
Abstract
Aims/hypothesis
Chronic hyperglycaemia aggravates insulin resistance, at least in part, by increasing the formation of advanced glycation end-products (AGEs). Methylglyoxal (MGO) is the most reactive AGE precursor and its abnormal accumulation participates in damage in various tissues and organs. Here we investigated the ability of MGO to interfere with insulin signalling and to affect beta cell functions in the INS-1E beta cell line.
Methods
INS-1E cells were incubated with MGO and then exposed to insulin or to glucose. Western blotting was used to study signalling pathways, and real-time PCR to analyse gene expression; insulin levels were determined by radioimmunoassay.
Results
Non-cytotoxic MGO concentrations inhibited insulin-induced IRS tyrosine phosphorylation and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) pathway activation independently from reactive oxygen species (ROS) production. Concomitantly, formation of AGE adducts on immunoprecipitated IRS was observed. Aminoguanidine reversed MGO inhibitory effects and the formation of AGE adducts on IRS. Further, the insulin- and glucose-induced expression of Ins1, Gck and Pdx1 mRNA was abolished by MGO. Finally, MGO blocked glucose-induced insulin secretion and PI3K/PKB pathway activation. These MGO effects were abolished by LiCl, which inhibits glycogen synthase kinase-3 (GSK-3).
Conclusions/interpretation
MGO exerted major damaging effects on INS-1E cells impairing both insulin action and secretion. An important actor in these noxious MGO effects appears to be GSK-3. In conclusion, MGO participates not only in the pathogenesis of the debilitating complications of type 2 diabetes, but also in worsening of the diabetic state by favouring beta cell failure.
Similar content being viewed by others

Introduction
The hallmark of type 2 diabetes is insulin resistance coupled to pancreatic beta cell failure. In the process leading to overt type 2 diabetes, several key insulin target tissues, such as the liver, muscle and fat, become resistant to the actions of the hormone. In the pathogenesis of insulin resistance dysregulation occurs at multiple steps in the complex network of the insulin signalling pathways [1].
Recent studies have shown that beta cells are targets of insulin action. Indeed, insulin is thought to act in an autocrine/paracrine manner to regulate beta cell mass [2] and function [3], and to modulate several cellular processes such as gene expression [4] and Ca2+ flux [5].
In a mouse model with beta cell specific knockout of the insulin receptor (IR), a decrease in glucose-stimulated insulin release and a progressive impairment of glucose tolerance are observed, suggesting that defects in insulin signalling at the level of the beta cell may contribute to alterations in hormone secretion found in type 2 diabetes [6]. Moreover, disruption of insulin signalling in the beta cell at the level of the insulin receptor substrates IRS-1 [7] or IRS-2 [8] leads, respectively, to altered secretion and beta cell proliferation. Finally, the IR appears to be involved in the modulation of the responses of beta cells to glucose as shown, for example, by studies in MIN-6 cells, in which a decrease in IRs using siRNA inhibits the accumulation of preproinsulin, Pdx1 and Gck mRNA normally seen at high glucose concentration [9].
To summarise, insulin resistance may not only affect the functioning of the ‘classical’ insulin target tissues, such as muscle, fat and liver, but also that of beta cells. This organismal insulin resistance could be a consequential factor in the pathogenesis of type 2 diabetes. The mechanisms underlying the deleterious effects of reduced insulin signalling on hormone synthesis and secretion of beta cells are poorly defined. Genetic and acquired factors are responsible for insulin resistance in hormone target tissues, while chronic hyperglycaemia further impairs insulin action by different mechanisms. In particular, chronic hyperglycaemia promotes the endogenous non-enzymatic glycoxidation of proteins, lipids and nucleic acids, leading to the formation of advanced glycation end-products (AGEs) [10]. Intracellularly formed α-ketoaldehydes, such as methylglyoxal (MGO), are an essential source of intracellular AGEs and abnormal MGO accumulation has been implicated in the development of damage in various tissues and organs [11]. MGO is formed non-enzymatically by dephosphorylation of triose phosphates and, under physiological conditions, is degraded into d-lactate by glyoxalase 1 [12].
While evidence exists that AGEs interfere with the complex pathways governing insulin signalling and secretion, it is not known which specific AGEs are the culprits and which pathways are affected. We previously reported that human glycated albumin treatment of cells and dietary AGEs given to mice induce insulin resistance in vitro [13] and in vivo [14], respectively, via a selective protein kinase C alpha activation. We have also shown that MGO induces insulin resistance in skeletal muscle cells through direct chemical modification of IRS proteins [15]. Finally, MGO is thought to exert deleterious effects on insulin secretion [16–21]. However, the molecular mechanisms involved in the effect of MGO on the regulation of insulin release remains to be clarified.
Given the mounting evidence for a major role of beta cells in the pathogenesis of type 2 diabetes, we investigated the ability of MGO to interfere with insulin signalling in the INS-1E beta cell line, and we evaluated its potential damaging impact on beta cell functions.
Methods
Materials
Media, sera and antibiotics for cell culture were from Invitrogen (Paisley, UK). Protein electrophoresis and western blot reagents were from Bio-Rad (Richmond, VA, USA). Enhanced chemiluminescence reagents and protein-A sepharose were from Pierce (Rockford, IL, USA). Insulin was from NovoNordisk (Bagsværd, Denmark). The antibodies used are listed in the electronic supplementary material [ESM] Table 1. Chloromethyl-2′7′-dichlorofluorescein diacetate (CM-DCF) was from Molecular Probes (Eugene, OR, USA). [2-14C]MGO (2.035 GBq/mmol) was manufactured by American Radiolabeled Chemicals (St Louis, MO, USA). Insulin was measured by radioimmunoassay (Insulin Rat RIA kit; Linco Research, St Louis, MO, USA). MGO, aminoguanidine (AG), and all other reagents were from Sigma (St Louis, MO, USA).
INS-1E cell culture
The insulin-secreting INS-1E rat beta cells (Passages 90–120) were cultured as previously described [22]. Cells were starved for 16 h in serum-free medium containing 0.2% (wt/vol.) BSA, pretreated or not with MGO, washed once with starvation medium and then exposed or not to 100 nmol/l insulin.
Incorporation of [2-14C]MGO
The incorporation of [2-14C]MGO into INS-1E cells was determined as previously described [15]. Details are provided in the ESM.
Measurement of production of ROS
The intracellular reactive oxygen species (ROS) were measured by CM-DCF fluorescence as previously described [15]. Details are provided in the ESM.
Cell viability assay
Cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described [23]. Details are provided in the ESM.
RNA isolation and real-time quantitative RT-PCR
Total RNA extraction, cDNA synthesis and real-time PCR analysis were performed as described previously [24]. Procedures and primer sequences used for amplification are provided in the ESM Methods and ESM Table 2, respectively.
Immunoblot analysis
INS-1E cells were washed and solubilised as previously described [22]. Details for immunoprecipitation, immunoblots and antibodies used are provided in the ESM.
Measurement of insulin secretion
Insulin secretion was assessed as previously described [25]. Details are provided in the ESM.
Statistical methods
Data were analysed with Statview software (Abacus Concepts, Piscataway, NJ, USA) by one-factor analysis of variance. p values ≤0.05 were considered as statistically significant.
Results
Incorporation of [2-14C]MGO into INS-1E cells
To determine whether the intracellular accumulation of MGO in INS-1E cells is potentially involved in impairment of insulin action, and to evaluate its impact on beta cell function, a time course of MGO incorporation into INS-1E cells was performed. The cells were incubated from 0 to 30 min with [2-14C]MGO (37 kBq/assay) at concentrations ranging from 0.25 to 1.0 mmol/l, and thereafter extensively washed with PBS. We found that within 30 min only 12.5 ± 1.5% of the labelled MGO was associated with the cells (Fig. 1a). Assuming that the cell-associated MGO is indeed intracellular, this would mean that at a concentration of 0.5 mmol/l, approximately 60 μmmol/l of this α-ketoaldehyde is within the cells. Similar observations have been reported concerning the incorporation of MGO into rat L6 skeletal muscle cells [15], and into rat aortic smooth muscle cells [26].

Effects of MGO in INS-1E cells. a After incubation of INS-1E cells with different amounts of labelled MGO (from 0.25 to 1 mmol/l) in starvation medium, the media were eliminated at the indicated times. The plated cells were washed three times with ice-cold PBS to remove non-specifically bound MGO, and thereafter lysed in 100 mmol/l NaOH. The radioactivity incorporated into the cells was measured by liquid scintillation counting (MGO: diamond, 0.25 mmol/l; square, 0.5 mmol/l; triangle, 1.0 mmol/l) (n = 3). b For the measurement of ROS production, INS-1E cells were treated with different concentrations of MGO (from 0.25 to 1 mmol/l) for 30 min (white bars) or 60 min (black bars). ROS production was determined with the fluorescent probe CM-DCF (n = 3). H2O2 is included as a control. c INS-1E cells were treated with different concentrations of MGO for 30 min, 1 h, 6 h and 24 h and thereafter their viability was measured using an MTT colorimetric assay. The results represent the mean±SD of four independent experiments
Role of MGO in production of ROS in INS-1E cells
To determine whether MGO leads to enhanced ROS production in INS-1E cells, intracellular ROS levels were measured before and after treatment of cells with increasing MGO concentrations (from 0.25 to 1.0 mmol/l) for different times (from 0 to 60 min). As shown in Fig. 1b, treatment of INS-1E cells with MGO at concentrations between 0.25 and 1.0 mmol/l does not lead to a marked ROS production. As a control, INS-1E cells were treated for 40 min with 70 μmmol/l H2O2, which induces approximately a 9.5-fold increase in ROS production.
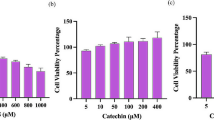
Effects of MGO treatment on INS-1E cell viability
INS-1E cells were exposed to MGO at concentrations ranging from 0.05 to 1.00 mmol/l, and for time periods of 30 min to 24 h. Thereafter, cell viability was measured using the MTT assay. As shown in Fig. 1c, under the conditions tested MGO failed to alter INS-1E cell viability.
Effects of MGO on insulin signalling in INS-1E cells
To investigate a possible role of MGO in the impairment of insulin action, different proximal insulin signalling steps were investigated in MGO-treated cells. INS-1E cells were exposed to different concentrations of MGO for 30 min and then incubated or not with insulin for 10 min. The insulin-stimulated tyrosine phosphorylation of IR was unchanged in MGO-treated cells (data not shown). In contrast, insulin-dependent IRS tyrosine phosphorylation was severely reduced in cells preincubated for 30 min with MGO (Fig. 2a). Indeed, MGO leads to a dose-dependent inhibition of insulin-stimulated IRS tyrosine phosphorylation. This decrease in tyrosine phosphorylation was not accompanied by changes in the level of IRS-1/2 protein, suggesting that a functional defect is responsible for the reduced phosphorylation. To evaluate the consequences of the deficient tyrosine phosphorylation of IRS, we first looked at the ability of IRS to dock with the PI3K p85 subunit. To this end, we measured the levels of p85 subunits in IRS immunoprecipitates obtained from INS-1E cells treated with 0.5 mmol/l MGO for 30 min, and in untreated controls. As shown in Fig. 3a–c insulin induced a marked increase in the amount of p85 subunit co-immunoprecipitating with IRS-1/2 (~threefold higher than the unstimulated level), and this p85 PI3K–IRS association correlated with the level of IRS-1 and IRS-2 tyrosine phosphorylation. As expected from its inhibitory effect on IRS tyrosine phosphorylation, MGO virtually abolished complex formation between p85 and IRS. Importantly, the reduced p85–IRS-1/2 interaction was not associated with changes in the levels of the IRS proteins (Fig. 3a–c). When the carbonyl scavenger, AG, was added before and during the incubation with MGO, the inhibitory actions of the α-ketoaldehyde on these early insulin signalling steps were prevented (Fig. 3a–c).

Effect of MGO on insulin signalling in INS-1E cells. INS-1E cells were incubated for 30 min without or with MGO at different concentrations (from 0.05 to 1 mmol/l) and then stimulated for 10 min with 100 nmol/l insulin. a The cells were then solubilised and extracts were immunoprecipitated with anti-IRS-1/2 antibodies followed by western blot analysis with antibodies to phosphotyrosine, IRS-1 and IRS-2. b,c Aliquots of the cell extracts were subjected to western blot analysis with antibodies to phospho-PKB and PKB. d INS-1E cells were treated with 0.05 or 0.1 mmol/l MGO for 24 h and aliquots of the extracts were subjected to western blot analysis with antibodies to phospho-PKB and PKB. Results are representative of three separate experiments. White bars, without insulin treatment; black bars, with insulin treatment; error bars show SD; *p < 0.05, **p < 0.01

Effect of MGO on insulin-induced activation of the PI3K pathway in INS-1E cells. a INS-1E cells were incubated for 30 min without or with 0.5 mmol/l MGO in the absence or presence of 4 mmol/l AG and then stimulated for 10 min with 100 nmol/l insulin. Thereafter, the cells were solubilised, the extracts were immunoprecipitated with anti-IRS-1/2 antibodies and subjected to western blot analysis with antibodies to phosphotyrosine (pTyr), p85, IRS-1 and IRS-2. IRS-1 and −2 tyrosine phosphorylation (b) and their association with p85 (c) were analysed by densitometry. Aliquots of the extracts were subjected to western blot analysis with antibodies (d) to phospho-PKB (pThr308-PKB) and PKB or (e) to phospho-GSK-3 (pSer-GSK-3) and GSK-3. The western blots were analysed by densitometry. Results shown are representative of three separate experiments. White bars, without insulin treatment; black bars, with insulin treatment; error bars show SD; *p < 0.05, **p < 0.01
Next we looked at events downstream of PI3K. As shown in Fig. 2b, insulin-induced PKB phosphorylation on Thr308 was significantly reduced (approximately 1.8-fold) after a 30 min treatment with 0.25 mmol/l MGO, and was nearly absent after a 0.5 mmol/l MGO exposure. Note that, whilst low MGO concentrations (0.05 and 0.1 mmol/l) had no effect on PKB phosphorylation after 30 min (Fig. 2c), incubation with the same concentrations for 24 h blocked insulin-induced PKB phosphorylation on Thr308 (Fig. 2d). To further evaluate the MGO-induced defect in the PI3K–PKB cascade, the kinase downstream of PKB was analysed. In beta cells, GSK-3 is present in α and β isoforms, which are both inhibited by PKB phosphorylation on the N-terminal serine residue (Ser21 in GSK-3α and Ser9 in GSK-3β). As illustrated in Fig. 3e, PKB-induced serine phosphorylation of GSK-3 was prevented by MGO. As was the case for PKB phosphorylation, the inhibitory effect of MGO on GSK-3 phosphorylation occurred without changes in GSK-3 protein level. As expected, AG added before and during the MGO incubation prevents the inhibition of PKB and GSK-3 phosphorylation (Fig. 3d,e).
Formation of MGO adducts on IRS in INS-1E cells
MGO is known to react with free amino groups and thiols to form AGE protein adducts, thereby altering protein function. We investigated whether incubation of INS-1E cells with MGO (0.5 mmol/l for 30 min, and 0.05 or 0.1 mmol/l for 24 h) was associated with formation of AGE–IRS adducts, more specifically N ε-(carboxyethyl)lysine (CEL) and argpyrimidine adducts. As shown by western blots of immunoprecipitated IRS, incubation of INS-1E cells with MGO resulted in the formation of CEL– (Fig. 4a,b) and argpyrimidine–IRS adducts (Fig. 4c,d). To confirm MGO-driven adduct generation, we examined whether AG blocks IRS modification observed after MGO treatment. Figure 4a,c shows that this is indeed the case.

Detection of CEL adducts on IRS immunoprecipitated from INS-1E. INS-1E cells were treated with 0.5 mmol/l MGO for 30 min without or with AG (a,c) or treated with 0.05 or 0.1 mmol/l MGO for 24 h (b,d). Thereafter, the cells were solubilised, the extracts were immunoprecipitated with anti-IRS-1/2 antibodies and subjected to western blot analysis with antibodies to IRS-1, CEL (a,b) and to argpyrimidine (argpyr) (c,d)
Effects of MGO on insulin-induced gene expression in INS-1E cells and on their glucose-induced insulin secretion
To gain further insight into the deleterious MGO action, we evaluated the expression profile of different genes important for beta cell functioning. Pancreatic duodenal homeobox-1 (PDX-1) is a key transcription factor for pancreas development and beta cell functions, which regulates, for example, the expression of the genes encoding insulin and glucokinase. In addition, insulin modulates PDX-1 expression and action [27]. Moreover, it was recently proposed that glucose regulates the levels of PDX-1 via the reciprocal actions of GSK-3 and PKB kinase [28]. To investigate whether MGO modulates gene expression, INS-1E cells were incubated with 0.5 mmol/l MGO for 30 min and then stimulated with insulin. Treatment of INS-1E cells with insulin alone induced a significant increase in Pdx1, Ins1 and Gck mRNA expression compared with the basal condition (approximately 1.8-, 1.6- and 3.0-fold, respectively). Interestingly, insulin-induced expression of these mRNAs was abolished by MGO (Fig. 5a). As expected, AG present before and during the incubation with MGO restores insulin stimulatory action on expression of these genes (Fig. 5a). Importantly, incubation of INS-1E cells with low MGO concentrations (0.05 and 0.1 mmol/l) for 24 h also inhibited insulin's stimulatory action on gene expression (Fig. 5b). Next we investigated the effects of MGO on insulin secretion. As shown in Fig. 5c, insulin secretion by INS-1E cells was significantly increased upon exposure to 20 mmol/l glucose, but this glucose action was abolished when cells were treated with 0.5 mmol/l MGO for 30 min. A similar inhibition of glucose-induced insulin secretion was seen with cells treated with 0.05 and 0.1 mmol/l MGO for 24 h (Fig. 5d). As expected, AG blocks MGO inhibition of glucose-stimulated insulin secretion (Fig. 5c). Taken together, our data demonstrate that MGO treatment of INS-1E cells hampers their glucose responsiveness.

Effect of MGO on insulin-induced gene expression in INS-1E cells and on their glucose-induced insulin secretion. INS-1E cells were incubated (a) for 30 min without or with 0.5 mmol/l MGO in the absence or presence of 4 mmol/l AG, or (b) for 24 h with 0.05 or 0.1 mmol/l MGO. Thereafter they were stimulated for 30 min with 100 nmol/l insulin. Total RNA was extracted and Pdx1, Ins1 and Gck mRNA was measured by quantitative RT-PCR using 18S as internal standard. INS-1E cells were incubated (c) for 30 min without or with 0.5 mmol/l MGO or (d) for 24 h with 0.05 or 0.1 mmol/l MGO, and thereafter stimulated for 30 min with 2.8 or 20 mmol/l glucose. Released insulin was measured by radioimmunoassay. Results shown are representative of three separate experiments. Error bars show SD; *p < 0.05, **p < 0.01, ***p < 0.001
Effects of MGO on glucose-induced signalling and gene expression in INS-1E cells
To determine whether MGO treatment influences glucose signalling and interferes with glucose action on gene expression, INS-1E cells were pretreated with 0.5 mmol/l MGO for 30 min or with 0.05 and 0.1 mmol/l MGO for 24 h, and thereafter exposed to 2.8 or 20 mmol/l glucose for 30 min. In the absence of MGO, incubation of INS-1E cells with 20 mmol/l glucose increased phosphorylation of the Thr308 residue of PKB. Pretreatment with MGO inhibits glucose-stimulated PKB phosphorylation without a detectable change in PKB protein level (Fig. 6a,b), thus demonstrating that MGO restrains glucose-induced activation of the PI3K/PKB pathway.

Effect of MGO on glucose-induced phosphorylation of PKB and on gene expression in INS-1E cells. INS-1E cells were incubated (a) for 30 min with 0.5 mmol/l MGO or (b) for 24 h with 0.05 or 0.1 mmol/l MGO, and thereafter stimulated for 30 min with 2.8 or 20 mmol/l glucose. Next the cells were solubilised and aliquots of the extracts were subjected to western blot analysis with antibodies to phospho-PKB or PKB and analysed by densitometry. INS-1E cells were incubated (c) for 30 min without or with 0.5 mmol/l MGO in absence or presence of 25 μmol/l LY294002 or (d) for 24 h with 0.05 or 0.1 mmol/l MGO, and thereafter they were stimulated for 30 min with 2.8 or 20 mmol/l glucose. Total RNA was extracted and Pdx1, Ins1 and Gck mRNA was measured by quantitative RT-PCR using 18 s as internal standard. Results shown are representative of three separate experiments. Error bars show SD; **p < 0.01
The MGO-produced impairment of glucose stimulatory action on the PI3K/PKB cascade led us to investigate the effect of the α-ketoaldehyde on glucose-induced mRNA expression of Pdx1, Gck and Ins1. In the absence of MGO, incubation with 20 mmol/l glucose for 30 min increased mRNA levels of these genes by 70%, 60% and 80%, respectively. In contrast, the glucose effect is abolished by MGO treatment. It appears that glucose action is mediated by the PI3K pathway as the PI3K inhibitor, LY294002, blocks it (Fig. 6c). Similarly, after a 24 h incubation with 0.05 mmol/l MGO, the glucose effect on expression of Pdx1 and Ins1 mRNA is reduced by 50% and 40%, respectively, and is almost completely blocked after a 24 h treatment with 0.1 mmol/l MGO (Fig. 6d).
Role of GSK-3 in MGO inhibitory action on biological responses of INS-1E cells
Recently, it was reported that mice overexpressing a constitutively active form of Gsk-3β (also known as Gsk3b) in beta cells showed a profound decrease in PDX-1 levels compared with control mice [29]. To determine whether GSK-3 activation is responsible for the deleterious effects of MGO, INS-1E cells were incubated for 30 min with 0.5 mmol/l MGO and then exposed to 2.8 or 20 mmol/l glucose in the presence of 40 mmol/l LiCl, a GSK-3 blocker [30]. In MGO-treated cells, GSK-3 phosphorylation in response to glucose is reduced with the likely concurrent increase in its activation, but LiCl treatment reverses this MGO inhibitory effect on GSK-3 phosphorylation (Fig. 7a). To approach the potential role of MGO-stimulated GSK-3 activation in the alteration of beta cell function, we investigated MGO effects on glucose-induced gene expression in the presence of LiCl. Glucose treatment leads to an increase in Pdx1, Ins1 and Gck mRNA levels by approximately 1.2-, 0.8- and 1.1-fold, respectively (Fig. 7b). In cells exposed to MGO the stimulatory action of glucose on gene expression is abolished. However, in MGO-treated cells LiCl incubation restores the increase in Pdx1, Ins1 and Gck mRNA produced by 20 mmol/l glucose. We then measured, in MGO-exposed cells, the effect of glucose on insulin secretion in the presence of LiCl. As shown in Fig. 7c, glucose-induced insulin secretion is robustly decreased by MGO incubation. Pretreatment with LiCl partially restores glucose-induced insulin secretion in the presence of MGO, indicating that MGO action on insulin secretion occurs, at least in part, through activation of GSK-3.

Role of GSK-3 in MGO inhibitory action on glucose effects in INS-1E cells. INS-1E cells were incubated for 30 min without or with 0.5 mmol/l MGO in absence or presence of 40 mmol/l LiCl, and thereafter stimulated for 30 min with 2.8 or 20 mmol/l glucose. a The cells were solubilised and phosphorylation levels of GSK-3 were analysed by western blot with specific antibodies. The western blots were evaluated by densitometry. b Total RNA was extracted and Pdx1, Ins1 and Gck mRNA were measured by quantitative RT-PCR using 18 s as internal standard. c Released insulin was measured by radioimmunoassay. Results shown are representative of three separate experiments. Error bars show SD; *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
Evidence exists in favour of the idea that beta cells could be targets for secreted insulin. Indeed, insulin is thought to stimulate beta cell proliferation and gene expression by activating the IR signalling cascade in an autocrine/paracrine fashion [2, 31]. Moreover, proinsulin synthesis and the beta cell responsiveness to glucose in terms of hormone secretion appear to be modulated by insulin signalling [2, 4]. Therefore, alterations of insulin signal transduction could lead to beta cell dysfunction contributing to the pathogenesis of type 2 diabetes. However, the molecular mechanisms through which defects in insulin signalling affect insulin synthesis and secretion remain largely unknown.
Increased production of AGEs and MGO, a precursor of AGEs, has been shown to contribute to insulin resistance in conditions associated to chronic hyperglycaemia [13, 15]. Here we show that exposure of INS-1E cells to MGO for short periods (30 min at 0.5 mmol/l) or prolonged periods (24 h at 0.05 and 0.1 mmol/l) impairs insulin-induced tyrosine phosphorylation of IRS. This occurs together with a reduction in p85–IRS association, PKB activation and serine phosphorylation of GSK-3. The decrease in tyrosine-phosphorylated IRS proteins is unlikely to be due to changes in IRS synthesis/degradation, as IRS protein levels were unaltered after these MGO treatments.
Generation of ROS is linked to MGO metabolism [26]. Further, oxidative stress has been implicated in the deterioration of insulin signalling in diabetes [32], in decreased glucose-stimulated insulin secretion, and in beta cell apoptosis [33]. Oxidative stress thus represents a risk factor for the development of diabetes. Under our experimental conditions we did not detect a significant increase in intracellular ROS accumulation and in cell death after a short or long exposure to MGO. Therefore, we would like to conclude that the MGO effects we observed in INS-1E cells are not mediated by ROS production. Our findings in INS-1E cells are consistent with those we obtained in L6 skeletal muscle cells, in which MGO impairs insulin signalling not by producing ROS, but by binding to IRS-1 and 2 and leading to a decrease in their tyrosine phosphorylation in response to insulin [15].
MGO is a reactive dicarbonyl molecule that can modify lysine and arginine residues in proteins. This also occurs in INS-1E cells, as incubation with MGO induces the formation of CEL– and argpyrimidine–IRS adducts. Interestingly, on rat IRS-1, proximal to YMXM motifs, which have been implicated in p85 binding, Lys602 near Y608MPM and Arg622 near Y628MPM are found [34, 35]. It is likely that adduct binding to Lys602 and/or Arg622 could lead to reduced tyrosine phosphorylation of IRS-1/2 resulting in decreased p85 anchoring by IRS. An alternative or additional scenario could be a similar modification of the Lys and/or Arg residues located within the IRS PH or PTB domains, which are essential for optimal IRS tyrosine phosphorylation by IR. As mentioned earlier, increased degradation and/or decreased synthesis of IRS seems to be improbable as protein levels are unchanged. Taken as a whole, our data gather grounds for belief in MGO-induced alteration of IRS resulting in a decrease in its docking function and hence perturbation of the downstream PI3K/PKB signalling node.
Several lines of evidence have been provided in favour of the idea that insulin modulates PDX-1 DNA-binding activity and insulin promoter activity [27]. PDX-1 is a key transcription factor involved in pancreas development and in modulation of the expression of several genes essential for beta cells, such as the insulin and glucokinase genes (Ins and Gck). It has been shown that both glucose and insulin stimulates PDX-1 binding activity to the insulin promoter via PI3K and mitogen activated protein kinase 11 (SAPK2/p38) [27, 36]. IR silencing in MIN-6 cells robustly suppressed the expression of Pdx1 mRNA, suggesting that insulin signalling plays a major role in the regulation of the expression and function of PDX-1 [9, 37]. PDX-1 also modulates the mRNA expression of Gck, which catalyses the first step of glycolysis, and hence regulates glucose responsiveness for insulin release [38]. Here we demonstrate in INS-1E cells that a short and prolonged exposure to MGO leads to impaired insulin-induced expression of the genes coding for PDX-1, insulin and glucokinase. Thus, MGO hampers the beneficial effect of insulin on the expression of genes involved in the preservation of the chief beta cell functions.
The MGO concentrations we have used are in the range of those applied by Brouwers et al. to mesenteric arteries [39], by Cook et al. to rat beta cells [17], and by Bento et al. to human retinal pigment epithelium cells [40]. Generally speaking these concentrations are intended to mimic the accumulation of MGO seen after chronic hyperglycaemia found in diabetes. Using, in INS-1E cells, either 0.5 mmol/l MGO for 30 min or between 0.05 and 0.1 mmol/l for 24 h, similar inhibitory effects were observed on insulin signalling without change in cell viability. Further, within 30 min incubation with 0.5 mmol/l MGO approximately 60 μmmol/l MGO is found to be associated with INS-1E cells. This is comparable with the results of a recent study in human retinal pigment epithelium cells [40].
Previous reports have shown that MGO alters insulin secretion [17, 19, 21]. Our current study on INS-1E cells further documents the inhibitory effect of MGO on glucose-induced insulin secretion, but clearly goes beyond by providing decisive insight into the mechanisms involved. While the issue remains controversial, facts have been presented suggesting that insulin exerts a positive effect on its exocytosis. Compatible with this notion, transfection of IRs in beta cell lines increases Ins mRNA levels and glucose-induced insulin secretion [41]. In mice, both conditional and general knockout for proteins of the insulin signalling cascade, such as IR, IRS-1 and IRS-2, led to impaired glucose-induced insulin release [6–8]. Irs1 transfection in bTC6-F7 cells increases glucose-induced insulin secretion [42], and isolated islets from Irs1 −/− mice present a reduced insulin content and an impaired secretory response to glucose [7]. Therefore, inhibition of insulin signalling at the IRS and/or at PKB level due to MGO exposure may be responsible for the defect in insulin secretion. Studies performed in mice with one Pdx1 allele inactivated showed that PDX-1 deficiency alters glucose-induced insulin secretion [43]. Further, increased AGE levels induced by chronic hyperglycaemia lead to islet dysfunction associated with reduced glucokinase levels in DBA/2 mice [44]. We found that, after incubation of INS-1E cells with MGO, high glucose failed to increase expression of Pdx1, Ins1 and Gck genes. This effect of glucose appears to be PI3K-dependent, since it is abolished by pretreatment with a PI3K inhibitor. Further, in MGO-treated cells no increased PKB and GSK-3 phosphorylation was observed in response to high glucose, supporting the idea that activation of the PI3K/PKB cascade has an important impact on the preservation of beta cell function. Indeed, our data dovetail with a previous work showing that the overexpression of a dominant-negative Pkb (also known as Akt) in beta cells leads to impaired insulin secretion [45].
Recent facts have been reported showing that GSK-3β activation is linked to beta cell failure in diabetic mouse models, and is involved in negative regulation of beta cell proliferation and mass [46, 47]. Moreover, GSK-3β activity is enhanced in insulin-resistant states, and in skeletal muscle from type 2 diabetic patients [48]. Studies in transgenic mice overexpressing a constitutively active form of Gsk-3β in beta cells revealed that it leads to a profound decrease in PDX-1 levels compared with control mice, probably due to reduced stability [29]. In MGO-treated INS-1E cells, GSK-3 phosphorylation in response to insulin is reduced with a likely concurrent increase in its activation.
Interestingly, the LiCl-induced restoration of GSK-3 phosphorylation and its ensuing inhibition reverses the repressive effect of MGO on glucose-induced insulin secretion in INS-1E cells. This suggests that GSK-3 activation may be responsible, at least in part, for the damaging MGO effects on beta cell function. The molecular mechanisms underlying GSK-3 action on insulin secretion are poorly defined and we cannot exclude the possibility that MGO action is partially due also to the direct binding of MGO to beta cell transcription factors and/or to proteins involved in the insulin secretion machinery.
To summarise, our present work strongly indicates that toxic reactive aldehydes, such as MGO, impact negatively on beta cell homeostasis. Hence, the deleterious actions of these molecules generated by metabolic dysregulation will worsen the diabetes disease process by favouring beta cell failure. The development of new strategies to improve insulin transduction in beta cells will allow the preservation and/or improvement of beta cell function in type 2 diabetic patients suffering from elevated circulating levels of toxic aldehydes due to chronic hyperglycaemia.
Abbreviations
- AGE:
-
Advanced glycation end-product
- AG:
-
Aminoguanidine
- CEL:
-
N ε-(carboxyethyl)lysine
- CM-DCF:
-
Chloromethyl-2′7′-dichlorofluorescein diacetate
- GLO:
-
Glyoxalase
- GSK-3:
-
Glycogen synthase kinase-3
- IR:
-
Insulin receptor
- KRBH:
-
Krebs–Ringer bicarbonate HEPES
- MGO:
-
Methylglyoxal
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PDX-1:
-
Pancreatic duodenal homeobox-1
- PI3K:
-
Phosphatidylinositol 3-kinase
- PKB:
-
Protein kinase B
- p85:
-
p85 Alpha regulatory subunit of PI3K
- ROS:
-
Reactive oxygen species
References
Gallagher EJ, LeRoith D, Karnieli E (2008) The metabolic syndrome – from insulin resistance to obesity and diabetes. Endocrinol Metab Clin North Am 37:559–579
Ohsugi M, Cras-Méneur C, Zhou Y et al (2005) Reduced expression of the insulin receptor in mouse insulinoma (MIN6) cells reveals multiple roles of insulin signaling in gene expression, proliferation, insulin content, and secretion. J Biol Chem 280:4992–5003
da Silva Xavier G, Varadi A, Ainscow EK, Rutter GA (2000) Regulation of gene expression by glucose in pancreatic β-cells (MIN6) via insulin secretion and activation of phosphatidylinositol 3′-kinase. J Biol Chem 275:36269–36277
Leibiger IB, Leibiger B, Moede T, Berggren PO (1998) Exocytosis of insulin promotes insulin gene transcription via the insulin receptor/PI-3 kinase/p70 S6 kinase and CaM kinase pathways. Mol Cell 6:933–938
Aspinwall CA, Qian WJ, Roper MG, Kulkarni RN, Kahn CR, Kennedy RT (2000) Roles of insulin receptor substrate-1, phosphatidylinositol 3-kinase, and release of intracellular Ca2+ stores in insulin-stimulated insulin secretion in beta-cells. J Biol Chem 275:22331–22338
Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR (1999) Tissue-specific knockout of the insulin receptor in pancreatic β cells creates an insulin secretory defect similar to that of type 2 diabetes. Cell 96:329–339
Kulkarni RN, Winnay JN, Daniels M et al (1999) Altered function of insulin receptor substrate-1-deficient mouse islets and cultured β-cell lines. J Clin Invest 104:R69–R75
Withers DJ, Gutierrez JS, Towery H et al (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391:900–904
da Silva Xavier G, Qian Q, Cullen PJ, Rutter GA (2004) Distinct roles for insulin and insulin-like growth factor-1 receptors in pancreatic beta-cell glucose sensing revealed by RNA silencing. Biochem J 377:149–158
Schalkwijk CG, Brouwers O, Stehouwer CD (2008) Modulation of insulin action by advanced glycation end products: a new player in the field. Horm Metab Res 40:614–619
Thornalley PJ (2007) Endogenous alpha-oxoaldehydes and formation of protein and nucleotide advanced glycation endproducts in tissue damage. Novartis Found Symp 285:229–243, discussion 243–246
Thornalley PJ (2003) Glyoxalase I-structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans 31:1343–1348
Miele C, Riboulet A, Maitan MA et al (2003) Human glycated albumin affects glucose metabolism in L6 skeletal muscle cells by impairing insulin-induced insulin receptor substrate (IRS) signaling through a protein kinase C alpha-mediated mechanism. J Biol Chem 278:47376–47387
Cassese A, Esposito I, Fiory F et al (2008) In skeletal muscle advanced glycation end products (AGEs) inhibit insulin action and induce the formation of multimolecular complexes including the receptor for AGEs. J Biol Chem 283:36088–36099
Riboulet-Chavey A, Pierron A, Durand I, Murdaca J, Giudicelli J, Van Obberghen E (2006) Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 55:1289–1299
Pi J, Bai Y, Zhang Q et al (2007) Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 56:1783–1791
Cook LJ, Davies J, Yates AP et al (1998) Effects of methylglyoxal on rat pancreatic beta-cells. Biochem Pharmacol 55:1361–1367
Best L, Thornalley PJ (1999) Trioses and related substances: tools for the study of pancreatic beta-cell function. Biochem Pharmacol 57:583–588
Benson RS, Best L, Sheader EA (2001) Cytotoxic action of methylglyoxal on insulin-secreting cells. Biochem Pharmacol 61:1381–1386
Sartori A, Garay-Malpartida HM, Forni MF et al (2008) Aminoacetone, a putative endogenous source of methylglyoxal, causes oxidative stress and death to insulin-producing RINm5f cells. Chem Res Toxicol 21:1841–1850
Dhar A, Dhar I, Jiang B, Desai KM, Wu L (2011) Chronic methylglyoxal infusion by minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley rats. Diabetes 60:899–908
El Ouaamari A, Baroukh N, Martens GA, Lebrun P, Pipeleers D, Van Obberghen E (2008) miR-375 targets 3′-phosphoinositide-dependent protein kinase-1 and regulates glucose induced biological responses in pancreatic beta-cells. Diabetes 57:2708–2717
Li L, El-Kholy W, Rhodes CJ, Brubaker PL (2005) Glucagon-like peptide-1 protects beta cells from cytokine-induced apoptosis and necrosis: role of protein kinase B. Diabetologia 48:1339–1349
Oriente F, Fernandez Diaz LC, Miele C et al (2008) Prep1 deficiency induces protection from diabetes and increased insulin sensitivity through a p160-mediated mechanism. Mol Cell Biol 28:5634–5645
Merglen A, Theander S, Rubi B, Chaffard G, Wollheim CB, Maechler P (2004) Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 145:667–678
Che W, Asahi M, Takahashi M et al (1997) Selective induction of heparin-binding epidermal growth factor-like growth factor by methylglyoxal and 3-deoxyglucosone in rat aortic smooth muscle cells: the involvement of reactive oxygen species formation and a possible implication for atherogenesis in diabetes. J Biol Chem 272:18453–18459
Wu H, Macfarlane WM, Tadayyon M, Arch JR, James RF, Docherty K (1999) Insulin stimulates pancreatic-duodenal homeobox factor-1 (PDX1) DNA-binding activity and insulin promoter activity in pancreatic beta cells. Biochem J 344:813–818
Humphrey RK, Yu SM, Flores LE, Jhala US (2010) Glucose regulates steady-state levels of PDX1 via the reciprocal actions of GSK3 and AKT kinases. J Biol Chem 285:3406–3416
Liu Z, Tanabe K, Bernal-Mizrachi E, Permutt MA (2008) Mice with beta cell overexpression of glycogen synthase kinase-3 beta have reduced beta cell mass and proliferation. Diabetologia 51:623–631
Jope RS (2003) Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci 24:441–443
Ohsugi M, Cras-Méneur C, Zhou Y, Warren W, Bernal-Mizrachi E, Permutt MA (2004) Glucose and insulin treatment of insulinoma cells results in transcriptional regulation of a common set of genes. Diabetes 53:1496–1508
Newsholme P, Haber EP, Hirabara SM et al (2007) Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol 583:9–24
Robertson RP (2004) Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem 279:42351–42354
White MF (1998) The IRS-signaling system: a network of docking proteins that mediate insulin and cytokine action. Recent Prog Horm Res 53:119–138
Van Obberghen E, Baron V, Delahaye L et al (2001) Surfing the insulin signaling web. Eur J Clin Invest 3:966–977
Macfarlane WM, Smith SB, James RF et al (1997) The p38/reactivating kinase mitogen-activated protein kinase cascade mediates the activation of the transcription factor insulin upstream factor 1 and insulin gene transcription by high glucose in pancreatic beta-cells. J Biol Chem 272:20936–20944
Kaneto H, Miyatsuka T, Kawamori D et al (2008) PDX-1 and MafA play a crucial role in pancreatic beta-cell differentiation and maintenance of mature beta-cell function. Endocr J 55:235–252
Wang H, Iynedjian PB (1997) Modulation of glucose responsiveness of insulinoma beta-cells by graded overexpression of glucokinase. Proc Natl Acad Sci USA 94:4372–4377
Brouwers O, Niessen PM, Haenen G et al (2010) Hyperglycaemia-induced impairment of endothelium-dependent vasorelaxation in rat mesenteric arteries is mediated by intracellular methylglyoxal levels in a pathway dependent on oxidative stress. Diabetologia 53:989–1000
Bento CF, Fernandes R, Ramalho J et al (2010) The chaperone-dependent ubiquitin ligase CHIP targets HIF-1α for degradation in the presence of methylglyoxal. PLoS One 5:e15062
Xu GG, Rothenberg PL (1998) Insulin receptor signaling in the beta-cell influences insulin gene expression and insulin content: evidence for autocrine beta-cell regulation. Diabetes 47:1243–1252
Xu GG, Gao ZY, Borge PD Jr, Wolf BA (1999) Insulin receptor substrate 1-induced inhibition of endoplasmic reticulum Ca2+ uptake in beta-cells. Autocrine regulation of intracellular Ca2+ homeostasis and insulin secretion. J Biol Chem 274:18067–18074
Brissova M, Shiota M, Nicholson WE et al (2002) Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J Biol Chem 277:11225–11232
Kooptiwut S, Kebede M, Zraika S et al (2005) High glucose-induced impairment in insulin secretion is associated with reduction in islet glucokinase in a mouse model of susceptibility to islet dysfunction. J Mol Endocrinol 35:39–48
Bernal-Mizrachi E, Fatrai S, Johnson JD et al (2004) Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest 114:928–936
Stein J, Milewski WM, Hara M, Steiner DF, Dey A (2011) GSK-3 inactivation or depletion promotes β-cell replication via down regulation of the CDK inhibitor, p27 (Kip1). Islets 3:21–34
Tanabe K, Liu Z, Patel S et al (2008) Genetic deficiency of glycogen synthase kinase-3 beta corrects diabetes in mouse models of insulin resistance. PLoS Biol 6:e37
Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR (2000) Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes 49:263–271
Acknowledgements
The research in Nice was supported by Inserm, Université de Nice Sophia Antipolis, Conseil Régional PACA and Conseil Général des Alpes-Maritimes, by the European Community FP6 EUGENE2 (grant LSHM-CT-2004-512013). F. Fiory was supported by a grant-in-aid from Servier (Neuilly-France) to E. Van Obberghen. The research in Naples was supported by the European Foundation for the Study of Diabetes (EFSD), the European Community’s PREPOBEDIA(201681) and by the Ministero dell’Università e della Ricerca Scientifica. The financial support of Telethon, Italy, to F. Beguinot is gratefully acknowledged. We thank A. Salvayre-Negre for antibodies to argpyrimidine.
Contribution statement
FF, AL, CM, JG and EVO were responsible for the conception and design of the article: FF, AL, CM, and JG performed the experiments. FF, CM, JG, FB and EVO analysed and interpreted data, and drafted the article: FF, AL, CM, JG, FB and EVO revised the paper critically for important intellectual content: All authors approved the final version.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article can be found at http://dx.doi.org/10.1007/s00125-011-2321-3
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
PDF 92 kb
ESM Table 1
PDF 111 kb
ESM Table 2
PDF 87 kb
Rights and permissions
About this article
Cite this article
Fiory, F., Lombardi, A., Miele, C. et al. Methylglyoxal impairs insulin signalling and insulin action on glucose-induced insulin secretion in the pancreatic beta cell line INS-1E. Diabetologia 54, 2941–2952 (2011). https://doi.org/10.1007/s00125-011-2280-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2280-8