glucose or with PAF was prevented by CV3988. Indeed, the PAF-induced inhibition of SIRT1 activity (350 ± 25 AFU/μg protein vs 690 ± 31 AFU/μg protein in control cells, p < 0.01) was almost completely abolished by CV3988 pre-treatment (580 ± 45 AFU/μg protein vs 690 ± 31 AFU/μg protein in control cells) (Fig. 6a). Similarly, the inhibition of SIRT1 activity in high-glucose-treated cells (390 ± 22 AFU/μg protein vs 690 ± 31 AFU/μg protein in control, p < 0.01) was also blocked by CV3988 pre-treatment (590 ± 29 AFU/μg protein vs 690 ± 31 AFU/μg protein) (Fig. 6a). Notably, SIRT1 protein level decreased after treatment with PAF or a high concentration of glucose (p < 0.01) (Fig. 6b). The SIRT1 protein level detected in EPCs treated with a high concentration of glucose was 55% that of the control AU (p < 0.01) and this returned to near basal level when early EPCs were pre-treated with CV3988 before high-glucose stimulation. Similarly, pre-treatment with CV3988 also prevented PAF-induced SIRT1 downregulation. The SIRT1 protein level was 41% (% of control AU, p < 0.01) in PAF-treated cells and 72% (% of control AU) in cells pre-treated with CV3988 before PAF stimulation (Fig. 6b).

Effect of PAF-R inhibitor on SIRT1 levels and activity in early EPCs. PBMCs (5 × 106 cells/ml medium) were incubated with PAF (50 ng/ml) for 6 h at 37°C in serum-free media or with media containing a high concentration of glucose (15 mmol/l). When the PAF-R antagonist was used, cells were pre-treated with CV3988 (10 μmol/l) for 2 h at 37°C. After 2 h the PAF-R antagonist was removed by washing the cells twice with HBSS-10 mmol/l HEPES and cells were then exposed to PAF or high-glucose treatment. Control cells were cultured for 6 h in serum-free media and in basal conditions up to 3 days. (a) SIRT1 activity determined by Fluor de Lys enzyme assay. (b) Western blot analysis of SIRT1 protein levels. Inset shows a representative image of western blot analysis (lane 1, control; lane 2, CV3988; lane 3, PAF 50 ng/ml; lane 4, CV3988 + PAF 50 ng/ml; lane 5, high-glucose; lane 6, CV3988 + high-glucose). (c) FACS computed counting of EPCs (CD34+KDR+) expressed as percentage of the number of gated cells. (d) Representative FACS plots and gating profile used for the quantification of CD34+KDR+ cells. Data are means ± SEM (n = 6); **p < 0.01 vs control; † p < 0.05 vs PAF or high-glucose
In agreement with previous studies [19], CD34+/KDR+ FACS computed counting showed a consistent decrease in cell number both in the presence of PAF (0.21 ± 0.005% vs 0.41 ± 0.005% in control, p < 0.01) and in the presence a high concentration of glucose (0.24 ± 0.01% vs 0.41 ± 0.005% in control, p < 0.01) (Fig. 6c, d). The detrimental effect of PAF or high glucose concentration on early EPC number was significantly ameliorated by pre-incubation with CV3988 before PAF or high-glucose treatment (p < 0.05) (Fig. 6c, d).
Discussion
This study offers novel insights into the relationship between the SIRT1 pathway and PAF signalling in early EPCs during hyperglycaemia. Here, we report the downregulation of SIRT1 and upregulation of PAF-R levels in early EPCs from patients with type 2 diabetes and, notably, in more marked fashion in those with poor glycaemic control than those with good glycaemic control.
Reduction in the number and angiogenicity of EPCs contributes to the development of vascular complications in diabetic patients [10, 27, 33, 34]. Indeed, in the pathological conditions characterised by impaired EPC mobilisation, such as diabetes mellitus, the angiogenic response to ischaemia may be compromised by the insufficient supply of EPCs to the ischaemic tissue [8, 35]. In-vitro and in-vivo studies have demonstrated that glucose toxicity, as determined by hyperglycaemia, is associated with impaired EPC proliferation, adhesion, incorporation into vascular structures and secretory activity [4, 8, 10, 36, 37]. Moreover, hyperglycaemia also enhances senescence and reduces the tube-forming ability of early and late EPCs [9]. However, the mechanism contributing to the reduction of circulating EPCs in type 2 diabetes, particularly in individuals with good and poor glycaemic control, has not yet been fully elucidated. Our observations that the EPC levels are significantly higher in diabetic patients with good glycaemic control than in those with poor glycaemic control, suggest that the function of EPCs could be improved by strict control of blood glucose, thereby preventing or ameliorating vascular complications in diabetic patients.
Several cellular processes, including insulin secretion, cell cycle and apoptosis, are strictly regulated by a family of mediators called sirtuins. SIRT1 is a positive regulator of insulin secretion and triggers glucose uptake and use [25]. The actions of SIRT1 may affect cellular pathways involved in ageing and metabolic diseases. In particular, SIRT1 plays an important role in glucose homeostasis, maintained within a narrow range under a variety of physiological conditions [25]. In recent years, it has become evident that SIRT1 is a key player of the EPC dysfunction in insulin resistance and metabolic syndrome [10, 25–27]. Indeed, a high glucose concentration impairs nicotinamide phosphoribosyltransferase (NAMPT) function and promotes production of reactive oxygen species, which may themselves reduce SIRT1 levels. SIRT1 downregulation is also responsible for high p53 acetylation and Jun NH2-terminal kinase (JNK) activation, which are related to monocyte activation and inflammation [27].
Importantly, in-vitro and in-vivo studies demonstrate that the reduction in the number of EPCs in the presence of hyperglycaemia or inflammatory stimuli plays a relevant role in lowering SIRT1 levels and activity [26, 27, 38]. Moreover, proteome and transcriptome profiles of EPCs exposed to high glucose concentrations also revealed changes in levels of proteins and genes involved in cell metabolism, the cell cycle and oxidative stress [26]. In particular, several proteins involved in the SIRT1 signalling cascade, such as SENP1, p53, NAMPT and RelA/p65, and their related genes, were found to be altered in high-glucose-treated EPCs [26]. An increased activation of the RelA/p65 subunit of NF-κB and release of proinflammatory cytokines are associated with decreased SIRT1 levels in monocyte–macrophage cell line [28]. To date, consistent knowledge is available about the multiple implication of SIRT1 in inflammation [25, 39].
In this paper, the involvement of the SIRT1 pathway in glucose homeostasis is substantiated by in-vitro studies showing the first evidence of a link between SIRT1 and PAF signalling in the pathogenic pathway of EPC glucose toxicity. Interestingly, the detrimental effects on EPC levels of direct in-vitro treatment with PAF or exposure to a high concentration of glucose, both associated with the downregulation of SIRT1, are abolished by the PAF-R antagonist, CV3988. EPCs release PAF, chemokines and thrombo-inflammatory mediators [15, 19]. In EPCs, the paracrine release of proinflammatory mediators and the expression of proinflammatory adhesion molecules are augmented by TNF-α [16], which, along with high glucose concentration, stimulates the biosynthesis of PAF through the activation of acetyl coenzyme A:lysoPAF acetyltransferase, a membrane-bound enzyme of the PAF biosynthetic remodelling route [20]. High glucose concentration also inhibits the PAF-acetylhydrolase (PAF-AH) activity in EPCs [19]. This enzyme either hydrolyses PAF (AH activity) or transfers acetyl groups from PAF to lysophospholipids (transacetylase activity) [40]. Furthermore, besides the downregulation of PAF catabolic enzyme genes (PAF-AH and PAFAH1B1) there is also an upregulation of genes involved in the production of phospholipase A2, which is a key enzyme involved in PAF biosynthesis as it produces lyso-PAF necessary for PAF production [19].
Our data show that SIRT1 protein levels and activity are decreased in individuals with type 2 diabetes to a higher extent in those with poor glycaemic control than in those with good glycaemic control. In patients with type 2 diabetes in whom glycaemic control is poor, a higher level of PAF-R protein is also observed. Obviously, differences might occur between the in-vitro biological regulation of SIRT1 in EPCs isolated from leucocyte-rich buffy coat of healthy individuals and that observed in patients with type 2 diabetes. Indeed, multiple direct or indirect regulatory mechanisms, not approached in this study, can take part in the complex signal transduction pathway of SIRT1 in glucose homeostasis and inflammation. Nevertheless, we unveil a link between SIRT1 and PAF signalling in the pathogenic pathway of glucose toxicity in EPCs. A major question is whether SIRT1 activation is a safe therapy for human diseases such as type 2 diabetes. However, the pathways involved in type 2 diabetes that are regulated by SIRT1 are very complex given the presence of various crucial intermediaries in metabolic tissues, which can be targeted for future prospects [41]. To date, the pharmacological search for active small SIRT1 inhibitors or activators is underway but the in-vivo effects of such compounds have not been fully established due to the intricacy of the regulatory mechanism and the multiple and overlapping biochemical pathways involved [25].
In conclusion, the results of this study confirm that SIRT1 is a regulator of EPC levels during altered glucose homeostasis and suggest that this pathway could be influenced by PAF/PAF-R signalling. Although the role of the PAF/PAF-R signalling cascade in the modulation of SIRT1 activity needs to be clearly defined with further studies, these findings provide further insight into the mechanisms by which EPCs exhibit impaired vascular repair and abnormal neovasculogenesis in diabetic patients.
Abbreviations
- AGEs:
-
Advanced glycation end-products
- AFU:
-
Arbitrary fluorescence units
- AH:
-
Acetylhydrolase
- AU:
-
Arbitrary units
- EBM-2:
-
Endothelial basal medium-2
- eNOS:
-
Endothelial nitric oxide synthase
- EPC:
-
Endothelial progenitor cell
- FBG:
-
Fasting blood glucose
- FOXO:
-
Forkhead box class O
- JNK:
-
Jun NH2-terminal kinase
- HBSS:
-
Hanks’ Balanced Salt Solution
- MAPK:
-
Mitogen activated protein kinase
- miR-126:
-
MicroRNA-126
- NAMPT:
-
Nicotinamide phosphoribosyltransferase
- NF-κB:
-
Nuclear factor-κB
- NO:
-
Nitric oxide
- PAF:
-
Platelet-activating factor
- PAF-R:
-
Platelet-activating factor receptor
- PAI-1:
-
Plasminogen activator inhibitor-1
- PBMC:
-
Peripheral blood mononuclear cell
- PGC-1α:
-
Peroxisome proliferator-activated receptor-γ coactivator-1α
- PGI2:
-
Prostaglandin I2
- PI3K:
-
Phosphatidylinositol 3-kinases
- SIRT1:
-
Silent information regulator 1
- t-PA:
-
Tissue plasminogen activator
- VEGF:
-
Vascular endothelial growth factor
References
Fadini GP, Losordo D, Dimmeler S (2012) Critical reevaluation of endothelial progenitor cell phenotypes for therapeutic and diagnostic use. Circ Res 110:624–637
Napoli C, William-Ignarro S, Byrns R et al (2008) Therapeutic targeting of the stem cell niche in experimental hindlimb ischemia. Nat Clin Pract Cardiovasc Med 5:571–579
Jarajapu YP, Grant MB (2010) The promise of cell-based therapies for diabetic complications: challenges and solutions. Circ Res 106:854–869
Tepper OM, Galiano RD, Capla JM et al (2002) Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation 106:2781–2786
Fadini GP, Boscaro E, de Kreutzenberg S et al (2010) Time course and mechanisms of circulating progenitor cell reduction in the natural history of type 2 diabetes mellitus. Diabetes Care 33:1097–1102
Loomans CJ, de Koning EJ, Staal FJ et al (2004) Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes 53:195–199
Liguori A, Fiorito C, Balestrieri ML et al (2008) Functional impairment of hematopoietic progenitor cells in patients with coronary heart disease. Eur J Haematol 80:258–264
Fadini GP, Miorin M, Facco M et al (2005) Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol 45:1449–1457
Chen YH, Lin SJ, Lin FY et al (2007) High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide-related but not oxidative stress-mediated mechanisms. Diabetes 56:1559–1568
Zhang J, Zhang X, Li H et al (2012) Hyperglycaemia exerts deleterious effects on late endothelial progenitor cell secretion actions. Diabetes Vasc Dis Res. doi:10.1177/1479164112444639
Meng S, Cao JT, Zhang B, Zhou Q, Shen CX, Wang CQ (2012) Downregulation of microRNA-126 in endothelial progenitor cells from diabetes patients, impairs their functional properties, via target gene Spred-1. J Mol Cell Cardiol 53:64–72
Li H, Zhang X, Guan X et al (2012) Advanced glycation end products impair the migration, adhesion and secretion potentials of late endothelial progenitor cells. Cardiovasc Diabetol 11:46
Chen J, Song M, Yu S et al (2010) Advanced glycation endproducts alter functions and promote apoptosis in endothelial progenitor cells through receptor for advanced glycation endproducts mediate overpression of cell oxidant stress. Mol Cell Biochem 335:137–146
Chen Q, Dong L, Wang L, Kang L, Xu B (2009) Advanced glycation end products impair function of late endothelial progenitor cells through effects on protein kinase Akt and cyclooxygenase-2. Biochem Biophys Res Commun 381:192–197
Zhang Y, Ingram DA, Murphy MP et al (2009) Release of proinflammatory mediators and expression of proinflammatory adhesion molecules by endothelial progenitor cells. Am J Physiol Heart Circ Physiol 296:H1675–H1682
Zhang Y, Herbert BS, Rajashekhar G et al (2009) Premature senescence of highly proliferative endothelial progenitor cells is induced by tumor necrosis factor-alpha via the p38 mitogen-activated protein kinase pathway. FASEB J 23:1358–1365
Seeger FH, Haendeler J, Walter DH et al (2005) p38 mitogen-activated protein kinase downregulates endothelial progenitor cells. Circulation 111:1184–1191
Balestrieri ML, Schiano C, Felice F et al (2008) Effect of low doses of red wine and pure resveratrol on circulating endothelial progenitor cells. J Biochem 143:179–186
Balestrieri ML, Giovane A, Milone L, Servillo L (2010) Endothelial progenitor cells express PAF receptor and respond to PAF via Ca(2+)-dependent signaling. Biochim Biophys Acta 1801:1123–1132
Balestrieri ML, Castaldo D, Balestrieri C, Quagliuolo L, Giovane A, Servillo L (2003) Modulation by flavonoids of PAF and related phospholipids in endothelial cells during oxidative stress. J Lipid Res 44:380–387
Balestrieri ML, Servillo L, Lee T (1997) The role of platelet-activating factor-dependent transacetylase in the biosynthesis of 1-acyl-2-acetyl-sn-glycero-3-phosphocholine by stimulated endothelial cells. J Biol Chem 272:17431–17437
Guarente L, Picard F (2005) Calorie restriction-the SIR2 connection. Cell 120:473–482
Brunet A, Sweeney LB, Sturgill JF et al (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015
Lemarié CA, ShbAt L, Marchesi C et al (2011) Mthfr deficiency induces endothelial progenitor cell senescence via uncoupling of eNOS and downregulation of SIRT1. Am J Physiol Heart Circ Physiol 300:H745–H753
Sanchez-Fidalgo S, Villegas I, Sanchez-Hidalgo M, de la Lastra CA (2012) Sirtuin modulators: mechanisms and potential clinical implications. Curr Med Chem 19:2414–2441
Balestrieri ML, Rienzo M, Felice F et al (2008) High glucose downregulates endothelial progenitor cell number via SIRT1. Biochim Biophys Acta 1784:936–945
de Kreutzenberg SV, Ceolotto G, Papparella I et al (2010) Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes 59:1006–1015
Yang SR, Wright J, Bauter M, Seweryniak K, Kode A, Rahman I (2007) Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am J Physiol Lung Cell Mol Physiol 292:L567–L576
Yang Z, Kahn BB, Shi H, Xue BZ (2010) Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem 285:19051–19059
Pfluger PT, Herranz D, Velasco-Miguel S, Serrano M, Tschöp MH (2008) Sirt1 protects against high-fat diet-induced metabolic damage. Proc Natl Acad Sci U S A 105:9793–9798
American Diabetes Association: Clinical practice recommendation (2011) Diabetes Care 25 (Suppl 1):S1–S147
van Ark J, Moser J, Lexis CP et al (2012) Type 2 diabetes mellitus is associated with an imbalance in circulating endothelial and smooth muscle progenitor cell numbers. Diabetologia 55:2501–2512
Krankel N, Adams V, Linke A et al (2005) Hyperglycemia reduces survival and impairs function of circulating blood-derived progenitor cells. Arterioscler Thromb Vasc Biol 25:698–703
Sica V, Williams-Ignarro S, de Nigris F et al (2006) Autologous bone marrow cell therapy and metabolic intervention in ischemia-induced angiogenesis in the diabetic mouse hindlimb. Cell Cycle 5:2903–2908
Fadini GP (2011) Is bone marrow another target of diabetic complications? Eur J Clin Invest 41:457–463
Humpert PM, Neuwirth R, Battista MJ et al (2005) Sdf-1 genotype influences insulin-dependent mobilization of adult progenitor cells in type 2 diabetes. Diabetes Care 28:934–936
Balestrieri ML, Balestrieri A, Mancini FP, Napoli C (2008) Understanding the immunoangiostatic CXC chemokine network. Cardiovasc Res 78:250–256
Fiorito C, Rienzo M, Crimi E et al (2008) Antioxidants increase number of progenitor endothelial cells through multiple gene expression pathways. Free Radic Res 42:754–762
Yoshizaki T, Schenk S, Imamura T et al (2010) SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab 298:E419–E428
Servillo L, Balestrieri C, Giovane A et al (2006) Lysophospholipid transacetylase in the regulation of PAF levels in human monocytes and macrophages. FASEB J 20:1015–1017
Pulla VK, Battu MB, Alvala M, Sriram D, Yogeeswari P (2012) Can targeting SIRT-1 to treat type 2 diabetes be a good strategy? A review. Expert Opin Ther Targets 16:819–832
Funding
This work was supported by a grant from Ministry of University and Research P.R.I.N./MIUR 2008 code 2008X2PNYX_002 (M. L. Balestrieri).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
MLB and RM were principal investigators and were responsible for the study conception and design. MLB, RM, GP and LS drafted the manuscript. MRR, PP and MB contributed to the collection of patients’ data. AE, NDO, AG and RC contributed to in-vitro experiments and confocal laser-scanning microscopy analysis. All authors were involved in analysis and interpretation of data and contributed to the critical revision of the manuscript. All authors provided final approval of the version to be published.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 164 kb)
ESM Table 1
(PDF 7 kb)
Rights and permissions
About this article
Cite this article
Balestrieri, M.L., Servillo, L., Esposito, A. et al. Poor glycaemic control in type 2 diabetes patients reduces endothelial progenitor cell number by influencing SIRT1 signalling via platelet-activating factor receptor activation. Diabetologia 56, 162–172 (2013). https://doi.org/10.1007/s00125-012-2749-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2749-0