
Abstract
Aims/hypothesis
Accumulating evidence suggests that diabetes increases the risk of dementia, but few studies have addressed possible mechanisms underlying this relationship. The aim of our study was to investigate the longitudinal association of glucose metabolism, insulin secretion and insulin action with the development of Alzheimer’s disease and vascular dementia.
Methods
The Uppsala Longitudinal Study of Adult Men is an ongoing observational study in Sweden in which 1,125 men aged 71 years and free from dementia underwent an OGTT and a euglycaemic insulin clamp between 1990 and 1995. During a median follow-up of 12 years, 257 persons developed dementia or cognitive impairment, of whom 81 had Alzheimer’s disease and 26 vascular dementia. Associations were analysed with the Cox proportional hazards method.
Results
Low early insulin response to oral glucose challenge, but not low insulin sensitivity, was associated with a higher risk of Alzheimer’s disease (HR for 1 SD decrease 1.32; 95% CI 1.02, 1.69) after adjustment for diabetes, blood pressure, body mass index, cholesterol, smoking and educational level. Low insulin sensitivity was associated with a higher risk of vascular dementia (HR for 1 SD decrease 1.55; 95% CI 1.02, 2.35), but not after multiple adjustments. Diabetes increased the risk of any dementia and cognitive impairment by 63%.
Conclusions/interpretation
In this community-based study, low early insulin response was associated with increased risk of subsequent Alzheimer’s disease, whereas low insulin sensitivity was not. Vascular dementia was not related to early insulin response. We suggest that glucometabolic disturbances are linked differentially to the pathogenesis of these two main dementia subtypes.
Similar content being viewed by others

Introduction
Alzheimer’s disease is the most common neurodegenerative dementia disorder, affecting a large proportion of populations worldwide, and it is increasing in prevalence [1]. Epidemiological studies and clinical observations have disclosed an association between diabetes mellitus and increased risk of cognitive impairment and dementia [2]. From some longitudinal studies, an association between insulin abnormalities and dementia has also been reported [3, 4]. However, the pathophysiological links underlying these associations are largely unknown and may involve different patterns of cerebral lesions [5]. Hyperglycaemia is accompanied by accumulation of advanced glycation end-products, oxidative stress and microvascular changes, which might lead to cerebral ischaemia. Insulin resistance and chronic peripheral hyperinsulinaemia may also lead to reduced insulin action in the brain [6]. In experimental studies, defective insulin signalling has been linked to cerebral accumulation of β-amyloid, a key neuropathological feature in Alzheimer’s disease [7].
Impaired insulin secretion and insulin resistance are the main determinants of the development of type 2 diabetes mellitus [8]. A low acute insulin response in an intravenous glucose tolerance test in midlife was associated with an increased risk of Alzheimer’s disease during a 32-year follow-up [9]. Furthermore, decreased peripheral insulin levels were associated with Alzheimer’s disease-related brain atrophy in a cross-sectional study [10], and such patients have also been found to have reduced insulin concentrations in the brain and the cerebrospinal fluid [11]. On the other hand, it has been suggested that features of the insulin resistance syndrome may imply an increase in the risk of vascular dementia, but the association with Alzheimer’s disease is still controversial [12, 13]. The relationship between insulin resistance as measured with the euglycaemic insulin clamp and incident dementia has not been assessed previously.
An OGTT, including measurement of the early insulin response, and a euglycaemic insulin clamp were performed in a population-based cohort of 1,125 men at age 71 years in Uppsala, Sweden. The aim was to clarify whether disturbances in glucose tolerance and insulin secretion and sensitivity would lead to increased risk of Alzheimer’s disease and vascular dementia later in life.
Methods
Study sample
A health survey entitled the Uppsala Longitudinal Study of Adult Men (ULSAM) was initiated in 1970. All men born in 1920–1924 living in the county of Uppsala were invited to participate and 2,322 (82%) agreed.
The baseline of the present study was the third examination cycle of the cohort (1991–1995). Of the 1,681 available 71-year-old men invited to take part in the investigation, 1,221 (73%) attended. Excluded from the present study were men with dementia at baseline (n = 5) and those who did not perform the OGTT and the euglycaemic–hyperinsulinaemic clamp (n = 91), leaving 1,125 participants for this analysis. All participants gave informed consent and the Ethics Committee of the Faculty of Medicine at Uppsala University approved the study.
Baseline examinations at age 71 years
An OGTT was performed by measuring the plasma concentrations of glucose, fasting insulin and insulin 30 and 120 min after ingestion of 75 g anhydrous dextrose. The early insulin response (EIR) was calculated as the ratio of the increment in plasma insulin to that in plasma glucose during the first 30 min of the OGTT [8]. Plasma concentrations of glucose were analysed by the glucose dehydrogenase method (Gluc-DH; Merck, Darmstadt, Germany). The concentrations of proinsulin and 32–33 split proinsulin were analysed using the two-site immunometric assay technique [14] in plasma samples that had been stored frozen (−70°C) since baseline. Insulin concentrations were also determined in these samples, using the Access Immunoassay System (Sanofi Pasteur Diagnostics; Redmond, WA, USA). The analyses were carried out at the Department of Clinical Biochemistry, Addenbrooke’s Hospital, Cambridge, UK [14].
Insulin sensitivity was determined by the euglycaemic insulin clamp method. This was performed as described by to DeFronzo et al. [15] with a slight modification: insulin was infused at a constant rate of 56 instead of 40 mU min−1 m−2) to achieve nearly complete suppression of hepatic glucose output [8]. The insulin-mediated glucose disposal rate, representing insulin sensitivity, calculated as the amount of glucose taken up during the last 60 min of the clamp procedure, is expressed as mg (kg body weight)−1 min−1. The OGTT and the euglycaemic clamp took place on separate days within 1 week. Measurements used in this study were made and analysed within a very short time (<1 week) after sampling, on a continuous basis during 1991–1995. A subgroup of 21 participants was investigated twice within 4–6 weeks to determine the combined effects of biological variation and measurement error on repeatedly measured variables. The intraclass correlation coefficients of the metabolic variables in ULSAM have been published previously [16].
Diabetes mellitus was defined as a fasting plasma glucose concentration >7.0 mmol/l or the use of pharmacological treatment for diabetes. Other observations made at age 71 years were a medical questionnaire, supine blood pressure measurements, BMI and serum cholesterol concentration. Apolipoprotein E (APOE) ε4 was genotyped by mini-sequencing in 860 participants. Smoking status (current smoker or non-smoker) and educational level (elementary school, secondary school or university) were assessed by interview questions.
Follow-up and assessment of dementia
The participants in ULSAM were invited to attend for cognitive testing on three occasions: at ages 71 (n = 986), 77 (n = 804) and 82 (n = 520) years. Testing at ages 71 and 77 years old included the Mini-Mental State Examination (MMSE) and Trail Making Tests A and B [17]. The tests used at age 82 were MMSE and a 7 min screening test that included clock drawing, enhanced cued recall, verbal fluency and temporal orientation. The diagnoses were not based on these test results. Participants with low performance were referred to the Uppsala geriatric memory clinic for a follow-up. Persons with cognitive impairment are less likely to attend follow-up examinations of a longitudinal study [18]. For this reason the available medical records of all ULSAM participants were reviewed, with 31 December 2005 as the censor date. The standardised review covered all outpatient clinics, including the Uppsala geriatric memory clinic, where persons may consult directly for memory problems, all hospitalisations, including those in the Geropsychiatric Department, and all primary care facilities, including community nursing homes and dementia group livings. Practically all medical care for the participants was provided in these settings. The outcome of interest was time to diagnosis of dementia.
The original work-up of participants with diagnosed specific dementia subtypes varied but always included a detailed medical history, relevant neuropsychological testing, physical examination and laboratory tests to rule out other systemic or brain diseases that might account for the deficits. A computed tomography (CT) scan was available in 87% of the patients with Alzheimer’s disease and 88% of those with vascular dementia. CT findings of up to two brain infarctions that were clinically silent, in the setting of dementia of slow, gradual onset and progression, were considered compatible with a diagnosis of Alzheimer’s disease (n = 5). Otherwise, CT scans in cases of Alzheimer’s disease were normal or showed atrophy or mild to moderate white matter changes.
To standardise any differences between the original work-ups, the records of all possible cases of dementia and cognitive impairment were reviewed, and the definitive diagnoses in the present study were confirmed by two experienced geriatricians, independently of each other and blinded to the baseline data. In the event of disagreement, a third geriatrician reviewed the case and the diagnosis was determined by majority decision. Our primary endpoint, Alzheimer’s disease, was diagnosed according to the criteria of the National Institute of Neurological and Communicative Diseases and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) [19] and the criteria from the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM IV) [20]. The secondary endpoint, vascular dementia, was diagnosed according to the criteria of the Alzheimer’s Disease Diagnostic and Treatment Centres (ADDTC) [21]. Further, we identified other types of dementia: mixed dementia, dementia with Parkinson’s disease, frontotemporal dementia, and dementia without sufficient information for a specific diagnosis. Cognitive impairment was defined as evidence of significant and persistent cognitive decline, but not severe enough to interfere with social functioning and activities of daily living. All these cases, together with Alzheimer’s disease and vascular dementia, were included in the group ‘any dementia or cognitive impairment’.
Dates of death were obtained from the continuously updated Cause of Death Register held by the Swedish National Board of Health and Welfare.
Statistical analysis
Logarithmic transformation was performed to achieve a normal distribution of the non-normally distributed variables (EIR, insulin, intact and 32–33 split proinsulin, glucose disposal and plasma glucose). The prognostic value for a change of 1 SD in a continuous variable regarding dementia incidence was investigated using Cox proportional hazards. Proportionality of hazards was confirmed by visual examination of Nelson–Aalen curves. Non-linear relations were excluded by examining incidence rates in quartiles of the independent variables. Observations were censored at death; date of migration from the county of Uppsala, as medical records would not be available after migration; date of dementia diagnosis; or at the censor date of 31 December 2005. The statistical software package STATA 8.2 (Stata Corporation, College Station, TX, USA) was used in all analyses.
The a priori level of significance was 0.05. All analyses were performed unadjusted and adjusted for age and the following cardiovascular risk factors as potential confounders for development of dementia: systolic blood pressure, serum cholesterol, BMI, smoking status, educational level and diabetes mellitus. Since insulin sensitivity is a modulator of the insulin response [21], we also adjusted EIR for insulin-mediated glucose uptake. The associations were stratified for APOE ε4 status in secondary analyses. Analyses were repeated in a subsample of men free from diabetes at baseline (n = 1,061) to avoid confounding of the observed associations by baseline diabetes or diabetes treatment.
Results
A total of 257 persons developed dementia or cognitive impairment, among whom 81 developed Alzheimer’s disease and 26 vascular dementias during a median follow-up period of 12 years. A total of 433 persons died and 23 persons migrated from the county of Uppsala. The baseline characteristics among the 71-year-old men are presented in Table 1. The incidence rate of Alzheimer’s disease was 7.1/1,000, of vascular dementia 2.5/1,000, and of any dementia and cognitive impairment 22.9/1,000 person-years at risk.
Prediction of Alzheimer’s disease
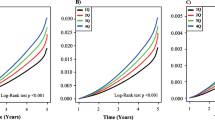
Low EIR was associated with a higher risk of Alzheimer’s disease in Cox proportional hazard analyses. Adjustment for age, diabetes, systolic blood pressure, serum cholesterol, BMI, smoking status and educational level fortified this association (Table 2). We examined the incidence rate of Alzheimer’s disease in quartiles of EIR and no obvious deviation from linearity was observed. The lowest risk was found in the highest quartile of EIR (Fig. 1). The cumulative incidence of Alzheimer’s disease in two groups with EIR, below and above the median, is presented in Fig. 2. High EIR was still significantly associated with lower risk of Alzheimer’s disease after adjustment for insulin sensitivity (HR for 1 SD decrease 1.25, 95% CI 1.01, 1.56). The crude association between impaired insulin secretion and Alzheimer’s disease was stronger in a subsample free from diabetes than in the whole cohort (HR 1.38, 95% CI 1.07, 1.78, unadjusted). Insulin sensitivity was not associated with the development of Alzheimer’s disease.

Incidence rates of Alzheimer’s disease by quartiles (1, lowest; 4, highest) of early insulin response in the OGTT at age 71 years. Vertical lines indicate 95% CIs

Nelson–Aalen plot of the cumulative incidence of Alzheimer’s disease during the median follow-up of 12 years in two groups of early insulin response (above and below the median) in the OGTT at age 71 years. Black line, early insulin response < median; grey line, early insulin response > median
A subset of patients were genetically tested for APOE ε4 status (n = 860). The presence of the APOE ε4 allele was found to increase the risk of developing Alzheimer’s disease (HR 2.41, 95% CI 1.48, 3.94). We found a strong association between EIR and the development of Alzheimer’s disease in APOE ε4-negative participants (HR 1.63, 95% CI 1.08, 2.55, p = 0.02) in the subsample free from diabetes. The association was weaker and no longer significant in APOE ε4-positive participants (HR 1.18, 95% CI 0.79, 1.76).
Prediction of vascular dementia
Low insulin sensitivity was associated with a higher risk of vascular dementia (Table 2). This association did not remain significant in the multivariable adjusted model. Neither low EIR nor APOE ε4 genotype was associated with a risk of vascular dementia.
Prediction of any dementia or cognitive impairment
Low EIR, high fasting plasma glucose, 2 h plasma glucose in the OGTT and diabetes mellitus were all significantly associated with a higher risk of overall dementia and cognitive impairment in the unadjusted model. Diabetes was a strong risk factor contributing to an elevated risk of approximately 60% in both the unadjusted and the multivariable adjusted model.
The possible associations between diabetes and specific dementia subtypes were not significant and are not reported because of the small number of diabetic men in these groups (n = 6 in the Alzheimer’s disease group and n = 4 in the vascular dementia group). Neither fasting specific insulin nor insulin propeptides were associated with any of the specified outcomes.
Discussion
In this community-based cohort of elderly men, a low early insulin response, but not low insulin sensitivity, was associated with an increased risk of Alzheimer’s disease during a median follow-up of 12 years. Low insulin sensitivity was associated with a higher risk of vascular dementia, but not after multiple adjustments.
The strengths of this study include the large population-based cohort, the long follow-up, the detailed classification into dementia subtypes and the detailed baseline characterisation of glucose metabolism, insulin secretion and insulin action. To our knowledge, the ULSAM cohort is the largest population that has been examined with the euglycaemic insulin method, the criterion standard for measurement of insulin resistance. The associations between impaired EIR and dementia were stronger in men without diabetes, eliminating the possibility that the diabetic state or its treatment at baseline influenced our results. We stated a priori that a complete description of our material regarding glucose and insulin metabolism was important. However, multiple independent analyses increase the risk of false positive results. It is difficult to estimate the risk of a type I error in the present study.
Since our primary interest was to study disease-specific mechanisms, strict criteria for dementia subtypes were important. A detailed clinical assessment and neuroimaging were available for the majority of the cases, making a specific dementia subclassification possible. Misclassification is an issue in any clinical study of dementia, but in our study it is likely to have been random with respect to baseline measurements. Even though we conducted reviews of medical records in all settings available and probably captured most of the cases, there is a risk, as in any epidemiological study, that some participants did not attend medical care, leading to underestimation of the true risk. Our homogeneous sample limits the possibility of generalising the results to women or other age and ethnic groups. Our findings need to be replicated in other cohorts.
In the present study, EIR was measured in an OGTT at 71 years. The findings of an association between impaired EIR and Alzheimer’s disease in the present study are in line with our previous report, in which the impaired acute insulin response measured at an intravenous glucose tolerance test at age 50 was associated with Alzheimer’s disease later in life [9]. Thus, this risk marker is stable at both middle and higher ages and can be detected in an OGTT, although this method is less reproducible than an intravenous glucose tolerance test. Furthermore, the association between low EIR and Alzheimer’s disease was independent of the degree of insulin sensitivity, which is a modulator of the insulin response [22]. Together, our previous and present studies emphasise the importance of insulin secretion in the development of Alzheimer’s disease.
The long-term association between dementia and insulin sensitivity was studied with the euglycaemic insulin clamp method. In previous studies assessing dementia risk, a cluster of clinical, metabolic and vascular risk factors has been used as a surrogate measurement of insulin resistance [12, 13, 23]. Such variables have limited sensitivity compared with the euglycaemic insulin clamp [24]. Hyperinsulinaemia (highest quartile) was associated with a higher risk of Alzheimer’s disease in a 5-year follow-up study [4], whereas both extremes of insulin distribution (lowest and highest sextile) increased the risk of dementia in a study with midlife assessment [3]. However, single fasting insulin estimates and cut-offs may not be sufficiently sensitive to identify insulin resistance [25].
In recent cross-sectional studies, the metabolic syndrome was more prevalent in patients with Alzheimer’s disease compared with controls [13, 23]. This may illustrate, however, that inactivity and an inadequate diet are consequences, rather than causes, of cognitive impairment and dementia. We did not find that insulin resistance and impaired glucose tolerance were associated with subsequent development of Alzheimer’s disease, which is in accordance with results from another large longitudinal cohort study with long follow-up [27].
The association between low EIR and Alzheimer’s disease was stronger in men without the APOE ε4 allele than in those with this allele, confirming our previous results from this population [9]. An impaired insulin response seems to be an important risk factor for Alzheimer’s disease in the absence of the high-risk allele. This observation is in line with an experimental study in which intravenous insulin administration facilitated cognitive functions only in patients without the APOE ε4 allele [27]. In contrast, another population study suggests synergistic effects of the presence of the APOE ε4 allele with type 2 diabetes on the risk of Alzheimer’s disease [28]. The question of how genetic predisposition modulates the association between insulin metabolism and dementia warrants further investigation.
Our results support the current information from neurobiological and neuropathological studies suggesting that insulin has important functions in the brain and may be involved in the pathogenesis of Alzheimer’s disease [6]. Although acute intravenous insulin facilitated memory functions in an experimental study, chronic hyperinsulinaemia and insulin resistance had negative effects [27]. Insulin may act directly on neurones and modulate energy metabolism in the hippocampus and the cortex [29]. Another possible mechanism could involve insulin-regulated β-amyloid secretion and extracellular degradation by competition for insulin-degrading enzyme [30]. Further, it has been hypothesised that insulin may regulate another key neuropathological feature in Alzheimer’s disease, tau hyperphosphorylation, through inhibition of glycogen synthase kinase 3 [31]. Impaired EIR could reflect an intracellular defect in a signal pathway common for pancreatic beta cells and brain neurones. Low EIR may lead to defective insulin signalling in the brain and affect neuronal growth and repair mechanisms. Further experimental studies are needed to explore these mechanisms and focus on the effects of insulin on neuronal cells. The lack of association between insulin sensitivity and Alzheimer’s disease in our study suggests that the mechanisms may lie among the other metabolic traits associated with the insulin-resistant state of type 2 diabetes.
Proinsulin is related to diabetes mellitus independently of EIR and insulin sensitivity [8]. However, we did not observe any association between proinsulin and dementia in this study. This suggests that conversion of proinsulin to insulin in the pancreatic beta cells does not reflect any processes associated with neuronal degeneration.
Decreased insulin sensitivity was associated with vascular dementia in the unadjusted analysis but not in multivariable analyses including traditional cardiovascular risk factors. However, as BMI and hypertension are strongly related to insulin resistance, it is difficult to judge the role of low insulin sensitivity in the causal pathway of vascular dementia with certainty. It is possible that in persons with decreased insulin sensitivity and factors closely related to this condition, vascular changes may lead to a clinical state of pure vascular dementia, as it is well established that insulin resistance is strongly associated with atherosclerotic alterations [32]. On the other hand, in patients with impaired early insulin secretion, amyloid-related mechanisms may predominate and lead to a dementia that we clinically classify as pure Alzheimer’s disease.
Our findings are important, as insulin secretion and action are the two key components interacting in impaired glucose tolerance and the development of type 2 diabetes mellitus [33]. A low early insulin response was associated with an increased risk of subsequent Alzheimer’s disease, whereas low insulin sensitivity was not. Vascular dementia was not related to a low early insulin response. We suggest that glucometabolic disturbances are differentially linked to the pathogenesis of these two main subtypes of dementia.
Abbreviations
- CT:
-
Computed tomography
- EIR:
-
Early insulin response
- MMSE:
-
Mini-Mental State Examination
- ULSAM:
-
Uppsala Longitudinal Study of Adult Men
References
Hebert LE, Beckett LA, Scherr PA, Evans DA (2001) Incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer Dis Assoc Disord 15:169–173
Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P (2006) Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 5:64–74
Peila R, Rodriguez BL, White LR, Launer LJ (2004) Fasting insulin and incident dementia in an elderly population of Japanese-American men. Neurology 63:228–233
Luchsinger JA, Tang MX, Shea S, Mayeux R (2004) Hyperinsulinemia and risk of Alzheimer’s disease. Neurology 63:1187–1192
Sonnen J, Larson E, Brickell K et al (2009) Different patterns of cerebral injury in dementia with or without diabetes. Arch Neurol 66
Neumann KF, Rojo L, Navarrete LP, Farías G, Reyes P, Maccioni RB (2008) Insulin resistance and Alzheimer’s disease: molecular links and clinical implications. Curr Alzheimer Res 2008(5):438–447
De Felice FG, Vieira MN, Bomfim TR et al (2009) From the cover: protection of synapses against Alzheimer’s-linked toxins: Insulin signalling prevents the pathogenic binding of A(beta) oligomers. Proc Natl Acad Sci U S A 10:1971–1976
Zethelius B, Hales N, Lithell HO, Berne C (2004) Insulin resistance, impaired early insulin response, and insulin polypeptides as predictors of the development of type 2 diabetes. Diabetes Care 27:1433–1438
Rönnemaa E, Zethelius B, Sundelöf J et al (2008) Impaired insulin secretion increases the risk of Alzheimer’s disease. Neurology 71:1065–1071
Burns JM, Donnelly JE, Anderson HS et al (2007) Peripheral insulin and brain structure in early Alzheimer disease. Neurology 69:1094–1104
Li L, Hölscher C (2007) Common pathological processes in Alzheimer disease and type 2 diabetes. Brain Res Rev 56:384–402
Kalmijn S, Foley D, White L et al (2000) Metabolic cardiovascular syndrome and risk of dementia in Japanese-American elderly men. The Honolulu–Asia Aging Study. Arterioscler Thromb Vasc Biol 20:2255–2260
Vanhanen M, Koivisto K, Moilanen L et al (2006) Association of metabolic syndrome with Alzheimer disease: a population-based study. Neurology 67:843–847
Sobey WJ, Beer SF, Carrington CA et al (1989) Sensitive and specific two-site immunoradiometric assays for human insulin, proinsulin, 65–66split and 32–33 split proinsulins. Biochem J 260:535–541
DeFronzo RA, Tobin JD, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237:E214–E223
Byberg L, Siegbahn A, Berglund L et al (1998) Plasminogen activator inhibitor-1 activity is independently related to both insulin sensitivity and serum triglycerides in 70-year–old men. Arerioscler Thromb Vasc Biol 18:258–264
Reitan RM (1958) Validity of the Trail Making test as an indicator of organic brain damage. Percept Mot Skill 8:271–276
Euser SM, Schram MT, Hofman A, Westendorp RG, Breteler MM (2008) Measuring cognitive function with age: the influence of selection by health and survival. Epidemiology 19:448–450
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlam M (1984) Clinical diagnosis of Alzheimer‘s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology 34:939–944
American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders: DSM-IV. American Psychiatric Association, Washington, DC
Chui HC, Victoroff JI, Margolin D, Jagust W, Shankle R, Katzman R (1992) Criteria for the diagnosis of ischemic vascular dementia proposed by the State of California Alzheimer’s Disease Diagnostic and Treatment Centers. Neurology 42:473–480
Kahn SE, Prigeon RL, McCulloch DK (1993) Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 42:1663–1672
Razay G, Vreugdenhil A, Wilcock G (2007) The metabolic syndrome and Alzheimer disease. Arch Neurol 64:93–96
Reaven GM (2004) Insulin resistance, cardiovascular disease, and the metabolic syndrome: how well do the emperor‘s clothes fit? Diabetes Care 27:1011–1012
Tuan CY, Abbasi F, Lamendola C, McLaughin T, Reaven G (2003) Usefulness of plasma glucose and insulin concentrations in identifying patients with insulin resistance. Am J Cardiol 92:606–610
Akomolafe A, Beiser A, Meigs JB et al (2006) Diabetes mellitus and risk of developing Alzheimer disease: results from the Framingham Study. Arch Neurol 63:1551–1555
Craft S, Newcomer J, Kanne S (1996) Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol Aging 17:123–130
Peila R, Rodriguez BL, Launer LJ (2002) Type 2 diabetes, ApoE gene, and the risk for dementia and related pathologies: the Honolulu–Asia Aging Study. Diabetes 51:1256–1262
Schulingkamp RJ, Pagano TC, Hung D, Raffa RB (2000) Insulin receptors and insulin action in the brain: review and clinical implications. Neurosci Biobehav Rev 24:855–872
Farris W, Mansourian S, Chang Y et al (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 100:4162–4167
Shubert M, Brazil DP, Burks DJ et al (2003) Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci 23:7084–7092
Laakso M, Sarlund H, Salonen R et al (1991) Asymptomatic atherosclerosis and insulin resistance. Arterioscler Thromb 11:1068–1076
Weyer C, Tataranni PA, Bogardus C, Pratley RE (2001) Insulin resistance and insulin secretory dysfunction are independent predictors of worsening of glucose tolerance during each stage of type 2 diabetes development. Diabetes Care 24:89–94
Acknowledgements
The study was supported by unrestricted grants from Uppsala University Hospital, Hjärnfonden, the Bertil Hållsten Reseach Foundation, Alzheimerfonden, The Swedish Research Council (2003-5546), DIADEM (QLK3-CT-2001-02362) APOPIS (contract no. LSHM-CT-2003-503330), Gamla Tjänarinnor, Emma Petterson, Ernfors Family Fund, and the Thuring and Sehlander Foundations.
Duality of interest statement
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rönnemaa, E., Zethelius, B., Sundelöf, J. et al. Glucose metabolism and the risk of Alzheimer’s disease and dementia: a population-based 12 year follow-up study in 71-year-old men. Diabetologia 52, 1504–1510 (2009). https://doi.org/10.1007/s00125-009-1393-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1393-9