
Abstract
The pattern recognition receptor or receptor for AGE (RAGE) is constitutionally expressed in a few cell types only. However in almost all cells studied so far it is induced by reactions known to initiate inflammation. Its biological activity seems to be mainly dependent on the presence of its various ligands, including AGE, S100-calcium binding protein/calgranulins, high-mobility group protein 1, amyloid-β-peptides and the family of β-sheet fibrils, all known to be elevated in chronic metabolic, malignant and inflammatory diseases. The RAGE pathway interacts with cytokine-, lipopolysaccharide-, oxidised LDL- and glucose-triggered cellular reactions by turning a short-lasting inflammatory response into a sustained change of cellular function driven by perpetuated activation of the proinflammatory transcription factor, nuclear factor kappa-B. RAGE-mediated persistent cell activation is of pivotal importance in various experimental and clinical settings, including diabetes and its complications, neurodegeneration, ageing, tumour growth, and autoimmune and infectious inflammatory disease. Due to RAGE’s central role in maintaining perpetuated cell activation, various therapeutic attempts to block RAGE or its ligands are currently under investigation. Despite broad experimental evidence for the role of RAGE in chronic disease, knowledge of its physiological function is still missing, limiting predictions about safety of long-term inhibition of RAGE × ligand interaction in chronic diseases.
Similar content being viewed by others


Introduction
The type I transmembrane receptor RAGE (receptor for AGEs) is an evolutionarily recent receptor belonging to the immunoglobulin superfamily. RAGE (also known as AGER) is located within the gene-dense major histocompatibility class III region on chromosome 6, which contains numerous genes involved predominantly in inflammatory and immune responses, such as TNFα (also known as TNF) and several complement components [1, 2]. Although its localisation links RAGE to genes involved in host response mechanisms and indicates a central role of RAGE in regulating immune responses and inflammation, it was originally described as a receptor for advanced glycation end-products and recognised as a cell surface receptor playing a pivotal role in diabetes and other metabolic diseases characterised by AGE accumulation [2–5]. AGE are the result of non-enzymatic glycation and glycoxidation processes, and are formed on proteins, lipids and nucleic acids in a pro-oxidant environment [6, 7]. Formation is enhanced with concomitant hyperglycaemia and delayed macromolecular turnover [7]. RAGE does not promote the uptake and removal of AGE, but AGE-ligation to RAGE induces inflammation through persistent activation of the proinflammatory transcription factor, nuclear factor kappa-B (NF-κB) [8, 9]. When Rage −/− mice were generated [10–12] and studied in different pathologies, it became apparent that RAGE is involved in a number of diseases of the innate immune system and RAGE was redefined as a promiscuous pattern recognition receptor (PRR), mediating immune and inflammatory responses, promotion and progression of cancer [13, 14], and micro- as well as macrovascular disease [2–5] by binding a variety of ligands released by inflammatory, stressed and damaged cells [8, 15, 16]. Subsequently, RAGE × ligand interaction has been associated with survival of RAGE-producing cells, production of cellular reactive oxygen species (ROS), increased levels of phosphorylated extracellular signal-regulated kinase (ERK)1/2, new synthesis of p65 (also known as RelA), NF-κB activation and sustained inflammation [8, 15, 17, 18]. RAGE activation of these cellular pathways is supposed to contribute to diabetes as well as to its complications. Thus, understanding the role of RAGE in inflammation, neurodegeneration and tumour biology (Fig. 1), as well as the various levels of regulating, promoting and controlling RAGE-mediated cell activation (Fig. 2) would play an important part in (1) deciphering the mechanisms that underlie the pathology of diabetes and its complications, (2) defining future therapeutic strategies and (3) explaining their potential pitfalls. In its first part, this review will therefore focus on the different levels of regulation of the RAGE pathway, while in the second part some examples for specific disease states are provided.

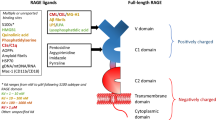
The biology of RAGE is driven by settings in which ligands accumulate and promote inflammation. RAGE is a multiligand receptor of the immunoglobulin superfamily of cell surface molecules and acts as a PRR for diverse ligands. Engagement of AGE, amyloid-β (Aβ) peptides, S100/calgranulins, HMGB1 and the β2-integrin MAC-1 contributes to a number of metabolic, neurodegenerative and inflammatory diseases and tumours by promoting cellular dysfunction and breaking immune tolerance. IκBα, inhibitor of NF-κB

RAGE-dependent cell activation is regulated at multiple levels. Summary of the various levels of regulation involved in the control of the RAGE pathway and its activation. C, ‘C’-type domain of immunoglobulin-like region; IκBα, inhibitor of NF-κB; V, ‘V’-type domain of immunoglobulin-like region

The multiple levels of regulation of the RAGE pathway
Regulation by ligand recognition
RAGE consists of a large extracellular part, a transmembrane domain and a 43 amino acid long cytoplasmic tail. The extracellular region is composed of three Ig-like domains: one V-type domain (V) and two C-type domains (C1, C2). The V and C1 domains seem to form an integrated unit, while C2 is attached to VC1 by a flexible linker and is fully independent [19]. The latter is likely to enable the receptor to bind to a wide spectrum of ligands, explaining its involvement in many pathophysiological settings. N-glycosylation of RAGE further modulates and enhances ligand recognition in two ways. While deglycosylation seems to sensitise RAGE to bind experimental AGE-modified proteins with an extremely high degree of modification by AGE (76–89% lysine residues modified) [20], carboxylated glycans on RAGE increase the affinity for S100-calcium binding protein (S100) and high-mobility group protein 1 (HMGB1) proteins [21, 22]. The quaternary structure of the RAGE extracellular domain, but also post-translational modifications of its primary structure, might account for the diversity of ligand recognition. It remains unknown whether differences in splice variants between humans and mice [23] might also influence ligand specificity in different diseases. Thus, the postulated promiscuity of RAGE, which would make it potentially able to bind almost damage-associated molecular patterns [24], does not in fact exist, since post-translational modifications affect the relative specificity of a given ligand in a given situation of cellular activation.
Regulation by ligand × RAGE interaction
Local and systemic inflammation induces the formation of ROS, oxidative and carbonyl stress, and glycooxidation, which in turn results in enhanced formation of AGE, advanced lipoxidation end-products (ALEs) and advanced oxidation protein products (AOPPs) [25–30]. When proteins were modified experimentally by similar processes to extreme, high extents, they were found to bind RAGE in vitro [26]. Thus, it remains to be studied whether AGE × RAGE interaction plays a significant role in vivo. Under hyperglycaemic conditions, AGE (but also ALE and AOPP) formation is increased and facilitates cellular activation [7], since any transient NF-κB activation by increased oxidative stress, high glucose, NEFA, oxidised LDL or other inflammatory stimuli upregulates RAGE expression [8].
In the late 1990s, S100A12 (EN-RAGE) was recognised to be another RAGE-binding protein [31], stimulating the field to study S100/calgranulin interaction with the receptor. So far S100A4, S100A6, S100A7, S100A8/9, S100A12, S100B and S100P have been identified to bind RAGE [32]. Ligands identified for RAGE also include amyloid-beta-peptides [33], HMGB1 (formerly known as amphoterin) [34] and surface molecules on bacteria and prions [8]. RAGE also binds to CD11b/CD18 (MAC-1) and promotes leucocyte extravasation into inflamed tissue [35], further putting RAGE in the context of innate immune responses.
In contrast to the long known role of AGE in late diabetic complications [36], these RAGE ligands have only recently been recognised to accumulate in diabetes and to contribute to its pathology. First evidence was provided by increased serum levels of S100A8/9 (migration inhibitory factor-related protein-8/14) in type 1 diabetic patients [37], as well as by elevated S100A12 plasma levels in type 2 diabetes [38]. In addition, increased serum levels of HMGB1 were associated with coronary artery disease in type 2 diabetes [39]. These findings were emphasised by the increased levels of circulating S100A8/A9 and increased accumulation of AGE, S100A8/A9 and HMGB1 observed in aortic lesions of diabetic Apoe −/− mice in a model of diabetes-accelerated atherosclerosis [40]. Thus, different members of the S100-protein family, HMGB1 and other ligands are providing a rationale for understanding disease-specific patterns of RAGE ligand-mediated cellular dysfunction. Experiments in streptozotocin-induced diabetic rats further demonstrated that diabetes increased amyloid-beta-peptide (1–40) levels in the brain, at least in part due to a decline in the brain-to-blood efflux of amyloid-beta-peptide (1–40) [41], extending the concept of ligand specificity to a local tissue specificity for RAGE × ligand interaction.
Furthermore, RAGE × ligand interaction is not only controlled by spatial and temporal patterns of ligand availability, but also on the level of affinity for the receptor. Carboxylated N-glycans on RAGE facilitate binding of HMGB1 [21] and mediate binding of S100A8/A9 to subpopulation of RAGE on colon cancer cells [22], but increased ligand binding to RAGE can also be observed upon glycation of its ligands [42]. N ɛ-carboxymethyllysine (CML)-modifications of S100A8 and S100A9 occur in inflammatory bowel disease and enhance RAGE-mediated sustained inflammation [42]. While the role of CML as a RAGE ligand is still being controversially discussed [43, 44] and some investigators have not been able to confirm that CML residues are ligands of RAGE [43], CML-modified S100-proteins may be important contributors in the progression of inflammation. This further supports the idea that an AGE-rich environment promotes complex ligand structures that enhance and amplify inflammation. However, it remains to be deciphered whether AGE modifications of RAGE ligands [42] or AGE formed by inflammatory cells via the myeloperoxidase system [45] are more important in maintaining inflammation.
Regulation by inducible RAGE expression
The functional role of RAGE is not only controlled by the presence and modifications of its ligands, but also at the level of RAGE gene expression itself. RAGE is constitutively produced during embryonic development, while its levels are downregulated in adult life. The only exception is the lung, in which RAGE is abundant throughout life. In other cells, however, RAGE expression is low under physiological conditions. RAGE is induced by inflammatory settings, since its transcription is controlled by several proinflammatory transcription factors, including SP-1, AP-2, NF-IL6 and NF-κB [8, 15, 16, 46]. Thus, a positive autoregulatory loop evolves, in which cytokines or other proinflammatory stimuli initiate NF-κB-activation, which in turn induces RAGE expression and subsequent RAGE-mediated perpetuated NF-κB activation [8, 9]. Ligation of RAGE results in a constantly growing and renewable pool of de novo synthesised, transcriptionally active p65, thereby amplifying the host response [9]. This feed-forward loop has been demonstrated in vitro and in vivo [9, 11] (Fig. 3). Consistently, endothelial cells cultured in the presence of highly glycated albumin (1 µg/ml) upregulate RAGE expression and activate proinflammatory gene expression upon induction with S100A8/A9 heterodimers (1 µmol/l), while the same concentration of S100A8/A9 does not alter gene expression in cells cultivated in the absence of glycated albumin [47]. It remains unknown whether alternate splicing of RAGE or shedding of RAGE contributes to the downregulation of this self-amplifying cell response. Remarkably, recent data indicate that soluble RAGE (sRAGE) binds to MAC-1 and thereby forms potent inflammatory complexes instead of limiting inflammation [48]. Another as yet unsolved mystery is the fact that pulmonary cells constitutively express RAGE without concomitant pulmonary inflammation.

RAGE converts acute cellular activation into a sustained cellular response. Schematic representation of RAGE functioning as a ‘master-switch’ by converting transient cellular activation into long-lasting cellular dysfunction: activation of NF-κB by a variety of stimuli results in increased NF-κB-regulated gene expression including RAGE induction [15]. Upregulation of RAGE provides an increasing number of binding sites for ligands and engagement of RAGE subsequently results in perpetuated NF-κB activation through de novo synthesis of p65 mRNA [9]. The increased availability of transcriptional active NF-κB neutralises inhibitor of NF-κB (IκBα)-driven negative auto-regulatory loops and perpetuates the cellular inflammatory response [9]. The mRNA insert is partly adapted from Fig. 10a in ref. [9]. d, days

Regulation by RAGE shedding and splicing
Cell activation stimulates inflammatory cells to release endogenous alarmins such as S100-proteins, HMGB1, heat shock proteins and IL1α via non-conventional secretory pathways. Binding of HMGB1 to RAGE induces RAGE shedding by A disintegrin and metalloprotease 10 (ADAM10) [49] and thus might represent one pathway for downregulating RAGE-mediated cell activation. So far, however, it is not known whether ADAM10-induced shedding of cellular RAGE is instrumental in downregulating surface RAGE and limiting RAGE-mediated inflammation or whether it only generates a biomarker for RAGE-dependent cell activation. It is also not known whether binding of other RAGE ligands also results in ADAM10-mediated RAGE shedding. However, proteolytic conversion of full-length RAGE into shedded RAGE is likely to account to a large extent for the sRAGE levels determined in plasma and serum of patients. In this context, it is most important to better understand sRAGE and its physiological function, since misinterpretations have hitherto contributed to a significant confusion in the literature.
Soluble RAGE, comprising the extracellular domain, but leaking from the transmembrane and cytoplasmic domain of RAGE, has been described in the plasma of men. Circulating sRAGE is either produced by receptor ectodomain shedding [49, 50] or represents splice variants of RAGE such as endogenous secreted RAGE (esRAGE) [51]. Comparison of splice variants between humans and mice revealed homologous and species-specific mechanisms for splicing of the RAGE gene, but also tissue-specific splice variant distribution and the absence of plasma sRAGE in mice [23, 52]. While diabetic mice demonstrated a significant increase in RAGE splice variants, the latter was not associated with any detectable level of sRAGE protein in murine plasma [23]. Since sRAGE can be readily detected in human plasma, this observation not only indicates important differences between human and murine RAGE [23], but further implies that sRAGE observed in humans is, in the main, produced by shedding mechanisms [49]. Since in vitro and experimental animal models have demonstrated that exogenous sRAGE functions as a decoy by preventing ligands from interacting with cellular RAGE, it was supposed that sRAGE might counteract RAGE-mediated cell activation [53]. However, the concentration of sRAGE found in diabetic patients with renal disease is ~1,200 pg/ml (<0.1 nmol/l). It seems questionable whether this is sufficient to scavenge accumulating ligands [42, 54], in particular, since sRAGE was previously shown to bind highly CML-modified AGE albumin prepared in vitro in a saturable and dose-dependent manner only at a K d of ~75 nmol/l [44]. Thus, the sRAGE concentration found in patients is approximately 750-fold lower than required to efficiently keep ligands away from cellular RAGE. Furthermore, in vitro studies have shown that only a minor part (~10%) of CML is capable of binding to RAGE, making sRAGE × CML interaction unlikely to be of physiological impact. Consistently, studies addressing the association between soluble forms of RAGE and disease are contradictory with regard to negative and positive correlations. In this context, sRAGE was shown to be associated with a greater prevalence of cardiovascular disease in type 1 and type 2 diabetic individuals [55] and with increased severity of renal disease in type 2 diabetic patients [56]. Hence sRAGE is an activation marker likely to reflect increased RAGE expression and shedding. Yet in the few studies in which sRAGE and esRAGE levels were determined in parallel, only sRAGE showed a significant correlation with albuminuria [56], while studies looking at the effects of various drugs such as thiazolidinediones, statins and ACE inhibitors on sRAGE levels revealed contradictory results [53, 57–60]. Since vascular disease is a local event and the RAGE pathway is controlled at several and, at least in part, tissue-specific levels, we believe that determining circulating sRAGE, which is influenced by shedding, splicing, degradation and the assay method employed, will not add to a better understanding of the molecular events promoting and perpetuating late diabetic complications.

Regulation at the level of downstream signalling
Although there is evidence that different ligands activate RAGE through different signal transduction cascades [61], the proximate mechanisms by which engagement of RAGE induces intracellular signalling have yet to be identified. While the cytoplasmic domain of RAGE lacks endogenous kinase activity or any other known motif involved in receptor signalling [62], in vitro and in vivo experiments using dominant negative RAGE devoid of the cytoplasmic tail indicate that the cytoplasmic domain is essential for intracellular signalling [15]. Experiments using in vitro produced, highly modified AGE albumin provide evidence that the cytoplasmic domain possibly interacts with diaphanous-1 and transduces the extracellular signals evoked into cellular signalling through activation the rho-GTPases RAC-1 and CDC42, reorganisation of the actin cytoskeleton, regulation of cell motility and migration [62]. Future studies, however, should show whether physiological ligand activation and/or cell-specific regulation mechanisms exist for the proposed RAGE-mediated multiple cellular signalling cascades, including RAC-1 and CDC42, NADPH-oxidase, ERK1/2(p44/p42)-, p38- and SAPK/JNK-MAP-kinases, phosphoinositol-3-kinase and the JAK/STAT pathway [8, 15, 17, 63]. A critical review of published data defining RAGE-dependent signalling pathways reveals that only a few approaches have studied those time points (e.g. >5 days) that would be most interesting for RAGE action [9].
Regulation by protein × protein interaction
Unstimulated endothelial cells are characterised by the expression of thrombomodulin (TM). The lectin domain of TM can bind HMGB1 and thereby prevent its interaction with RAGE [64]. Thus, binding of HMGB1 by TM might be a mechanism for terminating RAGE-mediated sustained inflammatory responses. In diabetes, TM production is reduced [65], which results in loss of this counter-regulatory pathway that is dependent on protein × protein interaction. Future studies need to show whether reconstitution of TM in experimental diabetes can reduce late diabetic complications by specifically interfering with the RAGE pathway.
Regulation by cell × cell interaction
The quaternary structure of RAGE further allows interaction with surface receptors integrated on to neighbouring cells, thus mediating cell × cell interaction as demonstrated for RAGE binding to the macrophage-1-glycoprotein (MAC-1; CD11b/CD18) [35]. During firm leucocyte arrest, the β2-integrins lymphocyte-function-associated-antigen-1 (LFA-1; αLβ; CD11a/CD18) and MAC-1 are expressed on the leucocyte surface and interact with their endothelial counter-receptors such as intercellular adhesion molecule 1 (ICAM-1) and RAGE. Besides its role as MAC-1 ligand, RAGE-mediated NF-κB activation regulates the gene expression of ICAM-1 and VCAM-1 [12]. In a model of type 2 diabetes, Apoe −/− db/db mice displayed RAGE-dependent enhanced production of VCAM-1 and tissue factor and matrix metalloproteinase-9 antigen/activity in aortic tissue, supporting a role of RAGE and its ligands in regulating leucocyte recruitment and adhesion in inflammatory processes. HMGB1-dependent mechanisms, which require the presence of neutrophil-expressed RAGE and MAC-1, further promote the recruitment of neutrophils at sites of inflammation [66]. These findings emphasise a growing role of RAGE in inflammatory processes and indicate that induction of RAGE in one cell is sufficient to induce inflammation in the neighbouring cells via ligand × RAGE interaction.

Regulation through inhibition of cellular defence mechanisms
In addition, the RAGE pathway is under control of a positive feed-forward loop due to a RAGE-mediated loss of cellular defence mechanisms, thus further enhancing perpetuated cell activation. Engagement of RAGE by highly modified AGE prepared in vitro results in the suppression of reduced glutathione and ascorbic acid levels [67]. Inhibition of glutathione is critical in maintaining activity of glyoxalase-1 (Glo1), the major cellular defence enzyme in the detoxification of precursors of AGE such as methylglyoxal, glyoxal and other alpha-oxoaldehydes [68–70]. Since alpha-oxoaldehydes represent the largest pool of reactive intracellular AGE, glyoxalase-1 has an important role in reducing the cellular AGE load [68]. Studies in the model organism Caenorhabditis elegans (C. elegans) have confirmed that overexpression of glyoxalase-1 not only prevents AGE formation, but also protects the animals from deleterious effects of oxidant stress as evidenced by increased longevity [70]. Hyperglycaemia, methylglyoxal, AGE and other RAGE ligands significantly reduce glyoxalase-1 expression and activity in cultured fibroblasts, endothelial cells and dorsal root ganglia cells, while inhibition or deletion of RAGE restored glyoxalase-1 in these cells [8]. This implies that engagement of RAGE not only results in increased cellular activation, but also in reduction of AGE-detoxifying mechanisms (Fig. 4). Impairment of these detoxifying mechanisms results in enhanced and perpetuated cell activation, ultimately limiting life span [70].

Ligand binding to RAGE impairs cellular defence mechanisms. Ligation of RAGE suppresses cellular detoxifying mechanisms such as glyoxalase-1 [8]. Subsequent accumulation of reactive metabolites promotes acute inflammatory responses and enhances RAGE-induced chronic inflammation. Aβ, amyloid beta; O −2 , superoxide
The role of RAGE in various pathological settings
RAGE in innate immune responses
In order to further define a potential role of RAGE in immunity and inflammation, interaction of ligands with cell surface RAGE was intercepted using recombinant sRAGE, RAGE-neutralising antibodies and/or transfection with plasmids overexpressing dominant negative RAGE. In addition, homozygous RAGE-deficient mice (Rage −/− mice) were generated [10]. Consistent with the proposed central role of RAGE in the immune response, it was demonstrated that RAGE inhibition or deletion protected mice from the lethal effects of septic shock caused by caecal ligation and puncture. Rage −/− mice further displayed reduced NF-κB activation in key target organs of septic shock, as well as a significant reduction of inflammatory cells migrating into the peritoneum [12]. In contrast, injection of exogenous AGE increased lethality and NF-κB activation in RAGE-bearing mice, but not in RAGE-deficient mice [71], further pointing to a causal relationship between RAGE ligands, RAGE signalling, NF-κB activation and lethality in septicaemia.
Rage −/− mice were also protected in colitis models [31, 42]. Members of the S100/calgranulin family released by inflammatory cells interact with cellular RAGE on endothelium, mononuclear phagocytes and lymphocytes, thereby inducing and sustaining cellular activation via RAGE [31]. Subsequent studies revealed that S100-proteins isolated from inflamed tissue of biopsies from patients with colitis were post-translationally modified by the defined AGE adduct CML [42]. These CML-modified S100 proteins, which by virtue of their proposed interaction with RAGE might have the capacity to drive and sustain the inflammatory response, seem to be primed for an exaggerated response to inflammatory stimuli [42]. Thus, inflammation is pronounced in situations where a tissue bed enriched with CML-mps and post-translational modifications is infiltrated by cells bearing RAGE. Besides AGE and S100-proteins, HMGB1 released from necrotic and activated inflammatory cells can exaggerate the innate immune response, particularly since HMGB1 × RAGE interaction has been shown to induce a proinflammatory phenotype in macrophages [15, 35, 46, 72, 73].

RAGE in adaptive immune responses
HMGB1 interaction with RAGE activates plasmacytoid dendritic cells and B cells in response to DNA-containing immune complexes and thus contributes to autoimmune pathogenesis [74]. Activation of innate immune responses may alter the way in which antigens are presented to T and other cells of the adaptive immune response. Consistent with this paradigm, blockade of RAGE, either by sRAGE, anti-RAGE F(ab′)2 fragments or the selective expression of dominant negative RAGE in CD4+ T cells, suppressed experimental autoimmune encephalomyelitis (EAE) induced by myelin basic protein peptide and encephalitogenic T cells [75]. When Pasteurella pneumotropica-mediated EAE was induced in Rage −/− mice, they developed typical EAE symptoms and no significant protection was found compared with wild-type mice [12]. In models of delayed type hypersensitivity, Rage −/− mice elicited a normal inflammatory response [12]. Unexpectedly, treatment of Rage −/− mice with sRAGE blocked the inflammatory response to the same extent as in RAGE-bearing wild-type mice [12]. The finding that Rage −/− mice were capable of evoking a normal adaptive immune response contrasted with the efficacy with which sRAGE suppressed inflammation in Rage −/− and wild-type mice. The explanation is that sRAGE functions as a decoy, which prevents RAGE ligands from sequestering to other surface receptors such as the toll-like receptors 2 and 4, CD36, α7-nicotinic acetylcholine receptors and proteoglycans, all of which have been shown to bind to various RAGE ligands [8, 15, 76–78].
Other studies, however, indicate a direct effect of RAGE on mobilisation of dendritic cells [79] and T cell migration, localisation, activation and differentiation [35, 75, 80], whereas HMGB1 signalling through RAGE seems to be required for clonal expansion, survival and functional polarisation of naive T cells [81]. Furthermore, clonal populations of Rage −/− mice-derived T cells adoptively transferred into wild-type recipients showed reduced responses [82]. Syngeneic islet graft and islet allograft rejection were reduced in diabetic mice treated with small molecule RAGE inhibitors and in diabetic Rage −/− mice [80], suggesting a close link between RAGE and the pathogenesis of type 1 diabetes [15]. It is not known why RAGE participates in some, but not all adaptive immune responses. One might speculate that adaptive immune responses in different tissues induce different ligand and receptor expression patterns and therefore might activate RAGE-dependent and RAGE-independent cellular responses. Furthermore, there might be differences in the crosstalk between the adaptive and innate immune response depending on the model tested.
RAGE in non-diabetic atherosclerosis and ischaemia
Prolonged inflammation is suspected to promote arterial remodelling. Studies of human vascular tissue have shown increased endothelial RAGE expression at the mRNA and antigen level in non-diabetic patients with peripheral occlusive vascular diseases [83]. Besides, smooth muscle cells and nerves in the vessel wall showed constitutively high levels of RAGE expression that were unchanged with ageing or by the presence of vascular disease [83]. Consistent with a pathogenic role for ligand × RAGE interaction in non-diabetic atherosclerosis, sRAGE reduced atherosclerotic lesions and inflammation in normoglycaemic Apoe −/− mice without affecting plasma cholesterol and triacylglycerol levels [84–87]. Wild-type mice treated with sRAGE and Rage −/− mice also showed significantly reduced neointima expansion in femoral artery denudation-induced arterial injury models, whereas protection conferred by sRAGE was more prominent than that due to RAGE deletion [40, 88].
Pathophysiologically relevant concentrations of C-reactive protein have recently been shown to impair the antioxidant defence in endothelial progenitor cells (EPC) [89] and to promote EPC sensitivity toward oxidant-mediated apoptosis and telomerase inactivation via a RAGE-dependent mechanism [90, 91]. In contrast, HMGB1 × RAGE interaction can stimulate EPC migration, which is consistent with previous reports where low concentrations of exogenously administrated HMGB1 led, in a murine model of myocardial infarction, to recovery through regeneration of cardiomyocytes from resident cardiac c-KIT+ progenitor cells [92]. However, in patients with ST-segment elevation myocardial infarction, elevated serum HMGB1 levels were associated with adverse clinical outcomes [93]. In line with the latter observation, a recent study comparing ischaemia/reperfusion injury of the heart in healthy wild-type and Rage −/− mice demonstrated that infarct size and severity of tissue damage was dependent on HMGB1 × RAGE interaction, indicating that RAGE links necrotic cell death to sustained inflammation and tissue damage [94]. Interestingly, HMGB1/RAGE-mediated myocardial injury was even more pronounced under diabetic conditions [95]. Similarly, HMGB1-RAGE signalling has been shown to contribute to ischaemic brain damage [96]. Tissue damage in RAGE-bearing mice was reduced by HMGB1 box A, an antagonist of HMGB1 interaction with RAGE [94, 96]. These data indicate that the availability of RAGE binding sites, the concentration of HMGB1 and its binding affinity for RAGE dictate whether HMGB1 has a repairing or damaging function; they also imply that situations in which high concentrations of HMGB1 can sufficiently bind to RAGE counteract the regenerating potential of HMGB1.
RAGE in late diabetic complications
The RAGE-dependent mechanisms described in the various models of immune and inflammatory cellular responses also seem to be present in diabetes [2, 15]. Induction of diabetes in RAGE-transgenic and Rage −/− mice confirmed that RAGE contributes, at least in part, to the development of macro- and microvascular complications. In diabetes-associated enhanced atherosclerosis models, RAGE overexpression in transgenic mice was associated with increased vascular injury, while RAGE deletion conferred partial vascular protection [40]. Diabetic nephropathy, characterised by renal enlargement, glomerular hypertrophy, albuminuria and mesangial expansion, was significantly increased in diabetic RAGE transgenic mice and prevented by the putative AGE inhibitor (+/−)-2-isopropylidenehydrazono-4-oxo-thiazolidin-5-ylacetanilide (OPB-9195) [97]. In contrast, Rage −/− mice were largely protected from diabetic nephropathy [98, 99]. Similar changes were observed in diabetic neuropathy. Whereas RAGE-transgenic diabetic mice showed an increase in functional deficits such as delayed motor nerve conduction velocity, Rage −/− mice were partially protected from the diabetes-induced loss of neuronal function [11, 100]. In the diabetic retina, RAGE is predominantly upregulated in glia cells [101]. While the understanding of RAGE and its role in retinal dysfunction in diabetes and/or activation of pro-inflammatory pathways is less complete than in other organ systems, increasing evidence indicates that RAGE contributes to cellular perturbations in the retina [102–104]. Remarkably, in each of these models, protection from development of the pathological features was more profound in wild-type mice treated with sRAGE than in Rage −/− mice. In diabetic neuropathy for example, diabetic Rage −/− mice were only partly protected from a loss of pain perception, while administration of sRAGE to diabetic wild-type mice completely restored pain perception [11]. These observations further suggest that ligands sequestered by sRAGE are likely to interact with cellular structures different from RAGE and restrict its role inasmuch as RAGE seems to be only one of many receptors contributing to diabetes and its complications.

Open questions and future perspectives
The results from in vitro observations and experimental rodent models provide firm evidence that RAGE and its ligands are central to the onset and progression of diabetes and its complications. In line with this, Yan et al. recently proposed a model which places RAGE at multiple points in the pathogenesis of type 1 diabetes [15]. They speculate that (auto)-immune attacks on pancreatic beta cells lead to infiltration of inflammatory cells and increased expression and secretion of S100 proteins and HMGB1 by inflammatory cells. These are likely to damage islets, which consequently results in the development of hyperglycaemia, subsequent accumulation of AGE, AGE-dependent recruitment of RAGE to target organs and finally in micro- and macrovascular complications [15]. A comparable situation is conceivable in type 2 diabetes, in which various metabolic disturbances induce inflammatory cells to secrete RAGE ligands, resulting in low-grade inflammation and increased oxidative and carbonyl stress, all of which promote AGE formation and induction of RAGE expression in the respective organs [8]. However, clinical observations do not yet convincingly support the proposed central role of RAGE in experimental diabetes and raise the question of whether studies performed in rodents, particularly in monogenetic mouse models, can be readily translated to human disease. Association studies aiming to link diabetes and its complications with sRAGE and esRAGE plasma and serum levels are far from conclusive due to over- or misinterpretation of associations and supposed correlations. Immunhistochemical characterisation of human inflammatory cells, vascular tissues, kidneys, nerves and retina has clearly demonstrated the accumulation of AGE in human diabetic tissue and its correlation with the severity of disease [6, 7]. So far, however, only a few studies have provided firm evidence of colocalisation of AGE with RAGE, its effector NF-κB and NF-κB-controlled gene expression, as recently demonstrated in human sural nerve biopsies from diabetes patients [11]. In any case, these studies can only provide evidence of association, but do not prove a cause-to-consequence relation. Therefore, multiple approaches to identify polymorphisms of the RAGE gene have been studied in relation to diabetic complications. While some polymorphisms correlate with insulin resistance and early- or late-onset of diabetic complications [105–108], none have so far pointed to the possibility that RAGE is the most important factor in the pathology of human diabetes. Although mutations deleting RAGE function may not be compatible with life and therefore not found in cohort studies, the fact that Rage −/− mice are viable and do not show an overt phenotype contradict this hypothesis or point to important differences between mice (kept in a sterile environment) and human diabetes.
Since RAGE ligands also bind to other receptors and might additionally act via receptor-independent mechanisms, it seems more likely that the extent of hyperglycaemia-induced cellular dysfunction and subsequent tissue damage are determined by the combination of various proinflammatory ligands, PRRs and differentially activated cellular pathways. This might be further modulated by the local distribution of the different ligands and receptors, as well as by the ligand-binding affinities for RAGE and RAGE-related receptors. This view is emphasised by the observation that in rodent models sRAGE protects more efficiently from late diabetic complications than RAGE deletion [8]. The observation that AGE are sufficient to induce neuronal damage in the model organism C. elegans (which neither possesses a known RAGE homologue, nor NF-κB) and that this damage can be ameliorated by overexpression of glyoxalase-1 further supports this notion [70]. First clinical trials using small RAGE inhibitors for short-term intervention have recently been initiated and are likely to provide some answers to these questions in the near future [15]. However, until the physiological function of RAGE has been clearly defined, it seems wise to adopt a cautious approach to future therapeutic strategies involving long-term blockade of RAGE or its ligands.
Abbreviations
- ADAM10:
-
A disintegrin and metalloprotease 10
- ALE:
-
Advanced lipoxidation end-products
- AOPP:
-
Advanced oxidation protein products
- CDC42:
-
Cell division cycle 42-protein
- CML:
-
N ɛ-carboxymethyllysine
- EAE:
-
Experimental autoimmune encephalomyelitis
- EPC:
-
Endothelial progenitor cells
- ERK:
-
Extracellular signal-regulated kinase
- esRAGE:
-
Endogenous secreted RAGE
- HMGB1:
-
High-mobility group protein 1
- ICAM-1:
-
Intercellular adhesion molecule 1
- MAC-1:
-
Macrophage-1 glycoprotein (CD11B/CD18)
- MRP:
-
Migration inhibitory factor-related protein
- NF-κB:
-
Nuclear factor kappa-B
- PRR:
-
Pattern recognition receptor
- RAC-1:
-
Ras-related C3 botulinum toxin substrate 1
- RAGE:
-
Receptor for AGE
- ROS:
-
Reactive oxygen species
- S100:
-
S100-calcium binding protein
- sRAGE:
-
Soluble RAGE
- TM:
-
Thrombomodulin
- VCAM-1:
-
Vascular cell adhesion molecule 1
References
Hudson BI, Stickland MH, Grant PJ, Futers TS (2001) Characterization of allelic and nucleotide variation between the RAGE gene on chromosome 6 and a homologous pseudogene sequence to its 5′ regulatory region on chromosome 3: implications for polymorphic studies in diabetes. Diabetes 50:2646–2651
Schmidt AM, Yan SD, Yan SF, Stern DM (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 108:949–955
Neeper M, Schmidt AM, Brett J et al (1992) Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 267:14998–15004
Schmidt AM, Hori O, Brett J, Yan SD, Wautier JL, Stern D (1994) Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arterioscler Thromb 14:1521–1528
Yan SD, Stern D, Schmidt AM (1997) What's the RAGE? The receptor for advanced glycation end products (RAGE) and the dark side of glucose. Eur J Clin Invest 27:179–181
Bierhaus A, Hofmann MA, Ziegler R, Nawroth PP (1998) AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovasc Res 37:586–600
Thornalley PJ (1998) Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell Mol Biol (Noisy-le-grand) 44:1013–1023
Bierhaus A, Humpert PM, Morcos M et al (2005) Understanding RAGE, the receptor for advanced glycation end products. J Mol Med 83:876–886
Bierhaus A, Schiekofer S, Schwaninger M et al (2001) Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 50:2792–2808
Constien R, Forde A, Liliensiek B et al (2001) Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis 30:36–44
Bierhaus A, Haslbeck KM, Humpert PM et al (2004) Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J Clin Invest 114:1741–1751
Liliensiek B, Weigand MA, Bierhaus A et al (2004) Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest 113:1641–1650
Sparvero LJ, Asafu-Adjei D, Kang R et al (2009) RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J Transl Med 7:17
Gebhardt C, Riehl A, Durchdewald M et al (2008) RAGE signaling sustains inflammation and promotes tumor development. J Exp Med 205:275–285
Yan SF, Ramasamy R, Schmidt AM (2009) Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J Mol Med 87:235–247
Yan SF, Du Yan S, Ramasamy R, Schmidt AM (2009) Tempering the wrath of RAGE: an emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann Med: 1–15
Coughlan MT, Thorburn DR, Penfold SA et al (2009) RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol 20:742–752
Li JH, Wang W, Huang XR et al (2004) Advanced glycation end products induce tubular epithelial-myofibroblast transition through the RAGE-ERK1/2 MAP kinase signaling pathway. Am J Pathol 164:1389–1397
Dattilo BM, Fritz G, Leclerc E, Kooi CW, Heizmann CW, Chazin WJ (2007) The extracellular region of the receptor for advanced glycation end products is composed of two independent structural units. Biochemistry 46:6957–6970
Osawa M, Yamamoto Y, Munesue S et al (2007) De-N-glycosylation or G82S mutation of RAGE sensitizes its interaction with advanced glycation endproducts. Biochim Biophys Acta 1770:1468–1474
Srikrishna G, Huttunen HJ, Johansson L et al (2002) N-Glycans on the receptor for advanced glycation end products influence amphoterin binding and neurite outgrowth. J Neurochem 80:998–1008
Turovskaya O, Foell D, Sinha P et al (2008) RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis 29:2035–2043
Kalea AZ, Reiniger N, Yang H, Arriero M, Schmidt AM, Hudson BI (2009) Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J 23:1766–1774
Foell D, Wittkowski H, Roth J (2007) Mechanisms of disease: a ‘DAMP’ view of inflammatory arthritis. Nat Clin Pract Rheumatol 3:382–390
Kalousova M, Zima T, Tesar V, Dusilova-Sulkova S, Skrha J (2005) Advanced glycoxidation end products in chronic diseases—clinical chemistry and genetic background. Mutat Res 579:37–46
Marsche G, Semlitsch M, Hammer A et al (2007) Hypochlorite-modified albumin colocalizes with RAGE in the artery wall and promotes MCP-1 expression via the RAGE-Erk1/2 MAP-kinase pathway. Faseb J 21:1145–1152
Anderstam B, Ann-Christin BH, Valli A, Stenvinkel P, Lindholm B, Suliman ME (2008) Modification of the oxidative stress biomarker AOPP assay: application in uremic samples. Clin Chim Acta 393:114–118
Capeillere-Blandin C, Gausson V, Descamps-Latscha B, Witko-Sarsat V (2004) Biochemical and spectrophotometric significance of advanced oxidized protein products. Biochim Biophys Acta 1689:91–102
Witko-Sarsat V, Friedlander M, Capeillere-Blandin C et al (1996) Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int 49:1304–1313
Ahmed N, Thornalley PJ (2002) Chromatographic assay of glycation adducts in human serum albumin glycated in vitro by derivatization with 6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate and intrinsic fluorescence. Biochem J 364:15–24
Hofmann MA, Drury S, Fu C et al (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901
Leclerc E, Fritz G, Vetter SW, Heizmann CW (2009) Binding of S100 proteins to RAGE: an update. Biochim Biophys Acta 1793:993–1007
Deane R, Du Yan S, Submamaryan RK et al (2003) RAGE mediates amyloid-beta peptide transport across the blood–brain barrier and accumulation in brain. Nat Med 9:907–913
Huttunen HJ, Fages C, Rauvala H (1999) Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem 274:19919–19924
Chavakis T, Bierhaus A, Al-Fakhri N et al (2003) The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med 198:1507–1515
Jandeleit-Dahm K, Cooper ME (2008) The role of AGEs in cardiovascular disease. Curr Pharm Des 14:979–986
Bouma G, Lam-Tse WK, Wierenga-Wolf AF, Drexhage HA, Versnel MA (2004) Increased serum levels of MRP-8/14 in type 1 diabetes induce an increased expression of CD11b and an enhanced adhesion of circulating monocytes to fibronectin. Diabetes 53:1979–1986
Kosaki A, Hasegawa T, Kimura T et al (2004) Increased plasma S100A12 (EN-RAGE) levels in patients with type 2 diabetes. J Clin Endocrinol Metab 89:5423–5428
Yan XX, Lu L, Peng WH et al (2008) Increased serum HMGB1 level is associated with coronary artery disease in nondiabetic and type 2 diabetic patients. Atherosclerosis. doi:10.1016/j.atherosclerosis.2008.12.016
Soro-Paavonen A, Watson AM, Li J et al (2008) Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 57:2461–2469
Liu Y, Liu H, Yang J et al (2008) Increased amyloid beta-peptide (1–40) level in brain of streptozotocin-induced diabetic rats. Neuroscience 153:796–802
Andrassy M, Igwe J, Autschbach F et al (2006) Posttranslationally modified proteins as mediators of sustained intestinal inflammation. Am J Pathol 169:1223–1237
Buetler TM, Leclerc E, Baumeyer A et al (2008) N(epsilon)-carboxymethyllysine-modified proteins are unable to bind to RAGE and activate an inflammatory response. Mol Nutr Food Res 52:370–378
Kislinger T, Fu C, Huber B et al (1999) N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem 274:31740–31749
Anderson MM, Requena JR, Crowley JR, Thorpe SR, Heinecke JW (1999) The myeloperoxidase system of human phagocytes generates Nepsilon-(carboxymethyl)lysine on proteins: a mechanism for producing advanced glycation end products at sites of inflammation. J Clin Invest 104:103–113
Bierhaus A, Stern DM, Nawroth PP (2006) RAGE in inflammation: a new therapeutic target? Curr Opin Investig Drugs 7:985–991
Ehlermann P, Eggers K, Bierhaus A et al (2006) Increased proinflammatory endothelial response to S100A8/A9 after preactivation through advanced glycation end products. Cardiovasc Diabetol 5:6
Pullerits R, Brisslert M, Jonsson IM, Tarkowski A (2006) Soluble receptor for advanced glycation end products triggers a proinflammatory cytokine cascade via beta2 integrin Mac-1. Arthritis Rheum 54:3898–3907
Raucci A, Cugusi S, Antonelli A et al (2008) A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). Faseb J 22:3716–3727
Zhang L, Bukulin M, Kojro E et al (2008) Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J Biol Chem 283:35507–35516
Cheng C, Tsuneyama K, Kominami R et al (2005) Expression profiling of endogenous secretory receptor for advanced glycation end products in human organs. Mod Pathol 18:1385–1396
Hudson BI, Carter AM, Harja E et al (2008) Identification, classification, and expression of RAGE gene splice variants. Faseb J 22:1572–1580
Santilli F, Vazzana N, Bucciarelli LG, Davi G (2009) Soluble forms of RAGE in human diseases: clinical and therapeutical implications. Curr Med Chem 16:940–952
Nakamura K, Yamagishi S, Adachi H et al (2008) Serum levels of soluble form of receptor for advanced glycation end products (sRAGE) are positively associated with circulating AGEs and soluble form of VCAM-1 in patients with type 2 diabetes. Microvasc Res 76:52–56
Nin JW, Ferreira I, Schalkwijk CG et al (2009) Levels of soluble receptor for AGE are cross-sectionally associated with cardiovascular disease in type 1 diabetes, and this association is partially mediated by endothelial and renal dysfunction and by low-grade inflammation: the EURODIAB Prospective Complications Study. Diabetologia 52:705–714
Humpert PM, Djuric Z, Kopf S et al (2007) Soluble RAGE but not endogenous secretory RAGE is associated with albuminuria in patients with type 2 diabetes. Cardiovasc Diabetol 6:9
Forbes JM, Thorpe SR, Thallas-Bonke V et al (2005) Modulation of soluble receptor for advanced glycation end products by angiotensin-converting enzyme-1 inhibition in diabetic nephropathy. J Am Soc Nephrol 16:2363–2372
Nakamura K, Yamagishi S, Nakamura Y et al (2005) Telmisartan inhibits expression of a receptor for advanced glycation end products (RAGE) in angiotensin-II-exposed endothelial cells and decreases serum levels of soluble RAGE in patients with essential hypertension. Microvasc Res 70:137–141
Santilli F, Bucciarelli L, Noto D et al (2007) Decreased plasma soluble RAGE in patients with hypercholesterolemia: effects of statins. Free Radic Biol Med 43:1255–1262
Tan KC, Chow WS, Tso AW et al (2007) Thiazolidinedione increases serum soluble receptor for advanced glycation end-products in type 2 diabetes. Diabetologia 50:1819–1825
Donato R, Sorci G, Riuzzi F et al (2008) S100B's double life: intracellular regulator and extracellular signal. Biochim Biophys Acta 1793:1008–1022
Hudson BI, Kalea AZ, Del Mar Arriero M et al (2008) Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem 283:34457–34468
Lin L, Park S, Lakatta EG (2009) RAGE signaling in inflammation and arterial aging. Front Biosci 14:1403–1413
Abeyama K, Stern DM, Ito Y et al (2005) The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest 115:1267–1274
Isermann B, Vinnikov IA, Madhusudhan T et al (2007) Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med 13:1349–1358
Orlova VV, Choi EY, Xie C et al (2007) A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. Embo J 26:1129–1139
Bierhaus A, Chevion S, Chevion M et al (1997) Advanced glycation end product-induced activation of NF-kappaB is suppressed by alpha-lipoic acid in cultured endothelial cells. Diabetes 46:1481–1490
Thornalley PJ (2003) Glyoxalase I—structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans 31:1343–1348
Vander Jagt DL (2008) Methylglyoxal, diabetes mellitus and diabetic complications. Drug Metabol Drug Interact 23:93–124
Morcos M, Du X, Pfisterer F et al (2008) Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell 7:260–269
Humpert PM, Lukic IK, Thorpe SR et al (2009) AGE-modified albumin containing infusion solutions boosts septicaemia and inflammation in experimental peritonitis. J Leukoc Biol. doi:10.1189/jlb.1008646
Kokkola R, Andersson A, Mullins G et al (2005) RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol 61:1–9
Chavakis T, Bierhaus A, Nawroth PP (2004) RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect 6:1219–1225
Tian J, Avalos AM, Mao SY et al (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8:487–496
Yan SS, Wu ZY, Zhang HP et al (2003) Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat Med 9:287–293
Su X, Lee JW, Matthay ZA et al (2007) Activation of the alpha7 nAChR reduces acid-induced acute lung injury in mice and rats. Am J Respir Cell Mol Biol 37:186–192
Ohgami N, Nagai R, Ikemoto M et al (2002) CD36, serves as a receptor for advanced glycation endproducts (AGE). J Diabetes Complications 16:56–59
van Zoelen MA, Marieke AD, Yang H, et al. (2009) Role of Toll-like receptors 2 and 4, and the receptor for advanced glycation end products (RAGE) in HMGB1 induced inflammation in vivo. Shock 31:280–284
Manfredi AA, Capobianco A, Esposito A et al (2008) Maturing dendritic cells depend on RAGE for in vivo homing to lymph nodes. J Immunol 180:2270–2275
Chen Y, Akirav EM, Chen W et al (2008) RAGE ligation affects T cell activation and controls T cell differentiation. J Immunol 181:4272–4278
Dumitriu IE, Baruah P, Valentinis B et al (2005) Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J Immunol 174:7506–7515
Moser B, Desai DD, Downie MP et al (2007) Receptor for advanced glycation end products expression on T cells contributes to antigen-specific cellular expansion in vivo. J Immunol 179:8051–8058
Ritthaler U, Deng Y, Zhang Y et al (1995) Expression of receptors for advanced glycation end products in peripheral occlusive vascular disease. Am J Pathol 146:688–694
Bucciarelli LG, Wendt T, Qu W et al (2002) RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 106:2827–2835
Wendt T, Harja E, Bucciarelli L et al (2006) RAGE modulates vascular inflammation and atherosclerosis in a murine model of type 2 diabetes. Atherosclerosis 185:70–77
Park L, Raman KG, Lee KJ et al (1998) Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med 4:1025–1031
Nawroth P, Bierhaus A, Marrero M, Yamamoto H, Stern DM (2005) Atherosclerosis and restenosis: is there a role for RAGE? Curr Diab Rep 5:11–16
Sakaguchi T, Yan SF, Yan SD et al (2003) Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest 111:959–972
Fujii H, Li SH, Szmitko PE, Fedak PW, Verma S (2006) C-reactive protein alters antioxidant defenses and promotes apoptosis in endothelial progenitor cells. Arterioscler Thromb Vasc Biol 26:2476–2482
Chen J, Huang L, Song M, Yu S, Gao P, Jing J (2009) C-reactive protein upregulates receptor for advanced glycation end products expression and alters antioxidant defenses in rat endothelial progenitor cells. J Cardiovasc Pharmacol 53:359–367
Yan SD, Bierhaus A, Nawroth PP, Stern DM (2009) Endothelial precursor cells and CRP on the RAGE: activation or cell death? J Cardiovasc Pharmacol 53:349–351
Limana F, Germani A, Zacheo A et al (2005) Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res 97:e73–e83
Kohno T, Anzai T, Naito K et al (2009) Role of high-mobility group box 1 protein in post-infarction healing process and left ventricular remodelling. Cardiovasc Res 81:565–573
Andrassy M, Volz HC, Igwe JC et al (2008) High-mobility group box-1 in ischemia–reperfusion injury of the heart. Circulation 117:3216–3226
Andrassy M, Volz CH, Bianchi ME, Kaya Z, Bierhaus A, Katus HA (2008) The role of Hmgb1 in the development of diabetic cardiomyopathy. Circulation 118:S351–S352
Muhammad S, Barakat W, Stoyanov S et al (2008) The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci 28:12023–12031
Yamamoto Y, Kato I, Doi T et al (2001) Development and prevention of advanced diabetic nephropathy in RAGE-overexpressing mice. J Clin Invest 108:261–268
Wendt TM, Tanji N, Guo J et al (2003) RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am J Pathol 162:1123–1137
Tan AL, Forbes JM, Cooper ME (2007) AGE, RAGE, and ROS in diabetic nephropathy. Semin Nephrol 27:130–143
Toth C, Rong LL, Yang C et al (2008) Receptor for advanced glycation end products (RAGEs) and experimental diabetic neuropathy. Diabetes 57:1002–1017
Wang Y, Vom Hagen F, Pfister F et al (2008) Receptor for advanced glycation end product expression in experimental diabetic retinopathy. Ann N Y Acad Sci 1126:42–45
Barile GR, Schmidt AM (2007) RAGE and its ligands in retinal disease. Curr Mol Med 7:758–765
Hammes HP, Du X, Edelstein D et al (2003) Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat Med 9:294–299
Hammes HP, Hoerauf H, Alt A et al (1999) N(epsilon)(carboxymethyl)lysin and the AGE receptor RAGE colocalize in age-related macular degeneration. Invest Ophthalmol Vis Sci 40:1855–1859
Gaens KH, van Der Kallen CJ, van Greevenbroek MM, Feskens EJ, Stehouwer CD, Schalkwijk CG (2008) Receptor for advanced glycation end product polymorphisms and type 2 diabetes: the CODAM study. Ann N Y Acad Sci 1126:162–165
Prevost G, Fajardy I, Besmond C et al (2005) Polymorphisms of the receptor of advanced glycation endproducts (RAGE) and the development of nephropathy in type 1 diabetic patients. Diabetes Metab 31:35–39
Rudofsky G Jr, Isermann B, Schilling T et al (2004) A 63 bp deletion in the promoter of rage correlates with a decreased risk for nephropathy in patients with type 2 diabetes. Exp Clin Endocrinol Diabetes 112:135–141
Kankova K, Sebekova K (2005) Genetic variability in the RAGE gene: possible implications for nutrigenetics, nutrigenomics, and understanding the susceptibility to diabetic complications. Mol Nutr Food Res 49:700–709
Acknowledgements
This work was supported in part by grants from the Deutsche Forschungsgemeinschaft (SFB 405 to P. P. Nawroth), the Dietmar Hopp Foundation (to A. Bierhaus and P. P. Nawroth) and the Juvenile Diabetes Research Foundation (A. Bierhaus and P. P. Nawroth).
Duality of interest
The authors declare that there is no duality of interests associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bierhaus, A., Nawroth, P.P. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 52, 2251–2263 (2009). https://doi.org/10.1007/s00125-009-1458-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1458-9