
Abstract
Aims/hypothesis
The fat-derived hormone leptin plays a crucial role in the maintenance of normal body weight and energy expenditure as well as in glucose homeostasis. Recently, it was reported that the liver-derived protein, insulin-like growth factor binding protein-2 (IGFBP-2), is responsible for at least some of the glucose-normalising effects of leptin. However, the exact mechanism by which leptin upregulates IGFBP-2 production is unknown. Since it is believed that circulating IGFBP-2 is predominantly derived from the liver and leptin has been shown to have both direct and indirect actions on the liver, we hypothesised that leptin signalling in hepatocytes or via brain–liver vagal efferents may mediate leptin control of IGFBP-2 production.
Methods
To address our hypothesis, we assessed leptin action on glucose homeostasis and plasma IGFBP-2 levels in both leptin-deficient ob/ob mice with a liver-specific loss of leptin signalling and ob/ob mice with a subdiaphragmatic vagotomy. We also examined whether restoring hepatic leptin signalling in leptin receptor-deficient db/db mice could increase plasma IGFBP-2 levels.
Results
Continuous leptin administration increased plasma IGFBP-2 levels in a dose-dependent manner, in association with reduced plasma glucose and insulin levels. Interestingly, leptin was still able to increase plasma IGFBP-2 levels and improve glucose homeostasis in both ob/ob mouse models to the same extent as their littermate controls. Further, restoration of hepatic leptin signalling in db/db mice did not increase either hepatic or plasma IGFBP-2 levels.
Conclusions/interpretation
Taken together, these data indicate that hepatic leptin signalling and subdiaphragmatic vagal inputs are not required for leptin upregulation of plasma IGFBP-2 nor blood glucose lowering in ob/ob mice.
Similar content being viewed by others


Introduction
IGFs and IGF binding proteins (IGFBPs) play an important role in regulating glucose metabolism [1–3]. IGF-1 specifically has been found to influence blood glucose concentrations directly by stimulating glucose uptake in target cells and indirectly by increasing the sensitivity of tissues to insulin [1]. Effects of IGFs are modulated by IGFBPs through the ability of IGFBPs to sequester IGFs, but interestingly, several reports demonstrate that IGFBPs can activate cell surface receptors directly and stimulate cellular events independently of IGFs [1, 3, 4].
IGFBP-2 is the second most abundant plasma circulating IGFBP, but its physiological role remains poorly understood. Interestingly, mice overexpressing IGFBP-2 under the cytomegalovirus (CMV) promoter have mildly reduced postnatal body weight [5] and IGFBP-2 knockout mice have slightly increased body weight and body fat mass [6]. Moreover, mice overexpressing IGFBP-2 under its native promoter are protected from high-fat-diet-induced obesity and glucose intolerance and display increased insulin sensitivity [2], while IGFBP-2 knockout mice on a chow diet showed no alterations in glucose tolerance [6]. Further, in humans, low levels of IGFBP-2 are correlated with decreased insulin sensitivity and levels are also reduced in the presence of obesity [7]. Overall, there appears to be an inverse association between IGFBP-2 and adiposity-related insulin resistance, but much is still unknown about the exact nature of this relationship.
Leptin has major effects on maintaining normal body weight and glucose metabolism and has been shown to increase hepatic transcription of Igfbp2 when administered either peripherally or centrally to lipodystrophic mice [8]. Further, it was recently demonstrated that peripheral administration of leptin to leptin-deficient ob/ob mice potently upregulates both hepatic Igfbp2 mRNA and circulating IGFBP-2 levels [3]. Moreover, Hedbacker et al. [3] showed that administration of an Igfbp2-expressing adenovirus to ob/ob mice had similar glucose lowering effects as leptin in various mouse models of insulin resistance and insulin-deficient diabetes and these effects were independent of body weight loss. Interestingly, in a small cohort of leptin-deficient people, it was observed that these patients had about 50% lower levels of plasma IGFBP-2 than controls and these levels were increased after 6 months of leptin treatment [3]. These results indicate that leptin is involved in regulating circulating IGFBP-2 levels and that IGFBP-2 may play an intricate role in mediating the effects of leptin on metabolism. However, the exact site and mechanism by which leptin regulates IGFBP-2 production is still unclear.
The liver, which produces the long, signalling isoform of the leptin receptor [9–11], may be an important site for leptin action on increasing plasma IGFBP-2. It has been shown that increased hepatic Igfbp2 mRNA is associated with increased circulating IGFBP-2 [3, 12, 13], suggesting that the liver is a major source of plasma IGFBP-2. Further, subcutaneous administration of leptin to the periphery is able to increase both liver Igfbp2 mRNA [3, 8] and plasma IGFBP-2 levels [3]. Thus, leptin may act directly on the liver to increase production and secretion of IGFBP-2. Intriguingly, however, central intracerebroventricular (ICV) doses of leptin that do not increase peripheral leptin levels are also able to increase hepatic Igfbp2 transcription [8]. Consequently, we hypothesised that leptin may increase hepatic Igfbp2 expression by acting directly on leptin receptors in the liver or by acting on the brain and increasing liver expression of Igfbp2 via parasympathetic brain–liver vagal efferents. To test these hypotheses, we provided peripheral leptin treatment to ob/ob mice lacking hepatic leptin receptors and to ob/ob mice that had been subjected to a subdiaphragmatic vagotomy. In both cases, leptin was still able to reduce hyperglycaemia and hyperinsulinaemia while potently increasing circulating IGFBP-2 levels. Further, when we restored hepatic leptin signalling in db/db mice, which have a global loss of leptin signalling, neither hepatic nor plasma IGFBP-2 levels increased. Therefore, despite evidence suggesting an important role for the liver in mediating leptin-induced IGFBP-2 production, neither hepatic leptin signalling nor subdiaphragmatic vagal efferents are required for leptin to increase IGFBP-2 levels in the plasma of leptin-deficient ob/ob mice.
Methods
Animals
Male C57BL/6 mice (#000664), ob/ob mice (#000632), and db/db mice (#000697) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). For details on the generation of Lepr flox/flox Albcre ob/ob mice, see the electronic supplementary material (ESM). Mice were housed with a 12 h light–12 h dark cycle and had ad libitum access to water and a standard chow diet (#5015 Lab Diet, St Louis, MO, USA). db/db mice were treated intravenously with 1 × 109 plaque-forming units (pfu) of an adenovirus expressing the gene for either the long signalling isoform of the mouse leptin receptor (Ad-Lepr-b, kindly provided by M.G. Myers (University of Michigan) and C.J. Rhodes (University of Chicago) [14]) or β-galactosidase (Ad-β-gal) as a control. Subdiaphragmatic vagotomies were performed by the Jackson Laboratory. All procedures with animals were approved by the University of British Columbia Animal Care Committee and carried out in accordance with the Canadian Council on Animal Care guidelines.
PCR and RT-PCR analysis
Liver tissue was collected from mice and genomic DNA was extracted using DNeasy kits (Qiagen, Mississauga, Canada). RNA was isolated using either an RNeasy kit (Qiagen, Mississauga, ON, Canada) or TRI Reagent (Ambion, Streetsville, ON, Canada). Reverse transcription reactions were performed with a poly T primer using a Superscript First-Strand Synthesis kit (Invitrogen, Burlington, ON, Canada). See ESM for primer sequences.
Subdiaphragmatic vagotomy
Subdiaphragmatic vagotomies or sham surgeries were performed by the Jackson Laboratory in 3.5-week-old ob/ob mice using the ventral abdominal approach. See ESM for a brief description of the procedure.
Implantation of mini-osmotic pumps
Recombinant mouse leptin was from the National Hormone and Peptide Program (Torrance, CA, USA) and was reconstituted with sterile PBS. Mini-osmotic pumps (Alzet, Palo Alto, CA, USA) were loaded with recombinant mouse leptin or PBS (control) and pre-equilibrated according to manufacturer guidelines. The pumps were implanted subcutaneously in isoflurane-anaesthetised mice.
Plasma analyte analysis
Body weight, blood glucose, insulin, leptin and IGFBP-2 were measured following a 4 h fast unless specified otherwise. Blood glucose concentration was measured with a One Touch Ultra Glucometer (Life Scan, Milpitas, CA, USA) from the saphenous vein. Plasma insulin levels were measured using an Ultrasensitive Mouse Insulin ELISA (ALPCO, Salem, MA, USA), leptin levels were measured using a Mouse Leptin ELISA (Crystal Chem, Downers Grove, IL, USA) and circulating IGFBP-2 levels were measured using a Mouse/Rat IGFBP-2 ELISA (ALPCO).
Verification of vagotomy
Eight weeks post-surgery, the completeness of the subdiaphragmatic vagotomy was assessed using previously described methods [15]. The test was based on the satiety effect of cholecystokinin (CCK), which is known to be mediated by the vagus nerve [16]. For details of the procedure, see the ESM.
Statistical analysis
Data are represented as mean ± SEM and were analysed using a Student’s t test. Statistical significance was defined as p ≤ 0.05.
Results
Low leptin levels are able to normalise hyperglycaemia and hyperinsulinaemia independent of weight loss
Osmotic pumps delivering a continuous dose of 0, 0.2, 1 or 5 μg/day leptin were implanted subcutaneously into ob/ob mice for 28 days and body weight (Fig. 1a), fasting blood glucose (Fig. 1b) and fasting insulin levels (Fig. 1c) were monitored. Mice receiving the lowest dose of 0.2 μg/day leptin continued to gain weight at a rate comparable with PBS-treated ob/ob mice. Although this low dose had no effect on body weight gain or fasting insulin levels, an improvement in fasting blood glucose was observed. A higher dose of 1 μg/day leptin administered to the ob/ob mice resulted in no net body weight change and thus the maintenance of obesity. However, even with no weight loss, the 1 μg/day dose also resulted in reduced fasting blood glucose levels, normalising to wild-type levels by day 20 of leptin treatment. Further, fasting insulin levels were normalised to wild-type levels by day 28 of treatment. As expected, the maximal effect was seen at the highest dose of leptin (5 μg/day), which led to a 25% decrease in body weight by day 28 and blood glucose levels comparable with wild-type controls by day 5 of leptin treatment.

Continuous leptin administration in ob/ob mice leads to reversal of hyperglycaemia and hyperinsulinaemia independently of weight loss. ob/ob mice were treated with a continuous infusion of leptin via mini-osmotic pumps for 28 days. Pumps were implanted subcutaneously on day 0 following a 4 h fast. a Per cent change in body weight and (b) blood glucose levels following a 4 h fast were measured over the 28 day treatment period. White circles, 0 μg/day leptin; grey circles, 0.2 μg/day; black circles, 1 μg/day; black triangles, 5 μg/day; white squares, C57BL/6 controls. c Plasma insulin levels were measured on day 27 of leptin treatment. Male ob/ob mice were 7 weeks old at the time of pump implant. C57BL/6 male mice were untreated and age-matched to ob/ob mice. Data are expressed as mean ± SEM, n ≥ 7. *p ≤ 0.05 compared with 0 μg/day controls
Leptin increases plasma IGFBP-2 levels in ob/ob mice in a dose-dependent manner
Continuous leptin administration at doses of 0.2, 1 and 5 μg/day for 28 days resulted in a dose-dependent increase of plasma leptin levels in the leptin-deficient ob/ob mice (Fig. 2a). This increase in leptin resulted in a concomitant rise in plasma IGFBP-2 levels, which also depended on the dose of leptin given (Fig. 2b). All doses were able to increase IGFBP-2 levels significantly above levels found in untreated ob/ob mice, and the highest dose of 5 μg/day for 28 days induced IGFBP-2 levels comparable with those observed in wild-type mice.

Continuous leptin administration in ob/ob mice leads to increased plasma IGFBP-2 levels in a dose-dependent manner. ob/ob mice were treated with a continuous infusion of leptin via mini-osmotic pumps for 28 days. Pumps were implanted subcutaneously on day 0 following a 4 h fast. a Leptin levels and (b) plasma IGFBP-2 levels were measured on day 28 of leptin administration following a 4 h fast. Male ob/ob mice were 7 weeks old at the time of pump implant. C57BL/6 male mice were untreated and age-matched to ob/ob mice. Data are expressed as mean ± SEM, n ≥ 7. *p ≤ 0.05 compared with 0 μg/day controls
Leprflox/flox Albcre+ ob/ob mice have truncated hepatic leptin receptor transcripts
In order to examine whether leptin regulates IGFBP-2 as a result of direct effects on the liver, ob/ob mice with a hepatocyte-specific loss of leptin signalling were generated using a Cre recombinase (Cre)-lox approach. To do this, Lepr flox/flox mice with loxP sites flanking exon 17 were used. When crossed with mice expressing a cre transgene, Lepr flox/flox mice undergo Cre-mediated recombination, resulting in the Lepr ∆17 allele, which generates truncated leptin receptors that lack the signalling domain and are deficient in leptin-mediated signal transducer and activator of transcription 3 (STAT3) phosphorylation [17]. Further, when Lepr flox/flox mice were crossed with heat-shock-Cre line 1 mice, which ubiquitously express Cre, the resulting Lepr ∆17/∆17 mice were obese, hyperphagic, hyperglycaemic, hyperinsulinaemic and sterile, a phenotype reminiscent of leptin-receptor-deficient db/db mice [18]. Thus, Lepr flox/flox mice were crossed with Albcre + mice and ob/+ mice to generate mice with a hepatocyte-specific loss of leptin signalling on a leptin-deficient ob/ob background. Cre-mediated recombination in the Lepr flox/flox Albcre + ob/ob mice resulted in the generation of the Lepr ∆17 allele (Fig. 3a, b), which is restricted to hepatocytes [19]. Figure 3c, d shows that Cre-mediated recombination in hepatocytes was achieved as expected on the ob/ob background. The faint amplicon seen in Fig. 3d at ∼343 bp suggests that Cre-mediated recombination was not complete in the liver of Lepr flox/flox Albcre + ob/ob mice, but this amplicon is likely to have been contributed by non-hepatocytes in the liver, which do not express the Albcre transgene.

Schematic of the Leprflox and Lepr∆17 gene and protein structures. a Genomic structure of the Lepr flox gene where, in Lepr flox/flox Albcre + ob/ob mice, excision of exon 17 is generated by Cre-mediated recombination at loxP sites resulting in Lepr ∆17. b The protein structure of Leprflox is equivalent to the wild-type isoform b of the leptin receptor containing the Box 1 and 2 motifs required for signal transduction. The Lepr∆17 protein lacks the Box 1 and 2 motifs and is devoid of signalling function. c Genomic DNA was extracted from liver tissue of Lepr flox/flox Albcre − ob/ob and Lepr flox/flox Albcre + ob/ob mice and used as a template for PCR. Using primers MLepr101 and MLepr102 (see [a]), the Lepr flox allele has an expected product size of 1,369 bp and the Lepr ∆17 allele is predicted to produce a 952 bp product. d RNA was extracted from liver tissue of Lepr flox/flox Albcre ob/ob mice and used as a template for RT-PCR using primers G and 60 (see [a]), which flank exon 17 of the leptin receptor gene. The predicted product sizes are 343 bp for Lepr flox and 267 bp for Lepr ∆17
Hepatic leptin signalling is not required for leptin regulation of plasma IGFBP-2 levels
Before leptin treatment, lepr flox/flox Albcre + ob/ob mice and their ob/ob littermate controls not carrying the cre transgene were equally obese, hyperglycaemic and hyperinsulinaemic (Fig. 4). To determine whether hepatic leptin signalling plays a significant role in increasing plasma IGFBP-2 levels and normalising glucose homeostasis, female Lepr flox/flox Albcre + ob/ob and Lepr flox/flox Albcre − ob/ob mice were administered leptin (5 μg/day) for 2 weeks via mini-osmotic pumps. This dose of leptin was able to reverse hyperglycaemia and hyperinsulinaemia as the mice had normal blood glucose and insulin levels on day 14 of leptin therapy (Fig. 4b, c). Further, IGFBP-2 levels started to increase 2 days into leptin treatment and steadily increased until leptin therapy was ceased (Fig. 5a, b). Surprisingly, circulating leptin levels during leptin treatment were higher in the ob/ob mice lacking hepatic leptin receptor signalling domains compared with littermate ob/ob controls with full-length hepatic leptin receptors. This suggests that the leptin receptor signalling domain may play a role in leptin clearance by the liver. Despite this difference in plasma leptin levels, it is clear that leptin delivery via an osmotic pump still resulted in increased plasma leptin levels above pre-treatment levels in both the Lepr flox/flox Albcre + ob/ob and Lepr flox/flox Albcre − ob/ob mice and, by the last day of leptin treatment, plasma IGFBP-2 levels had risen by nearly tenfold above pre-leptin levels in both groups of mice (Fig. 5b). Further, in the Lepr flox/flox Albcre + ob/ob mice, the higher leptin levels resulted in significantly higher plasma IGFBP-2 levels than Lepr flox/flox Albcre − ob/ob mice at some time points. Even at a lower dose of leptin for a shorter duration, plasma IGFBP-2 levels still rose to the same extent in ob/ob mice with and without hepatic leptin signalling (Fig. 5c). Furthermore, we also measured plasma IGFBP-2 levels in mice with a hepatocyte-specific loss of leptin signalling on a lean background (Lepr flox/flox Albcre + mice), which have physiological leptin levels [19], and these mice also had plasma IGFBP-2 levels similar to their controls (Fig. 5d). Taken together, these data clearly demonstrate that, regardless of how much leptin is circulating in the plasma, hepatic leptin signalling is not required for leptin to increase plasma IGFBP-2 levels.

Leptin administration to Lepr flox/flox Albcre ob/ob mice leads to weight loss and reverses hyperglycaemia and hyperinsulinaemia. Female Lepr flox/flox Albcre ob/ob mice were treated with a continuous leptin infusion (5 μg/day) via mini-osmotic pumps for 14 days. Pumps were implanted subcutaneously on day 0 following a 4 h fast. a Body weight, (b) blood glucose and (c) plasma insulin levels following a 4 h fast were measured before, during and after leptin treatment. White circles, Lepr flox/flox Albcre − ob/ob controls; black circles, Lepr flox/flox Albcre + ob/ob mice. Data are expressed as mean ± SEM, n ≥ 5

Leptin increases plasma IGFBP-2 levels independent of hepatic leptin signalling. Female Lepr flox/flox Albcre ob/ob mice were treated with a continuous leptin infusion (5 μg/day) via mini-osmotic pumps for 14 days. Pumps were implanted subcutaneously on day 0 following a 4 h fast. a Plasma leptin and (b) plasma IGFBP-2 levels were measured following a 4 h fast, n ≥ 5. c Male Lepr flox/flox Albcre ob/ob mice were treated with a continuous leptin infusion (2 μg/day) via mini-osmotic pumps for 7 days and plasma IGFBP-2 levels were measured following a 4 h fast, n ≥ 3. White circles, Lepr flox/flox Albcre − ob/ob controls; black circles, Lepr flox/flox Albcre + ob/ob mice. d Plasma IGFBP-2 levels were measured in 7- and 17-week-old Lepr flox/flox Albcre male mice after a 4 h fast, n ≥ 8. White bars, Lepr flox/flox Albcre − controls; black bars, Lepr flox/flox Albcre + mice. All data are expressed as mean ± SEM. *p ≤ 0.05 compared with littermate controls
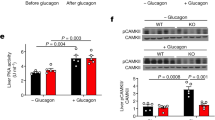
It is possible that, in our models of mice lacking hepatic leptin signalling, the Lepr∆17 protein is still able to respond to leptin and mediate hepatic Igfbp2 expression through a STAT3-independent pathway. Therefore, we used a different mouse model that does not depend on the Lepr ∆17 allele to test whether hepatic leptin signalling is involved in leptin-mediated increases in plasma IGFBP-2. We obtained db/db mice, which have a whole body loss of leptin signalling, and treated them with an adenovirus expressing the long, signalling isoform of the leptin receptor (Ad-Lepr-b) via the tail vein, which confers liver-selective expression of the adenoviral construct [20]. Using this method, we investigated whether restoring leptin signalling in the liver of db/db mice could increase hepatic or plasma IGFBP-2 levels. To confirm expression of Lepr-b in db/db mice after infection with Ad-Lepr-b, we performed RT-PCR on liver tissue using primers that flank the insertion mutation present in the Lepr gene of db/db mice. Figure 6a shows that, when we performed RT-PCR on liver from db/db mice infected with an adenovirus expressing β-galactosidase (Ad-β-gal), we obtained a PCR product at the expected size of ∼298 bp, which contains the db insertion mutation. In db/db mice infected with Ad-Lepr-b, the predominant PCR product was ∼192 bp, which is the expected product size for Lepr-b. This re-expression of Lepr-b resulted in functional leptin receptors since db/db mice treated with Ad-Lepr-b had significantly more hepatic phosphorylation of STAT3 than controls treated with Ad-β-gal (Fig. 6b, c). We next measured hepatic Igfbp2 mRNA levels in db/db mice treated with Ad-β-gal and compared this with C57BL/6 controls and found that indeed when whole body leptin signalling was absent, hepatic Igfbp2 transcript levels were <4% of C57BL/6 controls (Fig. 6d). Further, before virus treatment, both groups of db/db mice had plasma IGFBP-2 levels that were comparable with the low levels seen in ob/ob mice (Figs. 2b and 6e). Interestingly, even when hepatic leptin signalling was restored in db/db mice, neither hepatic nor plasma IGFBP-2 levels rose (Fig. 6d, e). These data clearly show that hepatic leptin signalling alone is not sufficient for mediating the effects of leptin in increasing hepatic or plasma IGFBP-2.

Restoration of hepatic leptin signalling in db/db mice does not normalise hepatic or plasma IGFBP-2 levels. Liver tissue was collected 26 days post-infection from db/db mice treated with an adenovirus expressing either β-galactosidase or the long signalling isoform of the leptin receptor, Lepr-b. a RT-PCR was performed to confirm the hepatic expression of wild-type Lepr-b. The predicted product sizes are 298 bp for the mutant db allele and 192 bp for wild-type Lepr-b. b Representative western blot showing phosphorylated (p-STAT3) and total STAT3 levels in livers of db/db-Ad-β-gal and db/db-Ad-Lepr-b mice. Quantification by densitometry analysis is shown in (c), n = 8. d Hepatic Igfbp2 transcript levels were measured by quantitative PCR, n ≥ 5. White bars, db/db-Ad-β-gal; black bars, db/db-Ad-Lepr-b; grey bars, C57BL/6 controls. e Plasma IGFBP-2 levels were measured following virus treatment, n = 6. White circles, db/db-Ad-β-gal; black circles, db/db-Ad-Lepr-b. All data are shown as mean ± SEM. *p ≤ 0.05 compared with db/db-Ad-β-gal
Effect of vagotomy on metabolic parameters in ob/ob mice
To investigate the role of vagal efferents on the ability of leptin to regulate plasma IGFBP-2 levels, we used ob/ob mice treated with a subdiaphragmatic vagotomy and sham-operated controls. The vagotomised ob/ob mice consumed less food and were less obese than the sham-operated ob/ob controls before leptin treatment (Fig. 7a, b). Further, while both groups of ob/ob mice were hyperglycaemic and hyperinsulinaemic, the vagotomised ob/ob mice had significantly higher fasting blood glucose levels and lower fasting insulin levels than sham-operated mice before the start of leptin treatment (Fig. 7c, d). Similar effects of a vagotomy on food intake, blood glucose and insulin levels have been previously reported [21–25].

Leptin treatment in ob/ob mice with a subdiaphragmatic vagotomy. Male ob/ob mice received a subdiaphragmatic vagotomy or sham surgery at 3.5 weeks of age. At 6 weeks old, mini-osmotic pumps were implanted delivering leptin (5 μg/day) for 14 days. a Body weight and (c) blood glucose levels were measured before, during and after leptin treatment. White circles, ob/ob-sham; black circles, ob/ob-vagotomy. b Overnight food intake and (d) fasting plasma insulin levels were measured on day 0 (Basal) and day 5 (Leptin) of leptin treatment. White bars, ob/ob-sham; black bars, ob/ob-vagotomy. Data are expressed as mean ± SEM, n ≥ 8. *p ≤ 0.05, compared with ob/ob-sham
Administration of leptin via mini-osmotic pumps (5 μg/day for 14 days) resulted in a similar rate of weight loss in both the sham-operated and vagotomised ob/ob mice (−4.1 ± 0.3% per day for sham-operated vs −3.9 ± 0.5% per day for vagotomised, p = 0.296). Remarkably, food intake, fasting blood glucose and fasting insulin levels were normalised in both vagotomised and sham-operated ob/ob mice after leptin treatment (Fig. 7), demonstrating that subdiaphragmatic vagal efferents are not required for leptin to normalise glucose metabolism in ob/ob mice. After 5 days of leptin administration, both groups of ob/ob mice had an equal rise in plasma leptin levels (Fig. 8a). This rise in leptin levels resulted in plasma IGFBP-2 levels that were over seven times higher than pre-leptin levels in both the vagotomised and sham-operated ob/ob mice (Fig. 8b), indicating that intact subdiaphragmatic vagal efferents are not required for leptin to increase IGFBP-2 production in ob/ob mice.

Leptin treatment leads to increased IGFBP-2 levels in ob/ob mice with a subdiaphragmatic vagotomy. Male ob/ob mice received a subdiaphragmatic vagotomy or sham surgery at 3.5 weeks old. At 6 weeks old, mini-osmotic pumps were implanted delivering leptin (5 μg/day) for 14 days. a Plasma leptin and (b) plasma IGFBP-2 levels following a 4 h fast on day 0 (basal) and day 5 (leptin) of leptin treatment were measured. White bars, ob/ob-sham; black bars, ob/ob-vagotomy. Data are expressed as mean ± SEM, n ≥ 8
To ensure that the subdiaphragmatic vagotomy was successfully achieved, food intake in response to CCK was examined in vagotomised and sham-operated mice after the cessation of leptin therapy. CCK is a satiety factor that can decrease food intake by acting on CCK1 receptors on the vagus nerve [26] and this effect is abolished after an abdominal vagotomy [16]. Food intake was significantly decreased by 26% in CCK-injected sham-operated ob/ob mice compared with saline-treated controls (Fig. 9a). Subdiaphragmatic vagotomy abolished the satiety effect of CCK, as there was no difference observed between food intake in the vagotomised mice that received CCK compared with the vagotomised mice that were saline-treated (Fig. 9a). Furthermore, we also measured stomach weight to verify vagotomy, as it has previously been reported that gastric distension is seen in vagotomised mice [15]. Indeed, the average empty stomach weight of vagotomised ob/ob mice was 1.75-fold greater than that of sham-operated controls (Fig. 9b, c). These data demonstrate that the vagotomies were successful.

Subdiaphragmatic vagotomy inhibits CCK-induced satiety in ob/ob mice and causes gastric distension. CCK was administered 8 weeks post-vagotomy and 2 weeks after cessation of leptin treatment. a Mice were fasted for 16 h and then given an intraperitoneal injection of saline or CCK (4 μg/kg). Mice were then given full access to food and food intake was measured after 30 min. Data are expressed as mean ± SEM, n ≥ 4. White bars, saline-treated controls; black bars, CCK-treated. b Empty stomach weight at time of tissue harvest and (c) images of full stomach at time of tissue harvest following a 16 h fast. White bars, ob/ob-sham; black bars, ob/ob-vagotomy. Data are expressed as mean ± SEM, n ≥ 8, *p ≤ 0.05
Discussion
The most well-established effects of leptin are its effects on reducing body weight by decreasing food intake and increasing energy expenditure. However, even in one of the first studies showing the effects of leptin in ob/ob mice, it was seen that, at doses too low to affect body weight, leptin could still reverse hyperglycaemia [27]. This observation has also been made recently by Hedbacker et al. [3] and in the present study (Fig. 1). Further, even in mouse models of type 1 diabetes, leptin can reverse hyperglycaemia independently of its effects on body weight and food intake [28–30]. Thus, one of the most potent effects of leptin in mice is actually in lowering glucose levels rather than in reducing body weight. Therefore, because of its glucose-lowering potential, it is of great interest to uncover the mechanism by which leptin potently reverses hyperglycaemia, even in the absence of weight loss.
It has been suggested that plasma IGFBP-2 may play a role in mediating the effects of leptin on glucose metabolism [3]. Leptin treatment has been shown to significantly increase circulating IGFBP-2 levels in ob/ob mice and, remarkably, IGFBP-2 administration via adenovirus was able to ameliorate diabetes in leptin-deficient ob/ob mice [3]. We saw that, even at the lowest dose of leptin (0.2 μg/day), which had effects on glucose homeostasis but not body weight, leptin was able to significantly increase IGFBP-2 levels compared with PBS-treated mice. The effect of leptin on circulating levels of IGFBP-2 was dose-dependent, and the highest dose of leptin treatment (5 μg/day) was able to increase plasma IGFBP-2 to levels found in normal, wild-type mice. These data further establish leptin as a potent regulator of plasma IGFBP-2 levels. However, the mechanism by which leptin increases plasma IGFBP-2 levels is still unknown.
The liver or the brain may be sites mediating the effects of leptin on increasing plasma IGFBP-2 levels [3, 8]. However, leptin was able to increase plasma IGFBP-2 levels in ob/ob mice lacking hepatic leptin signalling, indicating that hepatic leptin signalling is not required for leptin to increase levels of plasma IGFBP-2. Furthermore, when we re-expressed functional hepatic leptin receptors in hyperleptinaemic db/db mice, which have low plasma IGFBP-2 levels, neither hepatic nor plasma IGFBP-2 levels rose towards wild-type levels. Taken together, our data indicate that direct leptin action on the liver is neither required nor sufficient for mediating the effects of leptin on plasma IGFBP-2.
It is noteworthy that there were some time points where Lepr flox/flox Albcre + ob/ob mice treated with 5 μg/day leptin had significantly higher IGFBP-2 levels compared with controls, perhaps a reflection of the higher circulating leptin levels during leptin treatment. We speculate that the hepatic leptin receptor signalling domain may be involved in leptin clearance, resulting in the Lepr flox/flox Albcre + ob/ob mice having higher leptin levels during leptin treatment. Even though the kidney is reported to be the major site of leptin clearance [31, 32], it is possible that, at high levels of plasma leptin, such as those seen during continuous exogenous delivery of 5 μg/day leptin, the liver may be an important contributor to leptin clearance. Nonetheless, in both the Lepr flox/flox Albcre + ob/ob mice and the Lepr flox/flox Albcre − ob/ob controls treated with 5 μg/day leptin, leptin levels rose well above pre-treatment levels and, on the last day of leptin treatment, IGFBP-2 levels were nearly tenfold higher in both groups. In addition, Lepr flox/flox Albcre +mice, which had normal plasma leptin levels [19], also had normal plasma IGFBP-2 levels. Even hyperleptinaemic db/db mice with functional hepatic leptin receptors did not have altered IGFBP-2 levels compared with control db/db mice. Overall, our data show that, regardless of plasma leptin levels, direct leptin action on the liver is not required for leptin to increase plasma IGFBP-2.
Central leptin signalling appears to play an important role in mediating the effect of leptin on energy homeostasis and glucose metabolism. Mice with a neuron-specific knockout of leptin receptors have increased adiposity and hyperinsulinaemia [33], whereas mice with disrupted peripheral leptin signalling but intact central leptin receptors have normal body weight and glucose metabolism [34]. Furthermore, central leptin administration can modulate hepatic gene expression [8, 35–37]. Therefore, it is possible that leptin acts centrally to induce hepatic IGFBP-2 production and secretion via modulation of neural outputs to the liver. Interestingly, in leptin-deficient lipodystrophic mice, ICV delivery of leptin is able to upregulate Igfbp2 gene expression in the liver [8], which is suggested to be the main source of circulating IGFBP-2 [12, 13]. In addition, the central effects of leptin on modulating hepatic insulin sensitivity are reported to be mediated through the hepatic branch of the vagus nerve [14], which conveys parasympathetic innervation to the liver. To examine whether leptin acts on the brain to increase hepatic production of IGFBP-2 via brain–liver vagal communication, we treated vagotomised ob/ob mice with leptin. In spite of the initial metabolic differences in the vagotomised and sham-operated mice (Fig. 7), leptin treatment was able to increase circulating IGFBP-2 levels in both the vagotomised and sham-operated mice to an equal extent. Thus, even in the absence of subdiaphragmatic vagal innervations, leptin can still correct hyperglycaemia and hyperinsulinaemia in ob/ob mice, and brain–liver vagal efferents are not required for leptin to increase plasma IGFBP-2 levels.
While our data eliminate direct action of leptin on hepatocytes and brain–liver vagal efferents as the sole mediators of leptin-induced increases in plasma IGFBP-2, other possible mechanisms remain to be investigated. First, we did not eliminate the possibility that central leptin action can affect hepatic production and secretion of IGFBP-2 via sympathetic inputs to the liver, which remained intact in our mice. Further, while the liver is believed to be the major source of circulating IGFBP-2, other tissues may also contribute to plasma IGFBP-2 levels and these other tissues may play a bigger role when direct or indirect hepatic stimulation by leptin is absent. In fact, IGFBP-2 expression has been detected in several tissues, including kidney [38, 39], adrenal gland [40], brain [38, 41] and adipose [42, 43], all of which are known to secrete hormones and express the long, signalling isoform of the leptin receptor [9, 44, 45]. Visceral white adipose tissue in particular has recently been shown to express Igfbp2 mRNA in levels that correlate with plasma IGFBP-2 levels and, in the absence of leptin (ob/ob mice) or leptin signalling (db/db mice), Igfbp2 mRNA expression in visceral white adipose tissue is reduced [43]. Furthermore, another possibility is that leptin may indirectly increase hepatic and plasma IGFBP-2 by modulating levels of other hormones that in turn act on the liver to increase IGFBP-2 levels. While we have eliminated the most obvious mechanisms by which leptin might regulate hepatic and plasma IGFBP-2, there are other possibilities that remain to be addressed. Considering the remarkable effects of leptin and IGFBP-2 on ameliorating diabetes, more research should be done to better understand the regulation of plasma IGFBP-2 levels by leptin.
Abbreviations
- Ad-β-gal:
-
Adenovirus expressing the gene for β-galactosidase
- Ad-Lepr-b:
-
Adenovirus expressing the gene for Lepr-b
- ICV:
-
Intracerebroventricular
- CCK:
-
Cholecystokinin
- Cre:
-
Cre recombinase
- IGFBP-2:
-
IGF binding protein-2
- Lepr-b:
-
Leptin receptor, isoform b
- STAT3:
-
Signal transducer and activator of transcription 3
References
Wheatcroft SB, Kearney MT (2009) IGF-dependent and IGF-independent actions of IGF-binding protein-1 and -2: implications for metabolic homeostasis. Trends Endocrinol Metab 20:153–162
Wheatcroft SB, Kearney MT, Shah AM et al (2007) IGF-binding protein-2 protects against the development of obesity and insulin resistance. Diabetes 56:285–294
Hedbacker K, Birsoy K, Wysocki RW et al (2010) Antidiabetic effects of IGFBP2, a leptin-regulated gene. Cell Metab 11:11–22
Jones JI, Gockerman A, Busby WH Jr, Wright G, Clemmons DR (1993) Insulin-like growth factor binding protein 1 stimulates cell migration and binds to the alpha 5 beta 1 integrin by means of its Arg-Gly-Asp sequence. Proc Natl Acad Sci U S A 90:10553–10557
Hoeflich A, Wu M, Mohan S et al (1999) Overexpression of insulin-like growth factor-binding protein-2 in transgenic mice reduces postnatal body weight gain. Endocrinology 140:5488–5496
DeMambro VE, Clemmons DR, Horton LG et al (2008) Gender-specific changes in bone turnover and skeletal architecture in igfbp-2-null mice. Endocrinology 149:2051–2061
Ruan W, Lai M (2010) Insulin-like growth factor binding protein: a possible marker for the metabolic syndrome? Acta Diabetol 47:5–14
Asilmaz E, Cohen P, Miyazaki M et al (2004) Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J Clin Invest 113:414–424
Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim MH, Skoda RC (1996) Defective STAT signaling by the leptin receptor in diabetic mice. Proc Natl Acad Sci U S A 93:6231–6235
Fei H, Okano HJ, Li C et al (1997) Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci U S A 94:7001–7005
Cohen B, Novick D, Rubinstein M (1996) Modulation of insulin activities by leptin. Science 274:1185–1188
White ME, Leaman DW, Ramsay TG, Kampman KA, Ernst CW, Osborne JM (1993) Insulin-like growth-factor binding protein (IGFBP) serum levels and hepatic IGFBP-2 and -3 mRNA expression in diabetic and insulin-treated swine (Sus scrofa). Comp Biochem Physiol B 106:341–347
Delhanty PJ, Han VK (1993) The expression of insulin-like growth factor (IGF)-binding protein-2 and IGF-II genes in the tissues of the developing ovine fetus. Endocrinology 132:41–52
German J, Kim F, Schwartz GJ et al (2009) Hypothalamic leptin signaling regulates hepatic insulin sensitivity via a neurocircuit involving the vagus nerve. Endocrinology 150:4502–4511
Bluthe RM, Michaud B, Kelley KW, Dantzer R (1996) Vagotomy attenuates behavioural effects of interleukin-1 injected peripherally but not centrally. Neuroreport 7:1485–1488
Smith GP, Jerome C, Cushin BJ, Eterno R, Simansky KJ (1981) Abdominal vagotomy blocks the satiety effect of cholecystokinin in the rat. Science 213:1036–1037
Coppari R, Ichinose M, Lee CE et al (2005) The hypothalamic arcuate nucleus: a key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab 1:63–72
McMinn JE, Liu SM, Liu H et al (2005) Neuronal deletion of Lepr elicits diabesity in mice without affecting cold tolerance or fertility. Am J Physiol Endocrinol Metab 289:E403–E411
Huynh FK, Levi J, Denroche HC et al (2010) Disruption of hepatic leptin signaling protects mice from age- and diet-related glucose intolerance. Diabetes 59:3032–3040
Lam NT, Covey SD, Lewis JT et al (2006) Leptin resistance following over-expression of protein tyrosine phosphatase 1B in liver. J Mol Endocrinol 36:163–174
Louis-Sylvestre J (1983) Validation of tests of completeness of vagotomy in rats. J Auton Nerv Syst 9:301–314
Pocai A, Lam TK, Gutierrez-Juarez R et al (2005) Hypothalamic K(ATP) channels control hepatic glucose production. Nature 434:1026–1031
Zawalich WS, Zawalich KC, Rasmussen H (1989) Cholinergic agonists prime the beta-cell to glucose stimulation. Endocrinology 125:2400–2406
Teff KL, Townsend RR (2004) Prolonged mild hyperglycemia induces vagally mediated compensatory increase in C-peptide secretion in humans. J Clin Endocrinol Metab 89:5606–5613
Pocai A, Obici S, Schwartz GJ, Rossetti L (2005) A brain–liver circuit regulates glucose homeostasis. Cell Metab 1:53–61
Neary MT, Batterham RL (2009) Gut hormones: implications for the treatment of obesity. Pharmacol Ther 124:44–56
Pelleymounter MA, Cullen MJ, Baker MB et al (1995) Effects of the obese gene product on body weight regulation in ob/ob mice. Science 269:540–543
Yu X, Park BH, Wang MY, Wang ZV, Unger RH (2008) Making insulin-deficient type 1 diabetic rodents thrive without insulin. Proc Natl Acad Sci U S A 105:14070–14075
Wang MY, Chen L, Clark GO et al (2010) Leptin therapy in insulin-deficient type I diabetes. Proc Natl Acad Sci U S A 107:4813–4819
Denroche HC, Levi J, Wideman RD et al (2011) Leptin therapy reverses hyperglycemia in mice with streptozotocin-induced diabetes, independent of hepatic leptin signaling. Diabetes 60:1414–1423
Cumin F, Baum HP, Levens N (1996) Leptin is cleared from the circulation primarily by the kidney. Int J Obes Relat Metab Disord 20:1120–1126
Ceccarini G, Flavell RR, Butelman ER et al (2009) PET imaging of leptin biodistribution and metabolism in rodents and primates. Cell Metab 10:148–159
Cohen P, Zhao C, Cai X et al (2001) Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest 108:1113–1121
Guo K, McMinn JE, Ludwig T et al (2007) Disruption of peripheral leptin signaling in mice results in hyperleptinemia without associated metabolic abnormalities. Endocrinology 148:3987–3997
Fujikawa T, Chuang JC, Sakata I, Ramadori G, Coppari R (2010) Leptin therapy improves insulin-deficient type 1 diabetes by CNS-dependent mechanisms in mice. Proc Natl Acad Sci U S A 107:17391–17396
Sharma A, Bartell SM, Baile CA et al (2010) Hepatic gene expression profiling reveals key pathways involved in leptin-mediated weight loss in ob/ob mice. PLoS One 5:e12147
Liu L, Karkanias GB, Morales JC et al (1998) Intracerebroventricular leptin regulates hepatic but not peripheral glucose fluxes. J Biol Chem 273:31160–31167
Kita K, Nagao K, Taneda N et al (2002) Insulin-like growth factor binding protein-2 gene expression can be regulated by diet manipulation in several tissues of young chickens. J Nutr 132:145–151
Fornoni A, Rosenzweig SA, Lenz O, Rivera A, Striker GE, Elliot SJ (2006) Low insulin-like growth factor binding protein-2 expression is responsible for increased insulin receptor substrate-1 phosphorylation in mesangial cells from mice susceptible to glomerulosclerosis. Endocrinology 147:3547–3554
Ilvesmaki V, Liu J, Heikkila P, Kahri AI, Voutilainen R (1998) Expression of insulin-like growth factor binding protein 1-6 genes in adrenocortical tumors and pheochromocytomas. Horm Metab Res 30:619–623
Takeo C, Ikeda K, Horie-Inoue K, Inoue S (2009) Identification of Igf2, Igfbp2 and Enpp 2 as estrogen-responsive genes in rat hippocampus. Endocr J 56:113–120
Claudio M, Benjamim F, Riccardo B, Massimiliano C, Francesco B, Luciano C (2010) Adipocytes IGFBP-2 expression in prepubertal obese children. Obesity (Silver Spring) 18:2055–2057
Li Z, Picard F (2010) Modulation of IGFBP2 mRNA expression in white adipose tissue upon aging and obesity. Horm Metab Res 42:787–791
Kielar D, Clark JS, Ciechanowicz A, Kurzawski G, Sulikowski T, Naruszewicz M (1998) Leptin receptor isoforms expressed in human adipose tissue. Metabolism 47:844–847
Hoggard N, Mercer JG, Rayner DV, Moar K, Trayhurn P, Williams LM (1997) Localization of leptin receptor mRNA splice variants in murine peripheral tissues by RT-PCR and in situ hybridization. Biochem Biophys Res Commun 232:383–387
Acknowledgements
We thank C. Donald (University of British Columbia) for her technical assistance with the animal studies and S.C. Chua (Albert Einstein College of Medicine) for his generous contribution of the Lepr flox/flox mice. We would also like to thank M.G. Myers (University of Michigan) and C.J. Rhodes (University of Chicago) for providing the Ad-Lepr-b virus.
Funding
This work was funded by the Canadian Institutes of Health Research. FKH is supported by the Canadian Diabetes Association, HCD is supported by the Natural Sciences Research Council of Canada, and TJK is the recipient of a Senior Scholarship from the Michael Smith Foundation for Health Research.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors contributed to the conception and design or analysis and interpretation of data. Further, all authors contributed to drafting the article or revising it for important intellectual content. The approval of all authors was obtained before publication.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. Levi and F.K. Huynh contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 47.5 kb)
Rights and permissions
About this article
Cite this article
Levi, J., Huynh, F.K., Denroche, H.C. et al. Hepatic leptin signalling and subdiaphragmatic vagal efferents are not required for leptin-induced increases of plasma IGF binding protein-2 (IGFBP-2) in ob/ob mice. Diabetologia 55, 752–762 (2012). https://doi.org/10.1007/s00125-011-2426-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2426-8