
Abstract
Aim/hypothesis
High-dose aspirin treatment improves fasting and postprandial hyperglycaemia in patients with type 2 diabetes, as well as in animal models of insulin resistance associated with obesity and sepsis. In this study, we investigated the effects of aspirin treatment on inducible nitric oxide synthase (iNOS)-mediated insulin resistance and on S-nitrosylation of insulin receptor (IR)-β, IRS-1 and protein kinase B (Akt) in the muscle of diet-induced obese rats and also in iNos (also known as Nos2)−/− mice on high fat diet.
Methods
Aspirin (120 mg kg−1 day−1 for 2 days) or iNOS inhibitor (L-NIL; 80 mg/kg body weight) were administered to diet-induced obese rats or mice and iNOS production and insulin signalling were investigated. S-nitrosylation of IRβ/IRS-1 and Akt was investigated using the biotin switch method.
Results
iNOS protein levels increased in the muscle of diet-induced obese rats, associated with an increase in S-nitrosylation of IRβ, IRS-1 and Akt. These alterations were reversed by aspirin treatment, in parallel with an improvement in insulin signalling and sensitivity, as measured by insulin tolerance test and glucose clamp. Conversely, while aspirin reversed the increased phosphorylation of IκB kinase β and c-Jun amino-terminal kinase, as well as IRS-1 serine phosphorylation in diet-induced obese rats and iNos −/− mice on high-fat diet, these alterations were not associated with the improvement of insulin action induced by this drug.
Conclusions/interpretation
Our data demonstrate that aspirin treatment not only reduces iNOS protein levels, but also S-nitrosylation of IRβ, IRS-1 and Akt. These changes are associated with improved insulin resistance and signalling, suggesting a novel mechanism of insulin sensitisation evoked by aspirin treatment.
Similar content being viewed by others


Introduction
Diet-induced obesity is an experimental model of obesity and insulin resistance that is associated with a chronic inflammatory response and characterised by abnormal cytokine production, increased acute-phase reactants and other stress-induced molecules [1]. It is now well established that this inflammatory response is associated with the activation of intra-cellular inflammatory pathways that can negatively modulate insulin signalling, such as the IκB kinase β (IKK-β)/nuclear factor kappa B (NFκB), c-Jun amino-terminal kinase (JNK) and inducible nitric oxide synthase (iNOS) pathways [2–4].
High-dose aspirin treatment improved fasting and postprandial hyperglycaemia in patients with type 2 diabetes, as well as in animal models of insulin resistance associated with obesity and sepsis [5–7]. This improved insulin signalling was postulated to be secondary to blockage of the IKK-β/NFκB and JNK pathways, which prevent serine phosphorylation of IRS-1, a well-established mechanism of insulin resistance [8].
Recently, we and others reported that enhanced iNOS production is associated with obesity and inflammatory responses and can negatively modulate insulin signalling through S-nitrosylation of proteins involved in early steps of insulin signalling, such as insulin receptor (IR)-β, IRS-1 and protein kinase B (Akt) [3, 9]. Furthermore, pharmacological or genetic blockage of iNOS production/activity ameliorates insulin resistance associated with obesity or lipopolysaccharide treatment by inhibiting S-nitrosylation of these proteins [9, 10]. Interestingly, aspirin treatment can also decrease iNOS production and activity in several cell lines [11–13], but its effects on iNOS-induced insulin resistance in the muscle of obese rats has not yet been investigated.
In this study, we investigated the effects of aspirin treatment on iNOS-mediated insulin resistance and on S-nitrosylation of IR-β, IRS-1 and Akt in the muscle of diet-induced obese rats and also in iNos (also known as Nos2)−/− mice on a high-fat diet.
Methods
Materials
Antiphosphotyrosine, anti-IRβ, anti-IRS-1, anti-iNOS, anti-Akt, anti-pser307-IRS-1 and anti-phospho-JNK antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-p[ser473]Akt antibody was from Cell Signaling Technology (Beverly, MA, USA). Human recombinant insulin (Humulin R) was purchased from Eli Lilly (Indianapolis, IN, USA). Routinely used reagents were purchased from Sigma (St. Louis, MO), unless otherwise specified.
Animals
Male Wistar rats and C57BL/6 mice were obtained from the UNICAMP Central Animal Breeding Center (Campinas, São Paulo, Brazil). The iNos-knockout mice used were C57BL/6-backcrossed iNos –/– mice (C57BL/6-Nos2tm1Lau colony; Jackson Laboratory, Bar Harbor, ME, USA). Animals were allowed free access to standard rodent chow and water. Diet-induced obese animals were obtained by high-fat diet administration to one group of Wistar rats, to one group of C57BL/6 mice and to one group of iNos –/– mice. High-fat diet was initiated at 8 weeks of age and administered for 4 weeks; control animals were of the same age. The high-fat diet consisted of 55% energy derived from fat, 29% from carbohydrate and 16% from protein. Food was withdrawn 6 h before the experiments. The Ethics Committee of the University of Campinas approved all experiments involving animals.
Aspirin treatment
Aspirin (120 mg kg−1 day−1) or saline in equal volumes were given by oral gavage to diet-induced obese rats or mice for 2 days.
iNOS inhibitor treatment
Diet-induced obese rats received an intraperitoneal injection of the iNOS inhibitor l-N 6-(1-iminoethyl)lysine (l-NIL; 80 mg/kg body weight) or saline twice daily (every 12 h) for 2 days. This treatment protocol with l-NIL was adapted from a previously published procedure [14].
Insulin tolerance test
Rats were fasted for 6 h and submitted to a 30 min insulin tolerance test. Briefly, 1.5 IU/kg insulin was infused intraperitoneally in rats and glucose was measured at 0 (basal), 5, 10, 15, 20, 25 and 30 min thereafter. The glucose disappearance rate (K itt) was calculated from the formula 0.693/t 1/2, where t 1/2 stands for time for glucose to reach 50% of the basal value. Glucose t 1/2 was calculated from the slope of the least square analysis of blood glucose concentration during the linear phase of decline [15].
Euglycaemic–hyperinsulinaemic clamp studies
After 5 h of fasting, animals were anaesthetised i.p. with sodium pentobarbital (50 mg/kg body weight) and catheters were inserted into the left jugular vein (for tracer infusions) and carotid artery (for blood sampling), as previously described [16]. Each animal was monitored for food intake and weight gain for 5 days after surgery to ensure complete recovery. Food was removed for 12 h before the beginning of in vivo studies. A 120 min euglycaemic–hyperinsulinaemic clamp procedure was conducted in conscious, unrestrained, catheterised rats, as shown previously [16], with a prime continuous infusion of human insulin at a rate of 3.6 mU (kg body weight)−1 min−1 to raise the plasma insulin concentration to approximately 800–900 pmol/l. Blood samples (20 μl) were collected at 5 min intervals for the immediate measurement of plasma glucose concentration; 10% (vol./vol.) unlabelled glucose was infused at variable rates to maintain plasma glucose at fasting levels
IRβ, IRS-1 and iNOS immunoprecipitation
Anaesthetised mice were injected intraperitoneally either with saline or insulin (3.8 U/kg); 90 s later soleus muscles were removed and homogenised, as described below. Muscle lysates were incubated with anti-IRβ (0.3 mg/ml), anti-IRS-1 (1:1,000) or anti-iNOS (1:1,000) antibodies for 2 h and then incubated with protein A sepharose for a further 2 h. Beads were then washed with TRIS containing 1% (vol./vol.) Triton X-100 and phosphatase inhibitors, boiled for 5 min in Laemmli buffer and subjected to western blotting analysis [17, 18].
Western blot analysis
Muscle extracts, immunoprecipitates or biotinylated nitrosocysteines were subjected to sodium dodecyl sulphate polyacrylamide electrophoresis and immunoblotting was performed as described [17]. Immunoreactive bands were detected by the enhanced chemiluminescense method (RPN 2108; Amersham Biosciences - Sweden).
Detection of S-nitrosated proteins by biotin switch method
The biotin switch assay was performed essentially as previously described [19, 20]. Muscle tissue was extracted and homogenised in extraction buffer (250 mmol/l HEPES, pH 7.7, 1 mmol/l EDTA, 0.1 mmol/l neocuproine). After centrifugation at 9,000×g for 20 min, insoluble material was removed, extracts were adjusted to a concentration of 0.5 mg/ml protein and equal amounts were blocked with four volumes of blocking buffer (225 mmol/l HEPES, pH 7.7, 0.9 mmol/l neocuproine, 2.5% (vol./vol.) SDS and 20 mmol/l methyl-methanethiosulfonate [MMTS]) at 50°C for 30 min while being agitated. After blocking, extracts were precipitated with two volumes of cold acetone (−20°C), chilled at −20°C for 10 min, centrifuged at 2,000×g and 4°C for 5 min, washed with acetone, dried and finally resuspended in 0.1 ml HENS buffer (250 mmol/l HEPES, pH 7.7, 1 mmol/l EDTA, 0.1 mmol/l neocuproine and 1% (vol./vol.) SDS) per mg of protein. Until this point, all operations were carried out in the dark. A one-third volume of N-(6-(ciotinamido)hexyl)-3′-(2′-pyridyldithio)-propionamide (biotin-HPDP) 4 mmol/l and 2.5 mmol/l ascorbic acid was added, followed by incubation for 1 h at room temperature. Proteins were acetone-precipitated again and re-suspended in the same volume of HENS buffer.
For purification of biotinylated proteins, samples from the biotin switch assay were diluted with two volumes of neutralisation buffer (20 mmol/l HEPES, pH 7.7, 100 mmol/l NaCl, 1 mmol/l EDTA and 0.5% (vol./vol.) Triton X-100) and 15 μl of neutravidin–agarose/mg of protein in the initial extract were added, followed by incubation for 1 h at room temperature while being agitated. Beads were washed five times with washing buffer (20 mmol/l HEPES, pH 7.7, 600 mmol/l NaCl, 1 mmol/l EDTA and 0.5% Triton X-100) and incubated for 20 min at 37°C with elution buffer (20 mmol/l HEPES, pH 7.7, 100 mmol/l NaCl, 1 mmol/l EDTA and 100 mmol/l 2-mercaptoethanol) with gentle stirring. Supernatant fractions were collected, Laemmli buffer was added and proteins were separated by SDS-PAGE. Immunoblotting was performed as described above [19, 20].
Primary rat skeletal muscle cell isolation and culture
Male Wistar rats (90 g) were killed by cervical dislocation. The hindlimbs were quickly removed and used to prepare muscle cell culture, as described, previously [21]. The cells were cultured for the first 2 days in primary growth medium containing DMEM with 20% (vol./vol.) fetal calf serum and, thereafter, in fusion medium containing DMEM with 10% (vol./vol.) horse serum.
Salicylate treatment
The skeletal muscle cell cultures were divided in two different groups: control and salicylate (5 mmol/l). The cells were then incubated in DMEM containing all of the treatment substances described above.
Nitric oxide synthase activity assay
After 24 h, the cells were scraped off and homogenised in TRIS buffer (50 mmol/l, pH 7.4) containing l-citrulline (1 mmol/l), EDTA (0.1 mmol/l), dithiothreitol (1 mmol/l), leupeptin (10 μg/ml), soyabean trypsin inhibitor (10 μg/ml), aprotinin (2 μg/ml) and phenyl methyl sulfonyl fluoride (1 mmol/l). Homogenates were centrifuged (1,000×g) for 10 min and the supernatant fractions were passed over an ionic form, hydrogen, dry mesh column (Dowex 50WX8-200, 100–200; Dow Chemical, St Louis, MO, USA) to remove endogenous arginine. The samples (50 μl) were incubated in a modified TRIS buffer (50 mmol/l, pH 7.4) containing EGTA (1 mmol/l), flavin adenine dinucleotide (10 μmol/l), NADPH (1 mmol/l), BH4 (100 μmol/l) and 10 μmol/l l-arginine containing 100,000 cpm of l-[2,3,4,5-3H]arginine monohydrochloride (Amersham), previously equilibrated for 5 min at 37°C in a final volume of 100 μl. Pharmacological controls of enzymatic activity were performed in parallel and consisted of \( N^{\omega } \)-nitro-l-arginine methyl ester (l-NAME) addition (1 mmol/l) to the incubation medium. After 15 min, the reaction was stopped by adding 1 ml ice-cold buffer (pH 5.4) containing HEPES (20 mmol/l) and EDTA (1 mmol/l), followed by vortex mixing. The samples were then applied to a 0.6 ml Dowex 50WX8-200, pre-equilibrated with the stopping buffer. l-[2,3,4,5-3H]Citrulline was eluted and washed with 1 ml stopping buffer and radioactivity was determined by liquid scintillation counting. All measurements were made in duplicate. Protein concentrations were determined according to the method of Bradford [22] and the activity was expressed as pmol l-citrulline (mg protein)−1 min−1. The values were corrected for the amount of l-[2,3,4,5-3H]-citrulline found in the presence of l-NAME (1 mmol/l), which was added exogenously [23].
Statistical analysis
The results of blots are presented as direct comparisons of bands or dots in autoradiographs and were quantified by densitometry using Scion Image software (ScionCorp, Fredrick, MD, USA). Data were analysed by two-tailed unpaired Student’s t test or by repeated measures ANOVA (one- or two-way ANOVA), followed by post hoc analysis of significance (Bonferroni’s test) when appropriate, comparing experimental and control groups. The level of significance was set at p < 0.05.
Results
Effect of aspirin on insulin sensitivity and signalling and cytokine production
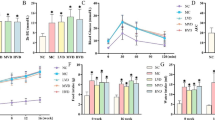
The animals receiving the high-fat diet (diet-induced obese rats) had higher food intake, a pattern not affected by administration of aspirin or l-NIL (Fig. 1a). As expected, these animals had an increase in body weight; however, treatment with aspirin or aspirin with l-NIL for 2 days did not change body weight compared with respective controls (Fig. 1b). Fasting insulin levels were higher in animals on the high-fat diet and were not changed by treatment with aspirin or aspirin plus l-NIL (Fig. 1c). Plasma glucose did not change significantly in any group of animals (Fig. 1d); however, insulin sensitivity, as determined by the glucose disappearance rate (Fig. 1e) or glucose clamp (Fig. 1f), was decreased in high-fat fed rats and showed an improvement after aspirin treatment.

Effect of aspirin on metabolic variables in diet-induced obese (DIO) rats. a Food intake (per 24 h), (b) body weight change, (c) fasting insulin and (d) fasting glucose during 2 days after aspirin (AAS) and/or l-NIL administration. Insulin sensitivity, as determined by (e) glucose disappearance rate (K itt) or (f) euglycaemic–hyperinsulinaemic clamp. Bars represent mean ± SEM from six to eight rats. *p < 0.05 vs control group; † p < 0.05 vs diet-induced obesity rats. C, control
Effect of aspirin on insulin signalling in diet-induced obese rats
An 80% reduction was observed in insulin-induced tyrosine phosphorylation of IRβ, as well as a 70% reduction in IRS-1 tyrosine phosphorylation, a 70% reduction in IRS-1 protein levels and an 80% reduction in Akt serine phosphorylation in muscle of diet-induced obese rats. These reductions were all reversed by aspirin treatment (Fig. 2a–d).

Effect of aspirin treatment on insulin signalling. Insulin-induced tyrosine phosphorylation (immunoblotting pY) of (a) immunoprecipitated IRβ and (b) IRS-1 in the muscle of control (C), diet-induced obese (DIO) and DIO rats after aspirin (AAS) treatment. c Protein levels in immunoblots (IB) of IRS-1 and (d) insulin-induced serine phosphorylation of Akt in the muscle of control, DIO and DIO rats after AAS treatment. Representative blots (a–d) are from experiments that were repeated independently, with similar results. α-Tubulin was used in immunoblotting as protein control. Bars represent quantification of blots as mean ± SEM from six to eight rats. *p < 0.05 vs control group; † p < 0.05 vs DIO rats
Effect of aspirin treatment on iNOS production/activity and S-nitrosylation of IRβ/IRS-1 and Akt in muscle of diet-induced obese rats
Inducible nitric oxide protein levels increased in the muscle of diet-induced obese rats, but the increase was blunted following aspirin treatment in the same animal group (Fig. 3a). We also investigated whether salicylate was able to modulate iNOS activity in cultured muscle cells of control rats. Results showed that salicylate treatment for 2 h reduced iNOS activity by ~50% (Fig. 3b).

Effects of aspirin on iNOS content/activity and S-nitrosylation of IRβ/IRS-1 and Akt in the muscle of diet-induced obesity (DIO) rats. a iNOS protein levels in immunoblots of gastrocnemius muscle of control (C), DIO and DIO rats after aspirin (AAS) administration. b iNOS activity was measured in control cultured muscle cells treated or not with salicylate (5 mmol/l), as described in Methods. c The biotin switch method was performed to detect S-nitrosylation of IRβ, (d) IRS-1 and (e) Akt in the muscle of control, DIO and DIO+AAS. Representative blots (a, c–e) are from experiments that were repeated independently, with similar results. α-Tubulin was used in immunoblotting as protein control. Bars represent quantification of blots as mean ± SEM of six to eight rats. *p < 0.05 vs control group; † p < 0.05 vs DIO rats
The enhanced iNOS production in high-fat fed rats was associated with an increase in S-nitrosylation of IRβ, IRS-1 and Akt. These increases were also reversed by aspirin treatment (Fig. 3c–e).
Effect of aspirin treatment on serine phosphorylaton of IRS-1 and JNK in the muscle of diet-induced obese rats
Increased JNK and IKKβ phosphorylation was observed in the muscle of diet-induced obese rats, accompanied by an increase in IRS-1 serine phosphorylation. After aspirin treatment there was a reduction in JNK and IKKβ phosphorylation, as well as in IRS-1 serine phosphorylation (Fig. 4a–c).

Effect of aspirin on JNK, IKK and IRS-1 serine phosphorylaton in the muscle of diet-induced obesity (DIO) rats. Representative immunoblots blots show (a) JNK and (b) IKKβ serine phosphorylation. c Ratio of IRS-1 serine phosphorylation/IRS-1 protein content ratio in the muscle of control (C), DIO and DIO rats after aspirin (AAS) administration, as shown by immunoblots and quantified in bar graph. Representative blots (a–c) are from experiments that were repeated independently, with similar results. α-Tubulin was used in immunoblotting as protein control. Bars represent quantification of blots as mean ± SEM of six to eight rats. *p < 0.05 vs control group; † p < 0.05 vs DIO rats
Effect of aspirin and l-NIL on Akt phosphorylation and on S-nitrosylation of IRS-1 and Akt in muscle of diet-induced obese rats
To show that aspirin attenuates insulin resistance in the muscle of diet-induced obese rats through the inhibition of S-nitrosylation of early steps of insulin action, we also investigated the effect of the iNOS inhibitor, l-NIL, either alone or in combination with aspirin, on insulin sensitivity, insulin signalling and S-nitrosylation of IRS-1 and Akt. Administration of l-NIL for 2 days resulted in improved insulin sensitivity, as demonstrated by an increase in glucose disappearance rate during the insulin tolerance test and also by an increase in glucose infusion rate during the glucose clamp. When l-NIL was added to aspirin, no further increase in insulin sensitivity was detected by either test (Fig. 5a, b). Insulin-induced Akt phosphorylation, which was decreased in diet-induced obese rats, showed a similar improvement after treatment with aspirin or l-NIL, both of which did not show any additional effect (Fig. 5c). S-nitrosylation of IRS-1 and Akt, which were increased in diet-induced obese rats, was similarly decreased after aspirin and/or l-NIL treatment (Fig. 5d, e).

Effects of aspirin and/or l-NIL treatment on Akt phosphorylation and S-nitrosylation of IRS-1 and Akt in muscle of diet-induced obesity (DIO) rats. a Glucose disappearance rate and (b) euglycaemic–hyperinsulinaemic clamp were determined 2 days after l-NIL and l-NIL plus aspirin (AAS) administration in DIO rats. c Representative immunoblots and quantification by bar graph of insulin-induced Akt serine phosphorylation in the muscle of controls (C), DIO rats and DIO rats 2 days after AAS, l-NIL or AAS plus l-NIL. d S-nitrosylation of IRS-1 and (e) Akt in the muscle after treatment as above (c). Representative blots (c–e) are from experiments that were repeated independently, with similar results. Bars represent quantification of blots as mean ± SEM of six to eight rats. *p < 0.05 vs control group; † p < 0.05 vs DIO rats
Effect of aspirin treatment on Akt phosphorylation and S-nitrosylation of IRS-1 and Akt in muscle of iNos −/− mice on high-fat diet
To investigate whether the effects of aspirin, associated with an inhibitor of iNOS, would also be reproduced in an iNos knockout animal, we investigated the effect of aspirin on insulin sensitivity and insulin signalling in muscle of iNos −/− mice on a high-fat diet.
Insulin sensitivity was determined using the glucose disappearance rate, which shows a good correlation with the gold standard glucose clamp, as previously described [15] and as also confirmed in this study with rats. As expected, control mice on a high-fat diet showed reduced insulin sensitivity (Fig. 6a). Interestingly, iNos −/− mice had higher insulin sensitivity than controls. When these animals were fed a high-fat diet for 2 months, they had a mild, but significant decrease in insulin sensitivity, as demonstrated by reduced glucose disappearance rate, when compared with control iNos −/− mice, but not when compared with wild-type mice on standard rodent chow. The treatment with aspirin showed no improvement in insulin sensitivity in iNos −/− mice on a high-fat diet (Fig. 6a).

Effect of aspirin on insulin sensitivity, Akt phosphorylation and S-nitrosylation of IRS-1 and Akt in muscle of iNos −/− mice on a high-fat diet. a Glucose disappearance rate in control and iNos knockout mice at 2 days after aspirin (AAS) administration. b Representative immunoblots blots, quantified as bar graphs, of insulin-induced Akt serine phosphorylation and (c, d) of S-nitrosylation of IRS-1 and Akt respectively in the muscle of control (C) and iNOS knockout mice 2 days after AAS administration. e JNK, (f) IKKβ and (g) IRS-1 serine phosphorylation in the muscle of animals treated as above (c, d) Representative blots (b–g) are from experiments that were repeated independently, with similar results. Bars represent quantification of blots as mean ± SEM of six to eight mice. *p < 0.05 vs control mice; † p < 0.05 vs DIO mice; ‡ p < 0.05 vs DIO iNos −/−; § p < 0.05 vs control iNos −/−
Insulin-induced Akt activation was moderately decreased in control animals, but only mildly decreased in iNos −/− mice on a high-fat diet; these alterations were reversed with aspirin treatment (Fig. 6b). As expected, there was an increase in S-nitrosylation of IRS-1 and Akt in the muscle of control mice on a high-fat diet, the increase being reversed by aspirin. However, we did not observe S-nitrosylation of IRS-1 or Akt in the muscle of iNos −/− mice on a high-fat diet (Fig. 6c, d).
As demonstrated in the muscle of rats on a high-fat diet, JNK and IKKβ phosphorylation increased in the muscle of control mice on a high-fat diet, accompanied by an increase in IRS-1 serine phosphorylation that was reversed by aspirin treatment (Fig. 6e–g). JNK and IKKβ phosphorylation were slightly increased, in parallel with an increase in IRS-1 serine phosphorylation, in the muscle of iNos −/− mice on a high-fat diet; these alterations were reversed by aspirin treatment (Fig. 6e–g).
Discussion
The glucose-lowering effects of aspirin, observed decades ago in patients with diabetes, are complex, possibly tissue-specific and probably not related to the inhibition by aspirin of cyclo-oxygenase enzymes [8]. Recently, new effects of aspirin have been described. Thus aspirin can directly inhibit IKKβ, downregulating inflammatory signalling through NF-κB in this pathway, which is implicated in developing insulin resistance [7]. Gene- and diet-induced obesity are associated with enhanced IKKβ activity in liver, which may induce serine phosphorylation of IRS-1, preventing its tyrosine phosphorylation by insulin [8, 24]. However, in muscle, data on the importance of IKKβ activation in the development of insulin resistance remain conflicting [25]. Another mechanism by which aspirin could prevent insulin resistance induced by obesity is through inhibition of JNK, another serine kinase associated with serine-phosphorylation of IRS-1 and therefore with insulin resistance. This mechanism has been described in HEK293 cells and observed in the muscle of septic and growth hormone-treated rats [5, 26, 27].
Our data show that, in the muscle of diet-induced obese rats and mice, aspirin was able to reduce IKKβ and JNK phosphorylation, and also IRS-1 serine phosphorylation. However, two results from our study suggest that these alterations were not crucial to the improvement in insulin action induced by aspirin. First, in diet-induced obese rats the effect of l-NIL, an inhibitor of iNOS, on insulin sensitivity was not synergised by aspirin treatment. Second, in iNos −/− mice on a high-fat diet, we saw an increase in IKKβ and JNK phosphorylation, as well as in IRS-1 serine phosphorylation. These increases were associated with only a mild reduction in insulin sensitivity, which was not reversed by aspirin treatment. These data suggest that, although aspirin reverses the increased IKKβ and JNK phosphorylation, as well as increased IRS-1 serine phosphorylation observed in diet-induced obese rats or in iNos −/− mice on a high-fat diet, these alterations have no parallel with the improvement in insulin action induced by this drug in muscle.
Insulin sensitivity was increased in iNos −/− mice, in parallel to an increase in insulin-induced Akt phosphorylation, suggesting that mechanisms related to iNOS may have a role in the control of physiological insulin sensitivity and not only in situations related to insulin resistance. When these mice were fed a high-fat diet for 2 months, they had a mild reduction in insulin sensitivity; however, the glucose disappearance rate under this diet was similar to that of wild-type mice on control rodent chow. As described above, in iNos −/− mice on a high-fat diet, aspirin did not improve insulin sensitivity, despite its improvement of insulin-induced Akt phosphorylation. These data suggest that minor changes in insulin signalling may have no influence on insulin sensitivity.
Although we did not investigate whether aspirin could attenuate insulin resistance in the liver of these obese animals, a previous report [28] has shown that iNOS content was enhanced in the liver of ob/ob mice, an enhancement associated with reduced protein levels of IRS-1 and IRS-2, which was prevented by iNOS blockage with l-NIL. This finding demonstrates that iNOS plays a role in fasting hyperglycaemia and contributes to hepatic insulin resistance in ob/ob mice. The effect of high-fat diet on fasting glucose levels was only modest in our model, and probably the importance of hepatic insulin resistance was only mild. In more prolonged high-fat diet treatment these effects of iNOS on hepatic insulin resistance would probably be more evident [28].
Several reports have shown that salicylates or aspirin inhibit the production of iNOS induced by cytokines in different cell types and suggest that one of the therapeutic actions of these drugs could be mediated by a reduction in iNOS content and nitric oxide production [11, 13]. This reduction in iNOS protein levels may be secondary to a decrease in NFκB activation [29]; however, a limitation of our study was that we did not directly measure NFκB activity in these experiments. Aspirin can also directly inhibit iNOS enzymatic activity, although some questions remain as to whether this occurs at therapeutic concentrations [12]. Our data show that aspirin reduced iNOS activity in cultured muscle cells and also reduced iNOS content in muscle of diet-induced obese rats. These data suggest that aspirin has a direct effect on muscle, reducing iNOS activity.
Recently, we demonstrated that iNOS induction in the muscle of diet-induced obese rats, ob/ob mice and lipopolysaccharide-treated mice is associated with enhanced nitric oxide production and enhanced S-nitrosylation of IRβ, IRS-1 and Akt [9, 10]. S-nitrosylation of IRβ and Akt was associated with reductions in their kinase activities, leading to downregulation of the IRβ/IRS-1/PI 3-kinase/Akt pathway [9, 30]. Mutated Akt1(C224S), in which cysteine 224 is substituted by serine, is resistant to nitric oxide donor-induced S-nitrosylation and inactivation, indicating that cysteine 224 is a major S-nitrosylation acceptor site. In vitro denitrosylation with reducing agent reactivated cellular Akt from nitric oxide donor-treated cells. In combination, these data demonstrated that S-nitrosylation of Akt is a specific post-translational modification that regulates its activity [30]. S-nitrosylation of IRS-1 is associated with its reduced protein levels in muscle, which may be secondary to degradation via the ubiquitin–proteasome system [9, 14]. Since the IRβ/IRS-1/Akt pathway plays a central role in metabolic actions of insulin in muscle, including stimulation of glucose uptake and glycogen synthesis, downregulation of this pathway in muscle by S-nitrosylation may be an important mechanism of iNOS-induced insulin resistance. The results of the present study reinforce the above data and show that, in iNos −/− mice on a high-fat diet, only a mild alteration in insulin sensitivity is observed. In addition, in these mice, while aspirin reduced IKKβ, JNK and IRS-1 serine phosphorylation, it did not improve insulin sensitivity. Similar results were observed in diet-induced obese rats treated with an iNOS inhibitor and aspirin. These data suggest that iNOS-induced insulin resistance is an important mechanism that modulates insulin sensitivity and that probably the beneficial effect of aspirin in insulin resistance is related to a reduction of iNOS protein levels and S-nitrosylation of IRβ, IRS-1 and Akt.
Insulin resistance is certainly a metabolic situation related to several mechanisms that act in parallel to downregulate insulin signalling. Our data reinforce the hypothesis that: (1) multiple mechanisms are involved in insulin resistance in diet-induced obese rats and mice; but that (2) iNOS-induced insulin resistance is an important mechanism modulating insulin signalling and insulin sensitivity in parallel; and (3) aspirin is able to improve this mechanism.
Recently, Dallaire and colleagues investigated whether targeted disruption of iNOS modulates the metabolic effect of rosiglitazone in obese mice [31]. Their results demonstrated that the iNOS/nitric oxide pathway is a critical modulator of peroxisome proliferator-activated receptor (PPAR)-γ activation and that invalidation of this pathway improves the efficacy of PPAR-γ agonism in an animal model of obesity and insulin resistance.
Taken together, our data show that aspirin treatment not only reduced iNOS protein levels, but also S-nitrosylation of IRβ, IRS-1 and Akt, which was related to improvements in insulin-induced tyrosine phosphorylation of IRβ and IRS-1, and serine phosphorylation of Akt. This suggests a novel mechanism of insulin sensitisation, evoked by aspirin treatment. These findings also suggest that inhibition of iNOS might be a major mediator of the insulin-sensitising effects of IKKβ inhibition.
Recently, in a double-masked, placebo-controlled trial, 20 obese non-diabetic adults were evaluated at baseline and after 1 month of salsalate treatment, a non-acetylated salicylate but with a lower bleeding risk. This proof-of-principle study demonstrates that salsalate reduces glycaemia and may improve inflammatory cardiovascular risk indexes in overweight individuals [32]. In addition, aspirin treatment improved glycaemic control of diabetic patients, in parallel with reductions in inflammatory response, including reduced levels of serum nitrite, which at least in part may be secondary to reduced iNOS activation [33].These new concepts are important for an understanding of how anti-inflammatory drugs attenuate insulin resistance, since direct therapeutic targets may be identified.
Abbreviations
- Akt:
-
Protein kinase B
- HENS:
-
HEPES, EDTA, neocuproine and SDS
- IKKβ:
-
IκB kinase β
- iNOS:
-
Inducible nitric oxide synthase
- IR:
-
Insulin receptor
- JNK:
-
c-Jun amino-terminal kinase
- l-NAME:
-
\( N^{\omega } \)-nitro-l-arginine methyl ester
- NFκB:
-
Nuclear factor kappa B
- l-NIL:
-
l- N 6-(1-iminoethyl)lysine
- PPAR:
-
Peroxisome proliferator-activated receptor
References
Sethi JK, Hotamisligil GS (1999) The role of TNF alpha in adipocyte metabolism. Semin Cell Dev Biol 10:19–29
Hirosumi J, Tuncman G, Chang L et al (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Perreault M, Marette A (2001) Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med 7:1138–1143
Yuan M, Konstantopoulos N, Lee J et al (2001) Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 293:1673–1677
Barreiro GC, Prattali RR, Caliseo CT et al (2004) Aspirin inhibits serine phosphorylation of IRS-1 in muscle and adipose tissue of septic rats. Biochem Biophys Res Commun 320:992–997
Hundal RS, Petersen KF, Mayerson AB et al (2002) Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest 109:1321–1326
Kim JK, Kim YJ, Fillmore JJ et al (2001) Prevention of fat-induced insulin resistance by salicylate. J Clin Invest 108:437–446
Gao Z, Zuberi A, Quon MJ, Dong Z, Ye J (2003) Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in tumor necrosis factor-treated cells through targeting multiple serine kinases. J Biol Chem 278:24944–24950
Carvalho-Filho MA, Ueno M, Hirabara SM et al (2005) S-nitrosylation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes 54:959–967
Carvalho-Filho MA, Ueno M, Carvalheira JB, Velloso LA, Saad MJ (2006) Targeted disruption of iNOS prevents LPS-induced S-nitrosylation of IRbeta/IRS-1 and Akt and insulin resistance in muscle of mice. Am J Physiol Endocrinol Metab 291:E476–E482
Cieslik K, Zhu Y, Wu KK (2002) Salicylate suppresses macrophage nitric-oxide synthase-2 and cyclo-oxygenase-2 expression by inhibiting CCAAT/enhancer-binding protein-beta binding via a common signaling pathway. J Biol Chem 277:49304–49310
Katsuyama K, Shichiri M, Kato H, Imai T, Marumo F, Hirata Y (1999) Differential inhibitory actions by glucocorticoid and aspirin on cytokine-induced nitric oxide production in vascular smooth muscle cells. Endocrinology 140:2183–2190
Wang Z, Brecher P (1999) Salicylate inhibition of extracellular signal-regulated kinases and inducible nitric oxide synthase. Hypertension 34:1259–1264
Sugita H, Fujimoto M, Yasukawa T et al (2005) Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. J Biol Chem 280:14203–14211
Bonora E, Moghetti P, Zancanaro C et al (1989) Estimates of in vivo insulin action in man: comparison of insulin tolerance tests with euglycemic and hyperglycemic glucose clamp studies. J Clin Endocrinol Metab 68:374–378
Prada PO, Zecchin HG, Gasparetti AL et al (2005) Western diet modulates insulin signaling, c-Jun N-terminal kinase activity, and insulin receptor substrate-1ser307 phosphorylation in a tissue-specific fashion. Endocrinology 146:1576–1587
Saad MJ, Araki E, Miralpeix M, Rothenberg PL, White MF, Kahn CR (1992) Regulation of insulin receptor substrate-1 in liver and muscle of animal models of insulin resistance. J Clin Invest 90:1839–1849
Tremblay F, Lavigne C, Jacques H, Marette A (2001) Defective insulin-induced GLUT4 translocation in skeletal muscle of high fat-fed rats is associated with alterations in both Akt/protein kinase B and atypical protein kinase C (zeta/lambda) activities. Diabetes 50:1901–1910
Jaffrey SR, Snyder SH (2001) The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE 2001: PL1
Martinez-Ruiz A, Lamas S (2004) Detection and proteomic identification of S-nitrosylated proteins in endothelial cells. Arch Biochem Biophys 423:192–199
Lynge J, Juel C, Hellsten Y (2001) Extracellular formation and uptake of adenosine during skeletal muscle contraction in the rat: role of adenosine transporters. J Physiol 537:597–605
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Pollock JS, Forstermann U, Mitchell JA et al (1991) Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci U S A 88:10480–10484
Cai D, Yuan M, Frantz DF et al (2005) Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 11:183–190
Rohl M, Pasparakis M, Baudler S et al (2004) Conditional disruption of IkappaB kinase 2 fails to prevent obesity-induced insulin resistance. J Clin Invest 113:474–481
Jiang G, Dallas-Yang Q, Liu F, Moller DE, Zhang BB (2003) Salicylic acid reverses phorbol 12-myristate-13-acetate (PMA)- and tumor necrosis factor alpha (TNFalpha)-induced insulin receptor substrate 1 (IRS1) serine 307 phosphorylation and insulin resistance in human embryonic kidney 293 (HEK293) cells. J Biol Chem 278:180–186
Prattali RR, Barreiro GC, Caliseo CT et al (2005) Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in growth hormone treated animals. FEBS Lett 579:3152–3158
Fujimoto M, Shimizu N, Kunii K, Martyn JA, Ueki K, Kaneki M (2005) A role for iNOS in fasting hyperglycemia and impaired insulin signaling in the liver of obese diabetic mice. Diabetes 54:1340–1348
Taylor BS, Geller DA (2000) Molecular regulation of the human inducible nitric oxide synthase (iNOS) gene. Shock 13:413–424
Yasukawa T, Tokunaga E, Ota H, Sugita H, Martyn JA, Kaneki M (2005) S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem 280:7511–7518
Dallaire P, Bellmann K, Laplante M et al (2008) Obese mice lacking inducible nitric oxide synthase are sensitized to the metabolic actions of peroxisome proliferator-activated receptor-gamma agonism. Diabetes 57:1999–2011
Fleischman A, Shoelson SE, Bernier R, Goldfine AB (2008) Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 31:289–294
Goldfine AB, Silver R, Aldhahi W et al (2008) Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci 1:36–43
Acknowledgements
This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s00125-017-4292-5.
Rights and permissions
About this article
Cite this article
Carvalho-Filho, M.A., Ropelle, E.R., Pauli, R.J. et al. Aspirin attenuates insulin resistance in muscle of diet-induced obese rats by inhibiting inducible nitric oxide synthase production and S-nitrosylation of IRβ/IRS-1 and Akt. Diabetologia 52, 2425–2434 (2009). https://doi.org/10.1007/s00125-009-1498-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1498-1