
Abstract
Aims/hypothesis
The role of Toll-like receptor 7 (TLR7), a sensor of viral and self RNA, in promoting autoimmune diabetes remains unclear. Our goal was to determine the effect of TLR7 stimulation on the priming and activation of diabetogenic CD8+ T cells.
Methods
We explored the effects of CL097 (TLR7/8 agonist) and immunoregulatory sequence 661 (IRS661, TLR7 inhibitor) on bone marrow-derived dendritic cells (BMDCs), diabetogenic CD8+ T cell function and autoimmune diabetes onset in NOD and 8.3 NOD T cell receptor transgenic mice (8.3 NOD mice).
Results
TLR7 stimulation of NOD BMDCs increased activation and production of proinflammatory cytokines. In vivo administration of CL097 activated T cells and dendritic cells and increased levels of proinflammatory cytokines and type 1/2 IFNs in NOD mice. In vivo antigen-specific cytotoxicity studies revealed enhanced cytotoxicity against islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP, an islet autoantigen) peptide pulsed targets in NOD mice treated with CL097 plus CD40 agonist. This combination treatment accelerated the onset of autoimmune diabetes in 8.3 NOD mice. Likewise, topical treatment of NOD mice with a TLR7 agonist accelerated diabetes onset. Spontaneous disease in 8.3 NOD mice and accelerated disease in CL097+CD40 agonist-treated 8.3 NOD mice were delayed by IRS661 treatment, which is associated with inhibition of the endogenous upregulation of IFN-α levels within the pancreatic lymph nodes.
Conclusions/interpretation
TLR7 stimulation accelerates the spontaneous onset of autoimmune diabetes in 8.3 NOD and NOD mice. Conversely, TLR7 inhibition prevents the early events associated with diabetogenesis.
Similar content being viewed by others

Introduction
Type 1 diabetes is an organ-specific autoimmune disease in which beta cells are destroyed by the immune system [1, 2]. Much of the beta cell destruction is mediated by self-reactive cytotoxic T lymphocytes (CTLs) [1, 2]. Both genetic and environmental factors play a pivotal role in the development of type 1 diabetes [3–5]. It has been suggested that Toll-like receptors (TLRs) may trigger the pathogenesis of type 1 diabetes [6, 7]. TLRs act as pathogen sensors that recognise distinct patterns on pathogens and influence the production of proinflammatory cytokines and chemokines in dendritic cells (DCs) and macrophages [8–10]. TLRs can also recognise self molecules such as endogenous RNA and DNA [11]. The inappropriate activation of TLRs results in DC maturation and has been proposed to drive the priming and activation of self-reactive CTLs [12].
Endosomal TLRs (TLR3, 7, 8 and 9) recognise RNA or DNA oligonucleotides and have been linked to the development of autoimmune disorders such as systemic lupus erythematosus, rheumatoid arthritis [12, 13] and psoriasis [14]. Slowly cleared apoptotic cells may stimulate endosomal TLRs. In the NOD mouse the clearance of apoptotic cells by macrophages is delayed [15, 16] and the death of beta cells promotes the activation and proliferation of diabetogenic CTLs [17]. Viruses may also precipitate type 1 diabetes in animal models and in humans [18, 19]. Coxsackie B viruses stimulate TLR7 and have been implicated as a trigger for the development of type 1 diabetes [20, 21]. A transgenic mouse model of type 1 diabetes in which lymphocytic choriomeningitis virus (LCMV) glycoprotein (GP) is expressed on beta cells and in which mice harbour a large population of LCMV GP specific CD8+ T cells, requires TLR3 or 7 stimulation by synthetic agonists or live virus for the induction of disease [22]. More recently, increases in IFN-α within the pancreatic lymph nodes as a result of release by plasmacytoid DCs (pDCs) have been shown to be critical for the initiation of spontaneous diabetes in NOD mice [23]. Why pDCs within the pancreatic lymph nodes of NOD mice contain high levels of IFN-α remains unclear. pDCs are a major source of IFN-α following viral infection, and self RNA may activate these cells through TLR7 [24]. We sought to further investigate the role of TLR7 in autoimmune diabetes in the NOD mouse. We studied the effect of TLR7 agonists and antagonists on the activation of diabetogenic T cells (responding to physiological levels of self antigen) and the development of diabetes using the NOD mouse model.
Methods
Animals and reagents
NOD mice (Jackson Laboratory, Bar Harbor, ME, USA) were bred in a specific-pathogen-free environment. The 8.3 NOD T cell receptor transgenic mice (8.3 NOD mice) [25] were kindly provided by P. Santamaria (Julia McFarlane Diabetes Research Centre, University of Calgary, Calgary, AB, Canada). All animal studies were approved by our institutional animal ethics committee.
The TLR agonists and inhibitors used in both the in vitro and in vivo experiments were: (1) TLR7 agonists: R837 (imiquimod; InvivoGen, San Diego, CA, USA), prAldaraTM/MC cream 5% (imiquimod) (GracewayTM/MC, Bristol, TN, USA) and loxoribine (LOX) (InvivoGen); (2) TLR7/8 agonist: CL097 (InvivoGen); (3) TLR9 agonist: CpG oligodeoxynucleotide (ODN) 1826 (5′-TCCATGACGTTCCTGACGTT-3′, phosphorothioate backbone) (IDT, San Diego, CA, USA); (4) TLR7 inhibitor: immunoregulatory sequence 661 (IRS661, described by Barrat et al. [26]) (IDT); and (5) control CpG ODN 1982 (5′-TCCAGGACTTCTCTCAGGTT-3′ phosphorothioate backbone) (IDT). Monoclonal anti-CD40 (CD40 agonist), agonistic antibody to CD40 (FGK45) was purified in-house.
Generation and stimulation/inhibition of mouse bone marrow-derived DCs
Bone marrow-derived DCs (BMDCs) were generated from murine bone marrow precursors in complete media (CM) consisting of RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS (vol./vol.), penicillin (100 U/ml) and streptomycin (100 μg/ml), as previously described [27]. Bone marrow precursor cells were flushed out from extracted femur bones of NOD mice and cultured for 6 days in CM. At days 1 and 3 of culturing, granulocyte-macrophage colony-stimulating factor (supernatant fractions from the X63 B cell line), IL-4 (1 ng/ml, Sigma-Aldrich, St Louis, MO, USA) and 10% FBS RPMI were added to the cultures. At day 6, immature BMDCs were stimulated with R837 (5 μg/ml; 20.8 μmol/l), LOX (170 μg/ml; 500 μmol/l), CL097 (1 μg/ml; 4.13 μmol/l) or CpG 1826 (5 μg/ml; 0.786 μmol/l) for 24 h. TLR7/8- or 9-activated BMDCs were incubated with IRS661 (9 μg/ml; 1.4 μmol/l) for 24 h.
Detection of activation/inhibition of NOD-derived BMDCs,CD4+ and CD8+ T cells, pDCs and conventional DCs in NOD mice
NOD BMDCs were stimulated with TLR agonists or inhibitors for 24 h, incubated with anti-Fc receptor (FcR) monoclonal antibody (2.4 G2) (American Type Culture Collection, Manassas, VA, USA) or with mouse IgG (Sigma-Aldrich) and analysed by FACSCalibur flow cytometer (BD Bioscience, San Jose, CA, USA) for CD11c+ DCs. Allophycocyanin (APC)- or phycoerythrin (PE)-conjugated anti-CD11c, and APC-conjugated anti-CD40 (all from BD Pharmingen, Mississauga, ON, Canada) were used to detect BMDC activation.
Female NOD or 8.3 NOD mice (6–8 weeks old) were injected s.c. with either CL097 (5 mg/kg) or CpG 1826 (5 mg/kg) to examine CD4+ and CD8+ T cells (24 h post-injection) or plasmacytoid and conventional DCs (6 h post-injection). For TLR7 inhibition, IRS661 (266 mg/kg) was administered s.c. 2 h prior to CL097 treatment. At 6 or 24 h post-injection, the axillary, mesenteric and pancreatic lymph nodes and the spleen were extracted, stained with fluorescent antibodies and analysed by FACS for CD4+ and CD8+ T cell, and plasmacytoid and conventional DC activation. DCs were isolated from lymph nodes and spleen using 1 mg/ml collagenase D (Roche Diagnostics, Mississauga, ON, Canada) supplemented with 40 g/ml DNase (Boehringer Mannheim, Laval, QC, Canada) in CM at 37°C for 60 min. The released cells were collected in CM with 0.1 mol/l EDTA buffer, incubated with anti-FcR monoclonal antibody, stained with fluorescent antibodies and analysed by FACS. Plasmacytoid DCs were identified as: B220+PDCA1+CD11clo whereas conventional DCs were identified as B220–PDCA1–CD11chi. CD4+ and CD8+ T cells were identified as B220− and either CD4+ or CD8+, respectively. Diabetogenic CD8+ T cells were identified as B220−CD8+NRP-V7+ using a tetramer (NRP-V7) that detects CTL specific for islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) peptide [28]. The following monoclonal antibodies were used for T cell and DC identification and activation in NOD mice: FITC-conjugated anti-CD4, peridinin–cholorophyll protein complex (PerCP)-conjugated anti-CD45R/B220, APC- or PE-conjugated anti-CD8, PE-conjugated NRP-V7 tetramer, FITC-conjugated anti-CD11c, PE-conjugated anti-PDCA1 (Cedarlane, Burlington, ON, Canada), FITC- or PE- conjugated anti-CD69, and APC-conjugated anti-CD40 (BD Pharmingen).
Cytokine assays
Supernatant fractions were collected from NOD BMDCs that had been stimulated with TLR agonists or inhibitors for 24 h. Sera were collected from NOD mice at 2 and 6 h post-injection with CL097 or CpG 1826 or in combination with IRS661. Cytokines were analysed using the mouse inflammation cytometric bead array (CBA) (BD Pharmingen). The CBA assay allows the simultaneous detection and quantification of soluble murine cytokines IL-6, IFN-γ, TNF-α and IL-12p70 and monocyte chemotactic protein-1 (MCP-1) in a single sample. IFN-α was measured using an IFN-α ELISA kit (PBL Biomedical Laboratories, Piscataway, NJ, USA).
Female NOD mice (3–4 weeks old) were injected s.c. with IRS661 (266 mg/kg), ODN 1982 (5 mg/kg) or CL097 (5 mg/kg) for 24 h before tissue extraction. The preparation and assessment of IFN-α production in lymph node homogenates were carried out using a modification of the method described by Li et al. [23]. Briefly, lymph nodes were extracted and homogenised into a single-cell suspension in 2% FBS PBS. Lymph node homogenates were lysed with radioimmunoprecipitation assay buffer (Roche Diagnostics; 50 mmol/l Tris-HCl [pH 7.4], 1% NP-40, 0.5% sodium deoxycholate, 150 mmol/l NaCl and 1 mmol/l EDTA) in the presence of a cocktail of protease inhibitors (complete protease inhibitor cocktail tablets) (Roche Diagnostics). Supernatant fractions from centrifuged lysates were removed and titrated for IFN-α levels by ELISA (R&D Systems, Minneapolis, MN, USA).
In vivo cytotoxicity assay
Female NOD mice (6–8 weeks old) were injected with CL097 (5 mg/kg; s.c.), CpG 1826 (5 mg/kg; s.c.) or CD40 agonist (10 mg/kg, i.p.) or in combination. The in vivo cytotoxicity assay was adapted from one described by Jarchum et al. [29]. Briefly, splenocytes from wild-type NOD mice were differentially labelled with 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE) by exposing to 0.3 μmol/l CFSE (CFSE LO) or 5 μmol/l CFSE (CFSE HI) for 10 min. The CFSE HI cells were pulsed with the target peptide, NRP-V7 (KYNKANVEL; islet cell mimotope of IGRP [28]; 1 μg/ml), and the CFSE LO cells were pulsed with the control peptide, TUM (KYQAVTTTL; 1 μg/ml), for 1 h. A 1:1 mixture of CFSE-labelled and pulsed splenocytes (1 × 107 CFSE HI with NRP-V7 peptide:1 × 107 CFSE LO with TUM peptide) was adoptively transferred i.v. into mice injected 24 h previously. On day 5 after adoptive transfer, splenocytes were collected and analysed by FACS. The numbers of target (CFSE LO) vs control (CFSE HI) cells recovered were determined and used to calculate the percentage of killing using the following formula: \( \% {\hbox{ specific lysis}} = {1} - \left[ {\left( {{\hbox{no}}.{\hbox{ of targets}}/{\hbox{no}}.{\hbox{ of control cells in injected animal}}} \right)/\left( {{\hbox{no}}.{\hbox{ of targets}}/{\hbox{no}}.{\hbox{ of control cells in control animal}}} \right)} \right] \times {1}00. \)
Induction, acceleration, inhibition and diagnosis of diabetes
Female 8.3 NOD mice (5–6 weeks old) were injected with CL097 (5 mg/kg, s.c.), CD40 agonist (10 mg/kg, i.p.) or IRS661 (266 mg/kg, s.c.), either alone or in combination. IRS661 was injected 2 h prior to CL097+CD40 agonist treatment in 8.3 NOD mice. Furthermore, IRS661 (266 mg/kg; s.c.) was injected only three times (at 6, 7 and 9 weeks of age) into 8.3 NOD mice in the long-term diabetes experiment. Topical R837 (prAldaraTM/MC cream 5%; 20 mg) was applied on the shaved back skin (1 cm2 treatment area) of female NOD mice (6–8 weeks old) weekly. Acceleration of diabetes was considered to occur within 14 days post-injection in 8.3 NOD mice. Blood glucose was measured in tail vein blood using Ascensia Contour glucostrips and glucometer (Bayer, Toronto, ON, Canada). Animals were considered to be diabetic when two consecutive blood glucose measurements exceeded 14 mmol/l.
Statistical analysis
A one-way ANOVA was used to calculate statistical significance with post hoc corrections for multiple comparisons where appropriate. A logrank test was applied to compare survival curves. All statistical analyses were conducted using Prism 3 GraphPad software (La Jolla, CA, USA).
Results
TLR7/9 agonists activate NOD-derived BMDCs and cause the secretion of proinflammatory cytokines and chemokines
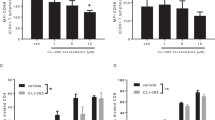
The NOD mouse provides a convenient model of autoimmune diabetes that allows analysis of the initial stages of the disease. TLR agonists activate DCs, and NOD DCs have altered reactivity compared with those in strains without autoimmune disease [30]. Antigen-presenting cell TLR mRNA levels (Tlr3, Tlr4 and Tlr5) are higher in NOD mice than in diabetes-resistant strains and increase further with disease onset [31]. To understand the role of TLR7 in the pathogenesis of autoimmune diabetes we screened small molecular TLR7 agonists for activation of BMDCs derived from the NOD mouse. NOD-derived BMDCs stimulated with the TLR7/8 agonist CL097 or either of the TLR7 agonists (R837 or LOX) showed increased levels of CD40 (Fig. 1a) and other markers of DC activation such as CD86 (data not shown), and these increases were comparable to those induced by the TLR9 agonist CpG 1826. A dose titration of each TLR7 agonist showed that CL097 was the most potent and effective activator of BMDCs in vitro and DCs in vivo in NOD mice (data not shown). Supernatant fractions collected from TLR7- and TLR9-stimulated BMDCs revealed secretion of proinflammatory cytokines (IL-6, TNF-α and IL-12p70) and a chemokine (MCP-1) (Fig. 1b–e). The TLR7/8 agonist CL097 induced the production of TNF-α and IL-6 but not IL-12p70 or MCP-1. Compared with the TLR7 agonists, the TLR9 agonist induced higher levels of IL-12p70 and MCP-1.

TLR7/9 activation of BMDCs from the NOD mouse. a NOD BMDCs were stimulated with optimal doses of different TLR7/8 and 9 agonists for 24 h. Flow cytometry was used to gate for CD11c+ DCs. The mean fluorescence intensity (MFI) of the activation marker CD40 was assessed to determine DC activation. b–e Culture supernatant fractions were collected (after 24 h) from TLR7/9-stimulated BMDCs and analysed by cytometric bead array for IL-6 (b), TNF-α (c), IL-12p70 (d) and MCP-1 (e). Data are representative of at least five independent experiments. Each TLR7, -7/8 or -9 agonist group was compared with the unstimulated control (DCs). *p < 0.05, † p < 0.006 by one-way ANOVA
CL097 induces the general activation of CD4+ and CD8+ T cells and pDCs and conventional DCs in NOD mice
We next examined the role of TLR7 activation in vivo by titrating CL097 in NOD mice. The s.c. or i.p. (data not shown) administration of CL097 (5 mg/kg) resulted in the general activation of CD4+ and CD8+ T cells in the axillary, mesenteric and pancreatic lymph nodes and in the spleen (Fig. 2a,b). CL097 administration also caused the general activation of pDCs and conventional DCs in the mesenteric and pancreatic lymph nodes and spleen (Fig. 2d,e). In addition, CL097 induced a statistically significant increase in serum levels of proinflammatory cytokines (IL-6, TNF-α and IL-12p70), a chemokine (MCP-1) and both type 1 and 2 IFNs (IFN-α and IFN-γ) compared with the negative control (Fig. 2c,f). Similar increases were observed in NOD mice injected with CpG 1826 (5 mg/kg, s.c. or i.p.; Fig. 2). Thus, treatment of NOD mice with TLR9 agonist (CpG 1826) or TLR7/8 agonist (CL097) activated a broad repertoire of CD4+ and CD8+ T cells. However, the TLR7/8 agonist was a more potent activator of pDCs than the TLR9 agonist (as determined by surface CD40 levels at 6 h post-injection). In addition, early (2 h post-injection) serum levels of IFN-α were higher following treatment with the TLR7/8 agonist in NOD mice. Similar responses to CL097 in terms of increases in IL-6, TNF-α and IL-12p70 were noted in non-diabetogenic C57BL/6 mice (data not shown).

CL097 induces the general activation of T cells and DCs, and increases proinflammatory cytokines/chemokine and type 1/2 IFNs in the NOD mouse. a CL097 or CpG 1826 was administered s.c. to the NOD mouse. At 24 h post-injection, the axillary, mesenteric and pancreatic lymph nodes (ALN, MLN and PLN) and spleen were extracted and analysed by flow cytometry. The activation marker CD69 was used to determine activation of CD4+ (a) and CD8+ (b) T cells. At 6 h post-injection, the activation marker CD40 was used to determine activation of pDCs (d, B220+PDCA1+CD11clo) and conventional DCs (e, B220–PDCA1–CD11chi). Serum levels of IL-6, TNF-α, IL-12p70, MCP-1 (c), IFN-α and IFN-γ (f) were measured at 2 h and 6 h following injection of CL097 (5 mg/kg) or CpG (5 mg/kg). Cytokines were measured by cytometric bead array or by ELISA. Data are representative of at least three independent experiments. CL097 or CpG 1826 groups were compared with the PBS group. *p < 0.05, † p < 0.005 by one-way ANOVA
CL097 and CD40 agonist stimulation induces efficient diabetogenic CTL function in NOD mice
We assessed whether CL097 could promote the priming and activation of functional diabetogenic CTLs in NOD mice in response to endogenous autoantigen using an in vivo antigen-specific cytotoxicity assay. Naive splenocytes from a NOD mouse were CFSE-labelled (CFSE HI or CFSE LO) and pulsed with a target peptide (NRP-V7, islet cell mimotope of IGRP, a critical islet autoantigen [28]) or a control peptide (TUM) and injected into treated NOD mice. We found that CL097 (5 mg/kg, s.c.) alone caused a modest specific lysis of the target peptide (∼25%; Fig. 3a–d). However, treatment with a combination of CL097 and CD40 agonist (10 mg/kg, i.p.) resulted in an increase of approximately twofold in the specific lysis of the IGRP-peptide-coated targets compared with CL097 treatment alone (Fig. 3a–d). Interestingly, the IGRP-specific lysis induced by the CL097+CD40 agonist combination was even greater than that induced by the CpG+CD40 agonist combination. CD40 ligand activation of DCs is essential for the generation of functional CTLs [32, 33]. The principal source of CD40 ligand is thought to be activated CD4+ T cells [33]. The efficient target lysis observed with combination therapy indicates that DC stimulation with CD40 in combination with TLR7/8 or TLR9 agonists induces efficient activation of diabetogenic CTLs in response to endogenous autoantigen in the NOD mouse.

Effect of CL097, CpG 1826 or CD40 agonist, alone or in combination as indicated, on diabetogenic CD8+ T cell function in NOD mice and on the general activation of polyclonal and diabetogenic CD8+ T cells in 8.3 NOD mice. Representative histograms of analysis of adoptively transferred cells recovered from spleen used to calculate the per cent specific lysis of naive (a), CL097+CD40 agonist (b) and CpG+CD40 agonist (c) treatment groups. d Summary of the per cent specific lysis of target peptide (NRP-V7) in the spleen in each treatment group, as assessed by flow cytometry. *p < 0.05, **p < 0.005 by one-way ANOVA. e CL097 or CpG 1826 were administered s.c. in 8.3 NOD mice. At 24 h post-injection, the axillary, mesenteric, and pancreatic lymph nodes (ALN, MLN and PLN) and spleen were extracted and analysed by flow cytometry. The activation marker CD69 was used to determine activation of polyclonal (IGRP tetramer–B220–CD8+) CD8+ T cells (grey bars) and diabetogenic (IGRP tetramer+B220–CD8+) CD8+ T cells (black bars) that recognise the islet antigen IGRP. Data are representative of at least two independent experiments. CL097 or CpG 1826 groups were compared with the corresponding cell types in the PBS group. Data are representative of three independent experiments. *p < 0.05, **p < 0.005 vs endogenous CD8+ T cells in the respective group; † p < 0.05 vs diabetogenic CD8+ T cells in the respective group; ‡ p < 0.005 by one-way ANOVA
Does the broad immune activation mediated by TLR7 or TLR9 agonists favour autoantigen presentation?
To further examine the effect of these agonists on the priming of diabetogenic T cells, we studied their effects on 8.3 NOD mice, animals that harbour a large population of diabetogenic CD8+ T cells specific for the islet antigen IGRP [28, 34, 35]. These mice have an accelerated onset of diabetes owing to increased frequencies of diabetogenic T cells and alterations in T cell regulation, but are useful tools to examine the cross-presentation of diabetogenic autoantigen. Furthermore, the islet autoantigen detected by CTLs in these mice is relevant and immunodominant in both mice and humans. Treatment of 8.3 NOD mice with CL097 or CpG 1826 resulted in the increased activation of both endogenous (non-transgenic) and diabetogenic CD8+ T cells in the axillary, mesenteric and pancreatic lymph nodes and spleen (Fig. 3e). Endogenous CD8+ T cell activation was greater in the axillary and mesenteric lymph nodes and spleen whereas diabetogenic CD8+ T cell activation was greater in the pancreatic lymph nodes, where pancreatic self antigen is presented. Thus both TLR7/8 and TLR9 agonists are able to induce the activation of both endogenous and diabetogenic T cell populations, and autoantigen-specific priming is favoured in the lymph nodes draining the target organ.
Treatment with CL097+CD40 agonist induces the acceleration of autoimmune diabetes in 8.3 NOD mice
The increased activity of functional diabetogenic CTLs in NOD mice treated with CL097+CD40 agonist suggests that this treatment combination could accelerate the development of autoimmune diabetes in 8.3 NOD mice. Treatment of 8.3 NOD mice with CL097+CD40 agonist caused a rapid acceleration of the development of autoimmune diabetes compared with the negative control (Fig. 4a). The administration of CL097 or CD40 agonist alone did not result in a significant acceleration of the development of autoimmune diabetes in 8.3 NOD mice, suggesting that promotion of diabetogenesis requires multiple stimuli.

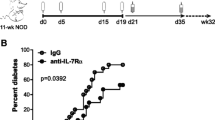
Effect of CL097 or imiquimod (TLR7 agonist), CD40 agonist and IRS661 (TLR7 inhibitor) on the onset of autoimmune diabetes in 8.3 NOD or NOD mice. a, b Diabetes survival curve at 14 days post-injection (PI) of 8.3 NOD mice. The 5–6-week-old female 8.3 NOD mice were treated once with CL097, CD40 agonist or IRS661 alone or in combination. IRS661 was injected 2 h prior to CL097+CD40 agonist treatment. The endpoint of accelerated diabetes was established to be within 14 days PI in 8.3 NOD mice. Black squares, control (n = 11); black diamonds, CL097 alone (n = 6); black triangles, CD40 agonist alone (n = 6); black circles, CL097+CD40 agonist (n = 13); black downward triangles, CL097+CD40 agonist+IRS661 (n = 12). p < 0.005 for CL097+CD40 agonist vs control, CL097+CD40 agonist+IRS661 vs control and CL097+CD40 agonist+IRS661 vs CL097+CD40 agonist by log-rank test on survival curves. c Repeated injection of IRS661 (three times only) delays the spontaneous onset of diabetes in 8.3 NOD mice. Diabetes incidence was measured PI (after first IRS661 dose) in these mice. Black squares, control (n = 13); black triangles, IRS661 (n = 14). p < 0.0001 for IRS661 vs control by log-rank test on survival curves. d Topical imiquimod was applied to the back dorsal skin of 6–8-week-old NOD females weekly. Blood glucose was monitored twice weekly and mice were termed diabetic after two consecutive blood glucose measurements of >14 mmol/l. Black squares, control (n = 22); black diamonds, imiquimod (n = 21). p < 0.05 for imiquimod vs control by log-rank test on survival curves

Effect of IRS661 on endogenous IFN-α production in the pancreatic lymph nodes of NOD mice. a Endogenous IFN-α levels in the pancreatic lymph node homogenates of naive female C57BL/6 (3–4 weeks old) and NOD mice (2, 3–4, 6 and 12 weeks old). b Endogenous IFN-α levels in the pancreatic lymph node homogenates of 3–4-week-old NOD mice injected once (and analysed 24 h post-injection) s.c. with IRS661, ODN 1982 or CL097. Data were obtained from two independent experiments. The CL097 group was compared with the C57BL/6 group. *p < 0.05, **p < 0.005 by one-way ANOVA
IRS661 inhibition of TLR7-induced effects in NOD and 8.3 NOD mice
To investigate whether TLR7 inhibition could be used therapeutically to alter the onset of type 1 diabetes we observed the effect of the TLR7 oligonucleotide inhibitor IRS661 on TLR7-stimulated NOD BMDCs and on TLR7 agonist-treated NOD mice. IRS661 was found to inhibit TLR7- (CL097, R837 and LOX) and partially inhibit TLR9- (CpG 1826) induced activation of NOD BMDCs (Electronic supplementary material [ESM] Fig. 1a–e). In addition, IRS661 (266 mg/kg, s.c.) inhibited the general activation of CD4+ and CD8+ T cells in the axillary, mesenteric and pancreatic lymph nodes and spleen of both CL097- and CpG 1826-injected NOD mice (ESM Fig. 2). Furthermore, IRS661 abrogated the elevations in serum levels of IL-6, TNF-α, IL-12p70, IFN-γ and MCP-1 in NOD mice treated with CL097 or CpG 1826 (ESM Fig. 3a–e). We next determined the effect of IRS661 on the TLR7 agonist-induced acceleration of diabetes development. A single treatment of IRS661 in 8.3 NOD mice injected with CL097+CD40 agonist blunted the acceleration of the development of autoimmune diabetes (Fig. 4b). Repeated treatment of 8.3 NOD mice with IRS661 (three times only) dramatically delayed the spontaneous onset of diabetes in these animals (Fig. 4c). Use of the control oligonucleotide, ODN 1982, did not affect the increased rate of development of diabetes in the CL097+CD40 agonist-treated mice ([36] and data not shown).
Topical R837 (imiquimod) accelerates the development of diabetes in NOD mice
To determine whether TLR7 agonists promote the development of diabetes in mice that harbour a more diverse repertoire of auto-reactive T cells, we used the TLR7 agonist R837 (imiquimod) on NOD mice. We applied the agonist topically as this is the form used clinically for the treatment of skin diseases. Topical treatment with imiquimod on the shaved dorsal back skin of female NOD mice resulted in a modest but significant increase (p = 0.02) in the development of autoimmune diabetes compared with the control (Fig. 4d). This acceleration in the development of autoimmune diabetes in NOD mice suggests that TLR stimulation with topical TLR7 agonists may promote the onset of autoimmune diabetes in susceptible individuals.
IRS661 inhibits endogenous IFN-α levels in the pancreatic lymph nodes in young prediabetic (3–4 weeks old) NOD mice
We assessed whether TLR7 inhibition affects the early events associated with the initiation of autoimmune diabetes in NOD mice. Li et al. [23] reported the presence of high endogenous IFN-α levels in the pancreatic lymph nodes of young prediabetic (3–4 weeks old) NOD mice, and these were associated with the onset of autoimmune diabetes. Similarly, we observed high endogenous IFN-α levels in the pancreatic lymph nodes in NOD mice of 3–4 weeks of age (Fig. 5a). This observation was specific to the pancreatic lymph nodes and was not found in other lymph nodes (axillary or mesenteric), spleen or pancreas in NOD mice (ESM Fig. 4). The treatment of young prediabetic NOD mice with IRS661 (TLR7 inhibitor) specifically decreased the endogenous IFN-α levels in the pancreatic lymph nodes (Fig. 5b and ESM Fig. 4). This decrease in endogenous IFN-α levels was not observed when NOD mice were treated with control oligonucleotide (ODN 1982). This demonstrates that TLR7 stimulation is involved in local IFN-α induction in the pancreatic lymph nodes.
Discussion
TLR7 signalling in conjunction with CD40 activation of DCs induced the activation of functional diabetogenic CTLs in the pancreatic lymph nodes of NOD mice. TLR7 and CD40 agonism accelerated the onset of autoimmune diabetes in 8.3 NOD mice. Inhibition of TLR7 signalling with oligonucleotide IRS661 prevented this acceleration of autoimmune diabetes and delayed the onset of spontaneous autoimmunity. Repeated application of the commercially available topical TLR7 agonist imiquimod resulted in acceleration of spontaneous autoimmune diabetes in NOD mice. Furthermore, inhibition of TLR7 signalling with IRS661 blocked the high endogenous IFN-α levels in the pancreatic lymph nodes of young prediabetic NOD mice, which was associated with the initiation of autoimmune diabetes in these mice. Taken together, these observations demonstrate that TLR7 stimulation prior to and at the time of early insulitis promotes autoimmunity in NOD mice.
Studies of TLR signalling in the development of autoimmune diabetes have revealed complex roles for this pathway. Abrogation of MyD88-mediated TLR signalling in NOD mice results in resistance to diabetes [37]. However, MyD88-deficient mice raised in a germ-free environment develop disease, indicating that MyD88 signalling is not required for diabetogenesis and that TLR-mediated changes in gut flora may prevent diabetes in a MyD88-independent fashion [37]. Previous work has demonstrated that exogenous TLR7 or TLR3 agonists increase IFN-α and pancreatic islet expression of MHC class I and transgenic islet antigens [38]. In this paper we show that administration of a single dose of TLR7/8 agonist+CD40 agonist in 8.3 NOD mice and repeated administration of TLR7 agonist in NOD mice accelerates the development of spontaneous diabetes. This provides a potential mechanism whereby repeated viral infections may precipitate diabetes [18]: coxsackie B virus, a virus previously associated with the onset of autoimmune diabetes in humans, stimulates myocardial autoimmunity through TLR7/8 activation [21]. Recent studies have shown that islet cells infected with human enterovirus activate DCs via an endosomal acidification-dependent mechanism [39], suggesting a role for TLR7 or TLR9 activation. The presence of TLR7 on antigen-presenting cells within islets of individuals with recent-onset diabetes has also been demonstrated [40].
Repeated topical application of a TLR7 agonist promotes diabetes. This suggests that repeated topical application of TLR7 agonists should be performed with caution in individuals at risk for autoimmune disease. Repeated high doses of parenteral TLR7 agonist prevent diabetes in NOD mice [41]. However, the relevance of repeated high-dose stimulation of TLR7 to human disease is unclear. Repeated administration of TLR9 agonist disrupts lymph node and splenic architecture [42] and repeated dosing in a rapid schedule induces TLR signalling tolerance [43]. Either mechanism may explain disease inhibition following repeated exposure to high doses of TLR7/8 or TLR9 agonists.
The agonist CL097 used in our studies stimulates both TLR7 and TLR8 and was chosen as it provided the most robust immune activation in vivo of the TLR7 agonists tested. TLR8 has been thought to be non-functional in the mouse. Recent data indicate that TLR8 activation may regulate TLR7 levels [44] and that TLR8 may be able to recognise viral DNA in the mouse [45]. It is thus possible that modulation of TLR8 activity contributes to the effects observed in our system. Collectively our data and the previous work indicate that different effects on autoimmunity are produced depending on the dose, route and frequency of TLR stimulation.
The demonstration that pancreatic lymph node upregulation of IFN-α levels by pDCs is critical for the onset of diabetes in the NOD mouse [23] suggests that endogenous stimulators of IFN-α, and in particular activators of pDCs, may participate in the initiation of diabetes. Tlr9 –/– NOD mice have a delayed onset of diabetes [36, 46], and this coincides with decreased IFN-α levels in the pancreatic lymph nodes of young Tlr9 –/– NOD mice [36], suggesting that endogenous TLR9 activation promotes diabetes onset through the activation of pDCs and the generation of pancreatic lymph node IFN-α. Comparison of the in vivo effects of TLR7 (CL097) and TLR9 (CpG 1826) agonists demonstrates that while both activate the immune system generally and diabetogenic T cells in particular, the TLR7 agonist is a more potent activator of pDCs and IFN-α (Fig. 2d–f). The inhibition of pancreatic lymph node IFN-α production by the TLR7 oligonucleotide inhibitor IRS661 further suggests that endogenous TLR7 stimulation promotes the initiation of diabetes. In murine models of systemic lupus erythematosus, TLR7-deficient animals demonstrate attenuated disease [47]. Our work suggests that TLR7 activation may also promote the development of diabetes. The demonstration of inhibition of diabetogenesis in 8.3 NOD mice and inhibition of IFN-α production in the pancreatic lymph nodes of NOD mice suggest that endogenous TLR7 stimulation at the initial stages of diabetes promotes disease.
IRS661 diminishes pancreatic IFN-α production in young NOD mice. Likewise, the TLR7 and TLR9 inhibitor chloroquine delays the onset of diabetes in NOD mice [36]. IRS661 was initially identified as a specific TLR7 inhibitor [26]. While IRS661 is much more effective at inhibiting TLR7 activation than TLR9 activation of NOD BMDCs in vitro (ESM Fig. 1a–e), this inhibitor also demonstrates modest TLR9 inhibitory effects in vivo (ESM Figs 2 and 3). Our observations thus support the further development of TLR7 and possibly TLR9 antagonists for the prevention and treatment of autoimmune diabetes.
Abbreviations
- BMDC:
-
Bone marrow-derived dendritic cell
- CBA:
-
Cytometric bead array
- CFSE:
-
5,6-Carboxyfluorescein diacetate succinimidyl ester
- CTL:
-
Cytotoxic T lymphocyte
- DC:
-
Dendritic cell
- IGRP:
-
Islet-specific glucose-6-phosphatase catalytic subunit-related protein
- IRS661:
-
Immunoregulatory sequence 661
- LOX:
-
Loxoribine
- MCP-1:
-
Monocyte chemotactic protein-1
- ODN:
-
Oligodeoxynucleotide
- pDC:
-
Plasmacytoid dendritic cell
- TLR:
-
Toll-like receptor
References
Anderson MS, Bluestone JA (2005) The NOD mouse: a model of immune dysregulation. Annu Rev Immunol 23:447–485
Castano L, Eisenbarth GS (1990) Type-I diabetes: a chronic autoimmune disease of human, mouse, and rat. Annu Rev Immunol 8:647–679
Akerblom HK, Vaarala O, Hyoty H, Ilonen J, Knip M (2002) Environmental factors in the etiology of type 1 diabetes. Am J Med Genet 115:18–29
Kaprio J, Tuomilehto J, Koskenvuo M et al (1992) Concordance for type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes mellitus in a population-based cohort of twins in Finland. Diabetologia 35:1060–1067
Redondo MJ, Yu L, Hawa M et al (2001) Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia 44:354–362
Beyan H, Buckley LR, Yousaf N, Londei M, Leslie RD (2003) A role for innate immunity in type 1 diabetes? Diab Metab Res Rev 19:89–100
Zipris D (2008) Innate immunity and its role in type 1 diabetes. Curr Opin Endocrinol Diab Obes 15:326–331
Janeway CA Jr, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20:197–216
Kaisho T, Akira S (2003) Regulation of dendritic cell function through toll-like receptors. Curr Mol Med 3:759–771
Kawai T, Akira S (2008) Toll-like receptor and RIG-I-like receptor signaling. Ann NY Acad Sci 1143:1–20
von Landenberg P, Bauer S (2007) Nucleic acid recognizing Toll-like receptors and autoimmunity. Curr Opin Immunol 19:606–610
Krieg AM, Vollmer J (2007) Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol Rev 220:251–269
Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A (2005) Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev 204:27–42
Lande R, Gregorio J, Facchinetti V et al (2007) Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449:564–569
O’Brien BA, Geng X, Orteu CH et al (2006) A deficiency in the in vivo clearance of apoptotic cells is a feature of the NOD mouse. J Autoimmun 26:104–115
O’Brien BA, Huang Y, Geng X, Dutz JP, Finegood DT (2002) Phagocytosis of apoptotic cells by macrophages from NOD mice is reduced. Diabetes 51:2481–2488
Turley S, Poirot L, Hattori M, Benoist C, Mathis D (2003) Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med 198:1527–1537
Hyoty H, Taylor KW (2002) The role of viruses in human diabetes. Diabetologia 45:1353–1361
Jun HS, Yoon JW (2003) A new look at viruses in type 1 diabetes. Diab Metab Res Rev 19:8–31
Drescher KM, Tracy SM (2008) The CVB and etiology of type 1 diabetes. Curr Top Microbiol Immunol 323:259–274
Triantafilou K, Orthopoulos G, Vakakis E et al (2005) Human cardiac inflammatory responses triggered by Coxsackie B viruses are mainly Toll-like receptor (TLR) 8-dependent. Cell Microbiol 7:1117–1126
Hamilton-Williams EE, Lang A, Benke D, Davey GM, Wiesmuller KH, Kurts C (2005) Cutting edge: TLR ligands are not sufficient to break cross-tolerance to self-antigens. J Immunol 174:1159–1163
Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO (2008) Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci USA 105:12439–12444
Ganguly D, Chamilos G, Lande R et al (2009) Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med 206:1983–1994
Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, Santamaria P (1997) Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med 186:1663–1676
Barrat FJ, Meeker T, Gregorio J et al (2005) Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med 202:1131–1139
Inaba K, Inaba M, Romani N et al (1992) Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med 176:1693–1702
Lieberman SM, Evans AM, Han B et al (2003) Identification of the β-cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci USA 100:8384–8388
Jarchum I, Baker JC, Yamada T et al (2007) In vivo cytotoxicity of insulin-specific CD8+ T cells in HLA-A*0201 transgenic NOD mice. Diabetes 56:2551–2560
Marleau AM, Summers KL, Singh B (2008) Differential contributions of APC subsets to T cell activation in nonobese diabetic mice. J Immunol 180:5235–5249
Mohammad MK, Morran M, Slotterbeck B et al (2006) Dysregulated Toll-like receptor expression and signaling in bone marrow-derived macrophages at the onset of diabetes in the non-obese diabetic mouse. Int Immunol 18:1101–1113
Grewal IS, Flavell RA (1996) The role of CD40 ligand in costimulation and T cell activation. Immunol Rev 153:85–106
Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ (1998) T cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 393:480–483
Anderson B, Park BJ, Verdaguer J, Amrani A, Santamaria P (1999) Prevalent CD8+ T cell response against one peptide/MHC complex in autoimmune diabetes. Proc Natl Acad Sci USA 96:9311–9316
Verdaguer J, Yoon JW, Anderson B et al (1996) Acceleration of spontaneous diabetes in TCR-β-transgenic nonobese diabetic mice by β-cell cytotoxic CD8+ T cells expressing identical endogenous TCR-α chains. J Immunol 157:4726–4735
Zhang Y, Lee AS, Shameli A et al (2010) TLR9 blockade inhibits activation of diabetogenic CD8+ T cells and delays autoimmune diabetes. J Immunol 184:5645–5653
Wen L, Ley RE, Volchkov PY et al (2008) Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455:1109–1113
Lang KS, Recher M, Junt T et al (2005) Toll-like receptor engagement converts T cell autoreactivity into overt autoimmune disease. Nat Med 11:138–145
Schulte BM, Kramer M, Ansems M et al (2010) Phagocytosis of enterovirus-infected pancreatic beta-cells triggers innate immune responses in human dendritic cells. Diabetes 59:1182–1191
Shibasaki S, Imagawa A, Tauriainen S et al (2010) Expression of toll-like receptors in the pancreas of recent-onset fulminant type 1 diabetes. Endocr J 57:211–219
Aumeunier A, Grela F, Ramadan A et al (2010) Systemic Toll-like receptor stimulation suppresses experimental allergic asthma and autoimmune diabetes in NOD mice. PLoS One 5:e11484
Heikenwalder M, Polymenidou M, Junt T et al (2004) Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat Med 10:187–192
Perry JA, Olver CS, Burnett RC, Avery AC (2005) Cutting edge: the acquisition of TLR tolerance during malaria infection impacts T cell activation. J Immunol 174:5921–5925
Demaria O, Pagni PP, Traub S et al (2010) TLR8 deficiency leads to autoimmunity in mice. J Clin Invest 120:3651–3662
Martinez J, Huang X, Yang Y (2010) Toll-like receptor 8-mediated activation of murine plasmacytoid dendritic cells by vaccinia viral DNA. Proc Natl Acad Sci USA 107:6442–6447
Wong FS, Hu C, Zhang L et al (2008) The role of Toll-like receptors 3 and 9 in the development of autoimmune diabetes in NOD mice. Ann NY Acad Sci 1150:146–148
Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ (2006) Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25:417–428
Acknowledgements
This work was supported by a grant from the Juvenile Diabetes Research Foundation and by the Canadian Institutes of Health Research to J. P. Dutz. A. S. Lee is supported by a Michael Smith Foundation of Health Research (MSFHR) Junior Graduate Studentship. J. P. Dutz is a MSFHR Senior Scholar and Senior Scientist at the Child and Family Research Institute. We are grateful to J. Cross in her assistance of the IFN-α ELISA and to other members of the J. P. Dutz laboratory for their comments and support.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
IRS661 inhibits TLR7-stimulated NOD BMDCs. a IRS661 was used to inhibit different TLR7 or 9 agonists. Flow cytometry was used to gate for CD11c+ DCs and the mean fluorescence intensity (MFI) of the activation marker CD40 was assessed to determine DC activation. b–e The supernatant fractions were collected (after 24 h) from treated BMDCs and analysed by cytometric bead array for IL-6 (b), TNF-α (c), IL-12p70 (d) and MCP-1 (e). Data are representative of at least three independent experiments. *p < 0.05, **p < 0.006 by one-way ANOVA (PDF 356 kb)
ESM Fig. 2
IRS661 inhibits both CL097- and CpG 1826-induced activation of CD8+ and CD4+ T cells in NOD mice. IRS661 (266 mg/kg, s.c) was injected 2 h prior to CL097 or CpG 1826 treatment in NOD mice. At 24 h post-injection, the axillary, mesenteric and pancreatic lymph nodes (LNs) and spleen were extracted and analysed by flow cytometry. The activation marker CD69 was used to determine activation of CD8+ and CD4+ T cells. Data are representative of at least three independent experiments. CL097 or CpG 1826 groups were compared with the PBS group (black stars). CL097+IRS661 or CpG+IRS661 groups were compared with CL097 or CpG 1826 groups, respectively (grey stars). *p < 0.05, **p < 0.001 by one-way ANOVA (PDF 288 kb)
ESM Fig. 3
IRS661 inhibits the increase in proinflammatory cytokines/chemokine and type 2 interferon induced by CL097 or CpG 1826 treatment in NOD mice. IRS661 was injected 2 h prior to CL097or CpG 1826 injection in NOD mice. Serum levels of IL-6 (a), TNF-α (b), IL-12p70 (c), MCP-1 (d) and IFN-γ (e) were measured at 2 or 6 h post-injection of CL097 or CpG 1826. Cytokines and the chemokine MCP-1 were measured by cytometric bead array. Data are representative of at least three independent experiments. The CL097 and CpG 1826 groups were compared with the PBS group (black stars). The CL097+IRS661 or CpG+IRS661 groups were compared with the CL097 or CpG 1826 groups (grey stars), respectively. *p < 0.05, **p < 0.005 by one-way ANOVA (PDF 391 kb)
ESM Fig. 4
Effect of IRS661 on endogenous IFN-α production in the axillary and mesenteric lymph nodes (LNs), spleen and pancreas of NOD mice. Endogenous IFN-α levels in the axillary (a) and mesenteric LNs (b), spleen (c) and pancreas (d) homogenates of 3–4-week-old NOD mice injected once (24 h post-injection) s.c. with IRS661 (266 mg/kg), ODN 1982 (5 mg/kg) or CL097 (5 mg/kg). Data were obtained from two independent experiments. The CL097 group was compared with the C57BL/6 group. **p < 0.005 by one-way ANOVA (PDF 275 kb)
Rights and permissions
About this article
Cite this article
Lee, A.S., Ghoreishi, M., Cheng, W.K. et al. Toll-like receptor 7 stimulation promotes autoimmune diabetes in the NOD mouse. Diabetologia 54, 1407–1416 (2011). https://doi.org/10.1007/s00125-011-2083-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2083-y