
Abstract
Aims/hypothesis
Recently, variants in the transcription factor 7-like 2 (TCF7L2) gene have been found to be consistently associated with type 2 diabetes in different populations. In this study, we hypothesized that TCF7L2 also contributed to genetic susceptibility for type 2 diabetes in a Chinese population.
Methods
We looked for new variants by direct sequencing of all exons and intron–exon junctions of TCF7L2 in 100 Chinese type 2 diabetic patients, and then we genotyped five single nucleotide polymorphisms (SNPs) by Snapshot technology in 1,000 Chinese individuals.
Results
By sequencing, we identified six SNPs (c.1,637C>A; c.1,674C>G; c.1,709G>A; c.1,846C>G; c.1,888C>T; and c.1,876T>G), and three of them led to non-synonymous polymorphisms (c.1,637C>A, His→Gln or Pro→Thr; c.1,674C>G, Pro→Arg; and c.1,709G>A, Ala→Thr). All of them are rare except c.1,637C>A, which had a frequency of 0.23 for the minor A allele in 98 sequenced individuals. In a case–control study, one of the newly discovered SNPs (c.1,637C>A), together with four reported ones (rs7903146, rs12255372, rs290487 and rs3814573) were genotyped. Comparison between allele and genotype frequencies of these SNPs in patients and controls showed marginal association for rs7903146 and rs290487 with type 2 diabetes (p = 0.063, OR 1.982, 95% CI 1.128–3.485; p = 0.071, OR 1.237, 95% CI 0.983–1.557, respectively). No association was found for rs12255372, rs3814573, c.1,637C>A and type 2 diabetes (p = 0.278–1.000).
Conclusions/interpretation
With the current sample size, we did not find any mutation in the coding sequence of TCF7L2 that confers a genetic risk for type 2 diabetes in a Chinese population, and did not replicate some of the major positive results obtained in other populations.
Similar content being viewed by others

Introduction
Recently, there have been reports that several single nucleotide polymorphisms (SNPs) and one tetranucleotide repeat polymorphism within the transcription factor 7-like 2 (TCF7L2) gene are strongly associated with genetic susceptibility for type 2 diabetes in Icelandic, Danish and European-American populations [1], and the results have been well replicated in other independent studies [2–13], including studies in Asian populations [7, 12, 13]. In fact, the TCF7L2 gene is regarded, so far, as the most influential gene in determining the genetic susceptibility for type 2 diabetes in human beings.
However, the previously well-replicated risk allele T of rs7903146 (T) was low (1–2%) in Asian populations compared with European and Sub-Saharan African populations (>20%) according to the dbSNP build 128 database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp, accessed April 2008). Furthermore, one study in China also showed low non-zero allele frequency of micro-satellite DG10S478 (3.9%) [14], making it difficult to use these genetic markers to study the impact of TCF7L2 on genetic susceptibility for type 2 diabetes in a Chinese population.
In this study, we hypothesized that: (1) given the fact that TCF7L2 was shown to be playing an important role in genetic susceptibility for type 2 diabetes in many other ethnic groups, it is reasonable to assume that this gene also has a big impact on genetic predisposition to type 2 diabetes in Chinese; (2) the risk alleles of TCF7L2 described so far are all within the non-coding region of this gene, and they may be markers, rather than causal variants, that are in linkage disequilibrium (LD) with real causal variants residing in the coding region of the TCF7L2 gene; (3) because of the potentially different genetic backgrounds between Chinese and other ethnic groups, markers in LD with causal variations in the TCF7L2 gene, or the causal variations themselves, are different from those which have been described in other ethnic groups.
In this study, we took a two-step approach to test these hypotheses. First, we sequenced, in a small sample of 100 individuals randomly selected from a patient group, all exons and intron–exon junctions of the TCF7L2 gene to look for other genetic variants/mutations. Second, using well-replicated makers reported in previous studies and new markers/mutations discovered by DNA sequencing in this study, we carried out a case–control study to assess the association of the TCF7L2 gene with type 2 diabetes in a Chinese population.
Methods
Patients and controls
All participants were of Northern Han Chinese ancestry residing in Beijing metropolitan area. Five hundred type 2 diabetic patients with a positive family history of type 2 diabetes were recruited from the outpatient clinics of Endocrinology and Metabolism at Peking University People’s Hospital in Beijing, China (age 49.4 ± 12.3 years; 289 men, 211 women). Diabetes mellitus was diagnosed in accordance with 1999 WHO criteria [15]. All patients were unrelated and had been diagnosed with type 2 diabetes after 30 years of age. The patients with high glutamic acid decarboxylase antibody levels (>99th percentile of the normal population) were excluded from this study. Known subtypes of diabetes (e.g. maturity onset diabetes of the young) were excluded by clinical criteria or genetic testing [16]. Five hundred non-diabetic control individuals were recruited from either spouses of diabetic patients receiving diabetes care at Peking University People’s Hospital, or individuals attending an annual health examination at a community hospital, according to the following criteria: Han Chinese, ≥45 years old, no family history of diabetes, with a normal OGTT and HbA1c <6%. All controls were unrelated (age 59.1 ± 9.4; 194 men, 306 women). The clinical features of the participants are summarised in Table 1. Diabetic patients were asked to stop taking long-acting sulfonylureas, and NPH, ultralente and glargine insulins at least 12 h before their fast and 2 h insulin examinations. Informed consent was obtained from all participants. The study protocol was approved by the Ethics Committee of Peking University People’s Hospital.
DNA sequencing
The DNA sequence of 17 exons, and the exon–intron boundaries of the TCF7L2 gene were obtained from Genbank sequences AJ270770 to AJ270778. The 17 exons of TCF7L2, together with intron–exon junctions, were amplified by PCR using oligonucleotides as shown in the Electronic supplementary material (ESM) Table 1. Among them, primers of exon 1, 2, 3, 6, 8, 14, 16 and 17B (part of exon 17) were designed by Primer Premier software version 5.0 (PREMIER Biosoft International, Palo Alto, CA, USA), and the others were obtained from a published article [17]. PCR conditions are shown in ESM Table 2. All the PCR products were then directly sequenced using a 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA). Sequence variants were confirmed by re-amplifying and re-sequencing.
SNP selection and genotyping for association study
For SNPs identified in our sequencing study, only SNPs with minor allele frequency (MAF) >5% were selected for further genotyping in the association study. As a result, only c.1,637C>A met this criterion. Other SNPs (rs12255372, rs7903146, rs3814573 and rs290487) selected for genotyping in this study were those found to be carrying an at-risk allele in previous studies reporting an association of TCF7L2 with type 2 diabetes in other ethnic groups [2–13, 18]. They were included in this study for purposes of replication. A Snapshot Technology Platform (Applied Biosystems) was used for genotyping. In total, five SNPs were genotyped. The overall success rate of genotyping was ≥96%, and the concordance rate was 100%, based on 30 duplicate comparisons for each SNP. There was 100% agreement between direct sequencing and genotyping results of SNP c.1,637C>A. There was no significant difference in genotyping success rate between patient and control groups.
Statistical analysis
Data are given as means ± SD for variables with normal distribution, and otherwise as medians (interquartile range). Variables not normally distributed were logarithmically transformed before statistical analysis. χ 2 tests were used to determine whether individual polymorphisms were in Hardy–Weinberg equilibrium. The differences of allele and genotype frequencies between the diabetic and the control individuals were analysed using Pearson’s χ 2 test. Logistic regression analysis was used to calculate ORs, 95% CIs and corresponding p values, after adjusting for sex, age and BMI as covariates. Since the aim of this study was to replicate in a Chinese population the genetic markers tested in other studies, we did not correct the p value for multiple testing. Multiple linear regression analysis was used to analyse the genotype–phenotype relationship under an additive genetic model. All statistical tests were performed using SPSS program version 11.5 for Windows (SPSS, Chicago, IL, USA). A p value <0.05 was considered statistically significant (two-tailed). Homeostasis model assessment (HOMA) of insulin resistance (HOMA-IR) and beta cell function (HOMA-beta) were calculated as described previously [19].
Power calculation was performed by Quanto software version 1.2.3 (University of Southern California, Los Angeles, CA, USA). According to a type 2 diabetes prevalence of 5.5% in China [20], and using an additive genetic model, for any SNP with an MAF of at least 20%, the sample size in our study had an 80% power at a p value of 0.05 to detect an effect size of 1.35. If TCF7L2 had an effect in a Chinese population similar to that observed in Europe (OR 1.56) [1], a sample size between 1,412 and 326 would be sufficient for any SNP with an MAF between 5 and 40%.
Pairwise LD coefficients (D′) between SNPs were calculated by Haploview software version 4.0 (Daly Lab at the Broad Institute, Cambridge, MA, USA)
Results
DNA sequencing
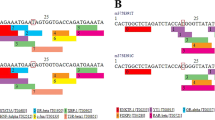
Successful sequencing was achieved for 98 samples. Six rare SNPs were identified (c.1,637C>A; c.1,674C>G; c.1,709G>A; c.1,846C>G; c.1,888C>T; and c.1,876T>G). All of them were in exon 17. No rs numbers have been assigned to these rare SNPs according to the dbSNP build 128 database. c.1,637C>A was a common polymorphism, with 23% of tested individuals carrying this polymorphism, whereas only one or two individuals carried minor alleles of the other five SNPs. Because of the existence of alternative splicing exons (exons 4, 13, 14, 15 and 16 totally expressed, exons 7, 9, 17 partially expressed), the transcripts of TCF7L2 can be divided into three kinds, STOP 1, STOP 2 and STOP 3, according to different COOH-terminal ends [17, 21]. STOP 1 refers to a short COOH-terminal end when exons 14 and 15 are transcribed together. STOP 2 contains a medium COOH-terminal end when both exons 14 and 15 are lacking. When only one of these two exons is transcribed, STOP 3 with a long COOH-terminal end is created. So, in STOP 1, all the six SNPs were in the 3′-untranslated region (UTR). In STOP 2, only c.1,637C>A led to non-synonymous mutation (His→Gln), and the others were in the 3′-UTR. In STOP 3, all the SNPs were in the coding region, and three of them led to amino acid changes (c.1,637C>A Pro→Thr; c.1,674C>G Pro→Arg; and c.1,709G>A Ala→Thr). There were 42 individuals carrying at least one mutation, and four individuals carrying two mutations. Those four individuals with two mutations had an early diagnosis of diabetes (<40 years old), and three of them were AA homozygotes of SNP c.1,637C>A. The other mutation carriers did not show special clinical features.
Association study
Table 2 summarises result of the case–control study. Hardy–Weinberg equilibrium was assessed in both the control and patient groups. The genotype frequencies of all SNPs were in accordance with Hardy–Weinberg equilibrium in the type 2 diabetes group as well as in the control group (p > 0.05). For LD, genotype data of both patient and control groups were used to estimate intermarker LD by measures of pairwise D′ and r 2. rs12255372 and rs7903146 were found to be in strong LD with each other (D′ = 0.816, r 2 = 0.116, Fig. 1, Table 3). The distribution of alleles and genotypes of all the SNPs were not significantly different between patient and control groups. However, after adjusting for sex, age and BMI, and using an additive genetic model, logistic regression analysis revealed that rs7903146 and rs290487 had a trend towards association with type 2 diabetes (rs7903146, p = 0.063, OR 1.982, 95% CI 1.128–3.485; rs290487, p = 0.071, OR 1.237, 95% CI 0.983–1.557).

Graphical representation of SNPs in Haploview LD graph of the TCF7L2 gene. Pairwise LD coefficients D′ × 100 are shown in each cell. The standard colour scheme of Haploview was applied for LD colour display; logarithm of odds (LOD) score ≥2 and D′ <1, shown in pink/red; LOD score <2 and D′ <1 shown in white
For the analysis of genotype–phenotype relationships, only non-diabetic participants were examined because treatment for diabetes may have distorted the relationship. We examined the potential influence of the four more prevalent SNPs (rs7903146, rs3814573, rs290487 and c.1,637C>A) on total cholesterol, total triacylglycerol, HDL- and LDL-cholesterol, fasting and postprandial plasma glucose, fasting and postprandial serum insulin, obesity index (as measured by BMI), insulin sensitivity (HOMA-IR) and insulin secretion (HOMA-beta). No association was observed for any of these metabolic traits with the four SNPs with or without adjustment for age and sex effects (data not shown).
Discussion
The discovery of variants of the TCF7L2 gene contributing strongly to genetic predisposition to type 2 diabetes is a breakthrough in the genetic study of type 2 diabetes. In the history of studying the genetics of type 2 diabetes, there has not been a single gene like TCF7L2 for which the original positive finding was widely replicated in different populations, suggesting a bigger impact of this gene on the pathogenesis of type 2 diabetes. Therefore, it is reasonable to postulate that TCF7L2 may also play an important role in the genetic susceptibility for type 2 diabetes in a Chinese population. However, since all the variants found to be associated with type 2 diabetes in the TCF7L2 gene were all in non-coding sequence, it is still not clear whether those variants are causal or in LD with real causal variants residing in the coding region of the TCF7L2 gene. Given the fact that risk alleles of SNPs in the TCF7L2 gene found in non-Asian populations are all rare in some Asian populations studied [18, 22], there is also a possibility that other frequent risk-carrying SNPs are unique to Asian or Chinese populations, because of the different evolutionary forces exerted on Asian or Chinese populations [23].
To test the hypothesis that the causal variations are located in the exons of TCF7L2, we sequenced all the exons of the gene. The six variants identified in 100 randomly selected patients were located in either the coding region or the 3′-UTR, depending on how the exons are expressed. All variants, except SNP c.1,637C>A in exon 17, are very rare. SNP c.1,637C>A was originally described by Groves et al. [24] in genetically unexplained neonatal diabetes mellitus and a study of TCF7L2 in maturity onset diabetes of the young population [17]. In these studies, no association was found between SNP c.1,637C>A and any specific clinical features of the individuals studied. In our case–control study, the frequency of the minor allele (allele A) of SNP c.1,637C>A was 0.25 in the patient group, and 0.27 in the control group. The difference between the two groups was not statistically significant (p = 0.278).
To test the hypothesis that SNPs in TCF7L2 associated with type 2 diabetes in other studies were also associated with type 2 diabetes in a Chinese population, we tested four SNPs in patients and controls. We have observed a trend towards association between two SNPs (rs7903146 and rs290487) and type 2 diabetes. Assuming the same effect size of rs7903146 and rs290487 in a Chinese population as was observed in Europe [1] and Taiwan [18], based on the observed MAF of these two markers in this study and the type 2 diabetes prevalence of 5.5% in China [20], power calculation using an additive genetic model showed that case samples of 1,508 and 622 would be necessary to achieve 80% power for rs7903146 and rs290487, respectively, indicating that the sample size in this study was underpowered in evaluating rs7903146 and rs290487. No associations were found between the other two SNPs (rs3814573 and rs12255372) and type 2 diabetes.
SNPs rs7903146 and rs12255372, which showed the strongest association with type 2 diabetes in the original study by Grant et al. [1], were well replicated by subsequent studies [2–13]. In those studies, the frequency of risk alleles (rs7903146 and rs12255372) were found to be more prevalent in non-Asian populations (MAF = 0.250–0.292 and 0.217–0.267, respectively), suggesting that the genetic background of Asians is different from that of other ethnic groups such as European, American, African and others, perhaps because of a positively selective sweep in this gene region [23]. In our study, these alleles were very rare in the control group (MAF = 0.0305 and 0.009, respectively), consistent with the Hap-Map (http://www.hapmap.org/, accessed April 2008) Chinese Han Population (MAF = 0.022, and almost 0, respectively), the Hong Kong Chinese population (rs7903146 MAF = 0.024), the Taiwan Chinese population (MAF = 0.0287, and 0.0040, respectively), and Japanese populations (MAF = 0.03–0.042 and 0.02–0.022, respectively). In studies carried out in the Asian population, the association of rs7903146 with type 2 diabetes was replicated in the Japanese population [12, 13], but not in the Hong Kong Chinese population [22] and Taiwan Chinese population [18]. The association of rs12255372 with type 2 diabetes was not replicated in the Taiwan Chinese population [18], and was contradictory in the Japanese population [12, 13]. In the study of the Hong Kong population [22], rs12255372 was not tested.
SNP rs3814573 and rs290487 were chosen for testing in this study on the basis of two additional published reports [4, 18]. In the study of Lehman et al. [4], the T allele of rs3814573 located in the intron 4 of TCF7L2 displayed the strongest association with increased age of onset of diabetes and was located in a haplotype that may carry a protective variant of TCF7L2. However, in our study, no protective effect was observed for this SNP. In the study of Chang et al. [18], the SNP rs290487 located in the intron 7 of TCF7L2 was the only SNP, among 20 SNPs studied, that was found to be associated with type 2 diabetes in the Chinese population in Taiwan (allele-specific p = 0.0021; OR 1.51 for the CC genotype [1.10–2.07], p = 0.0085). In our study, the frequency of minor allele C in the patient group was 0.39, almost identical to that in the Taiwan population. We have observed a trend towards association between rs290487 and type 2 diabetes (OR 1.237 [0.983–1.557], p = 0.071]. It should be noted that the sample size in Chang’s study (760 patients and 760 controls) was larger than ours.
In the study of the Hong Kong Chinese population [22], rs11196218, which located in a separate LD block in the intron 4 of TCF7L2, was found to be associated with type 2 diabetes. We did not test this SNP in the current study, but it is a marker worth testing in our future studies.
In summary, we did not find any mutation in the coding region of TCF7L2 that confers the genetic risk of type 2 diabetes in a Chinese population. In addition, with the current sample size, we did not replicate some of the major positive results obtained in other studies. In the future, a systematic study of other SNPs in or adjacent to TCF7L2 in a larger case–control study or meta-analysis of data from studies of TCF7L2 in the Chinese population are warranted to evaluate thoroughly the role of this important gene in the genetic predisposition to type 2 diabetes in the Chinese population.
Abbreviations
- HOMA-beta:
-
homeostasis model assessment of beta cell function
- HOMA-IR:
-
homeostasis model assessment of insulin resistance
- LD:
-
linkage disequilibrium
- MAF:
-
minor allele frequency
- SNP:
-
single nucleotide polymorphism
- UTR:
-
untranslated region
References
Grant SF, Thorleifsson G, Reynisdottir I et al (2006) Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 38:320–323
Damcott CM, Pollin TI, Reinhart LJ et al (2006) Polymorphisms in the transcription factor 7-like 2 (TCF7L2) gene are associated with type 2 diabetes in the Amish: replication and evidence for a role in both insulin secretion and insulin resistance. Diabetes 55:2654–2659
Humphries SE, Gable D, Cooper JA et al (2006) Common variants in the TCF7L2 gene and predisposition to type 2 diabetes in UK European Whites, Indian Asians and Afro-Caribbean men and women. J Mol Med 84:1005–1014
Lehman DM, Hunt KJ, Leach RJ et al (2007) Haplotypes of transcription factor 7-like 2 (TCF7L2) gene and its upstream region are associated with type 2 diabetes and age of onset in Mexican Americans. Diabetes 56:389–393
Scott LJ, Bonnycastle LL, Willer CJ et al (2006) Association of transcription factor 7-like 2 (TCF7L2) variants with type 2 diabetes in a Finnish sample. Diabetes 55:2649–2653
Cauchi S, Meyre D, Dina C et al (2006) Transcription factor TCF7L2 genetic study in the French population: expression in human beta-cells and adipose tissue and strong association with type 2 diabetes. Diabetes 55:2903–2908
Chandak GR, Janipalli CS, Bhaskar S et al (2007) Common variants in the TCF7L2 gene are strongly associated with type 2 diabetes mellitus in the Indian population. Diabetologia 50:63–67
van Vliet-Ostaptchouk JV, Shiri-Sverdlov R, Zhernakova A et al (2007) Association of variants of transcription factor 7-like 2 (TCF7L2) with susceptibility to type 2 diabetes in the Dutch Breda cohort. Diabetologia 50:59–62
Saxena R, Gianniny L, Burtt NP et al (2006) Common single nucleotide polymorphisms in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in nondiabetic individuals. Diabetes 55:2890–2895
Sladek R, Rocheleau G, Rung J et al (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445:881–885
Cauchi S, Achhab YE, Choquet H et al (2007) TCF7L2 is reproducibly associated with type 2 diabetes in various ethnic groups: a global meta-analysis. J Mol Med 85:777–782
Horikoshi M, Hara K, Ito C et al (2007) A genetic variation of the TCF7L2 gene is associated with risk of type 2 diabetes in the Japanese population. Diabetologia 50:747–751
Hayashi T, Iwamoto Y, Kaku K et al (2007) Replication study for the association of TCF7L2 with susceptibility to type 2 diabetes in a Japanese population. Diabetologia 50:980–984
Li WY, Luo TH, Zhang HL et al (2007) A study on the association between DG10S478 polymorphism and type 2 diabetes mellitus risk in Chinese population. J Diagn Concepts Pract 6:111–114
Alberti KG, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med 15:539–553
Han XY, Ji LN (2005) Contribution of MODY2 gene to the pathogenesis of Chinese early onset familial type 2 diabetes. Beijing Da Xue Xue Bao 37:591–594
Cauchi S, Vaxillaire M, Choquet H et al (2007) No major contribution of TCF7L2 sequence variants to maturity onset of diabetes of the young (MODY) or neonatal diabetes mellitus in French white subjects. Diabetologia 50:214–216
Chang YC, Chang TJ, Jiang YD et al (2007) Association study of the genetic polymorphisms of the transcription factor 7-like 2 (TCF7L2) gene and type 2 diabetes in the Chinese population. Diabetes 56:2631–2637
Matthews RD, Hosker JP, Rudenski AS et al (1985) Homeostasis model assessment: insulin resistance and beta cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
Gu D, Reynolds K, Duan X et al (2003) Prevalence of diabetes and impaired fasting glucose in the Chinese adult population: International Collaborative Study of Cardiovascular Disease in Asia (InterASIA). Diabetologia 46:1190–1198
Duval A, Rolland S, Tubacher E, Bui H, Thomas G, Hamelin R (2000) The human T cell transcription factor-4 gene: structure, extensive characterization of alternative splicings, and mutational analysis in colorectal cancer cell lines. Cancer Res 60:3872–3879
Ng MCY, Tam CTH, Lam VKL et al (2007) Replication and identification of novel variants at TCF7L2 associated with type 2 diabetes in Hong Kong Chinese. J Clin Endocrinol Metab 92:3733–3737
Helgason A, Pálsson S, Thorleifsson G et al (2007) Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet 39:218–225
Groves CJ, Zeggini E, Minton J et al (2006) Association analysis of 6,736 U.K. subjects provides replication and confirms TCF7L2 as a type 2 diabetes susceptibility gene with a substantial effect on individual risk. Diabetes 55:2640–2644
Acknowledgements
We thank all the patients and controls for agreeing to join this study. We thank SinoGenoMax (Beijing), Tiangen Biotech (Beijing) and the Beijing Genomic Institute for providing DNA sequencing services. We thank the Chinese National Human Genome Center (Shanghai) for providing genotyping services. This study was supported by the National Natural Science Foundation of China (30570873) and the National Basic Research Program of China (2006CB503900).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
Primers used to screen for mutations in the entire TCF7L2 coding sequence (PDF 24.6 KB)
ESM Table 2
PCR conditions for 17 fragments containing TCF7L2 gene exons (PDF 32.7 KB)
Rights and permissions
About this article
Cite this article
Ren, Q., Han, X.Y., Wang, F. et al. Exon sequencing and association analysis of polymorphisms in TCF7L2 with type 2 diabetes in a Chinese population. Diabetologia 51, 1146–1152 (2008). https://doi.org/10.1007/s00125-008-1039-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-008-1039-3