
Abstract
Aims/hypothesis
The neogenesis of islets from cultured human adult pancreatic tissue has been reported. The islet progenitors have been thought to be ductal cells. Since previous experiments have been ‘contaminated’ by a number of pre-existing islet cells, we examined their involvement in islet cell neogenesis.
Methods
Fresh human pancreatic cells with different purities of islet cells were grown in monolayer culture and labelled with bromodeoxyuridine. Transitional cells were analysed by double immunofluorescence staining. For purified ductal cell culture, pre-existing islets were eliminated on a magnetic cell separation system.
Results
We confirmed that less than 1% of the endocrine cells proliferated, mainly during the first 48 h of culture. However, a 10-fold larger proportion of the cells acquired a transitional phenotype by starting to coexpress the ductal marker cytokeratin 19 (CK19). These cells represented more than 10% of all endocrine cells after 1 day in culture, and 6% at 5 days of culture. Using magnetic cell sorting, we eliminated cells expressing neural cell adhesion molecule (N-CAM), after which we obtained 99.7% pure non-endocrine CK19-rich cell populations. These cell populations could be expanded in vitro. However, their endocrine differentiation capacity was severely reduced as compared with the original mixed cell cultures.
Conclusions/interpretation
These results suggest that islet neogenesis in this culture system at least partly represents the de-differentiation of islet cells into a duct-cell-like phenotype, with further re-differentiation in appropriate conditions. The plasticity of differentiated human pancreatic cell types may thus be an important mechanism of human pancreas regeneration.
Similar content being viewed by others
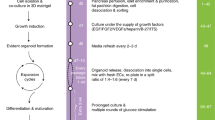


Introduction
Islet-cell transplantation in diabetic patients would greatly benefit from methods that increase the numbers of transplanted beta cells either in vitro prior to engraftment or through increased beta cell replication or neogenesis within the graft. New beta cells might be generated in two ways: by replication of differentiated islet cells; and by differentiation from stem/progenitor cells. Based on studies in mice, it has recently been proposed that replication of pre-existing beta cells is the predominant mechanism operating in adult animals [1]. However, the replication of human beta cells is extremely low, both during fetal development [2] and in adult islets [3]. Several studies suggest that neogenesis of islets could occur in the adult pancreas. However, the exact nature and identity of islet-cell precursors remains a puzzle. Experimental models in rodents have shown that beta cells can regenerate after tissue injury, apparently through the differentiation of precursor cells located within pancreatic ducts [4, 5]. Morphological observations also suggest the appearance of neogenic islet cells with ductal origin in the human pancreas [6]. There is direct evidence of islet cell differentiation from experimentally transplanted human ductal cells taken from young donors [7]. Furthermore, a recent analysis of the cellular composition of clinical islet grafts shows that long-term graft function correlates with the number of transplanted ductal cells [8]. In addition to this evidence of duct cells as the islet precursors, a number of studies suggest that a more primitive stem cell could reside within the pancreas or islets [9, 10].
We and others have shown the in vitro generation of endocrine cells from adult pancreatic ductal cell culture [11–13]. One problem of experiments performed with human pancreatic cells is the presence of pre-existing islet cells at the onset of culture. It has been proposed that bilateral transdifferentiation between human pancreatic exocrine and endocrine cells could occur in long-term culture [14]. Several other studies also demonstrate that differentiated islet cells retain the plasticity to de-differentiate when cultured either as a monolayer [15] or in three-dimensional structures [16]. Recently it has been shown that human adult islets can de-differentiate through epithelial-to-mesenchymal transition to generate proliferative vimentin-positive cells. By serum deprivation these precursor cells can re-differentiate into hormone-expressing islet-like clusters [17]. In the present study, using the previously reported culture model [13], we studied the replication and de-differentiation of pre-existing islet cells in monolayer culture, and applied magnetic cell sorting to estimate the contribution of these cells to in vitro islet neogenesis.
Subjects and methods
Tissue preparation and in vitro cultures
Human islets were isolated according to previously described methods [18, 19] in Uppsala, Sweden. After Ficoll gradient purification, the fractions with different purities of islet cells were collected and shipped on ice to Helsinki for subsequent cultures. Pancreatic tissues from 10 donors were used for these studies (mean age 53±14 years, range 30–67 years). All procedures were approved by institutional ethical committees in Sweden and Finland. The donors or their next of kin gave written informed consent to the use of their pancreas tissues.
As previously described, but with a minor modification [13], the fresh pancreatic cell clumps were first expanded into monolayers in six-well culture plates (Corning, Corning, NY, USA) in CMRL-1066 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal calf serum (PromoCell, Heidelberg, Germany). After 5–7 days of expansion, the media were changed for serum-free DMEM/F12 (National Public Health Institute, Helsinki, Finland) supplemented with ITS (5 mg/l insulin+5 mg/l transferrin+5 μg/l sodium selenite; Sigma, St. Louis, MO, USA), 2 g/l bovine serum albumin, 8 mmol/l glucose and 10 mmol/l nicotinamide (Sigma). The cells were then overlaid with Matrigel, a basement membrane preparation from Engelbreth–Holm–Swarm mouse tumour cells (BD Biosciences, Bedford, MA, USA), according to the manufacturer’s instructions with the exception of dilution (1:10) and overnight gelling at 37°C. Within 2–3 weeks after gel overlay, three-dimensional structures, cysts and cultivated human islet buds (CHIBs), which protruded from monolayers, were hand-picked for analysis. The remaining monolayer cells were collected using a solution of 0.05% trypsin and 0.02% EDTA (Invitrogen).
Bromodeoxyuridine labelling and immunocytochemical staining
To determine endocrine cell proliferation during the monolayer expansion period, the cultured cells were labelled with 10 μg/ml bromodeoxyuridine (BrdU; ZYMED, San Francisco, CA, USA) for 24 h at different time-points before fixation in 4% paraformaldehyde (PFA). The cells that were positive for endocrine marker chromogranin A and proliferation marker BrdU were expressed as a percentage of endocrine cells.
Cytospins were prepared from dissociated (0.05% trypsin and 0.02% EDTA) cell suspensions, which were spun to microscope slides (SuperFrostPlus, Menzel, Braunschweig, Germany) by centrifugation at 55 g for 8 min. Cytospin slides were fixed in 4% PFA for 15 min and rinsed in PBS before staining. Harvested cysts/CHIBs were fixed for 1 h in 4% PFA, rinsed with PBS, suspended in a 2% agarose–PBS solution and centrifuged at 220 g for 1 min to form compact pellets, which were then embedded in paraffin for sectioning.
Immunostaining was performed in the above preparations to identify various cell types using the following primary antibodies: guinea-pig anti-porcine insulin 1:100, rabbit anti-human glucagon 1:500, rabbit anti-human somatostatin 1:500, rabbit anti-human pancreatic polypeptide 1:500, rabbit anti-human chromogranin A (Chro A) 1:500, mouse anti-human cytokeratin 19 1:50, mouse anti-human vimentin 1:200, and mouse anti-BrdU 1:100 (all from DAKO, Carpinteria, CA, USA); mouse anti-synaptophysin 1:5 (Boehringer Mannheim, Mannheim, Germany); rabbit anti-neural cell adhesion molecule (N-CAM) 1:50 (kindly provided by V. Cirulli, The Whittier Institute for Diabetes, La Jolla, CA, USA). Non-specific binding was blocked by preincubation in 3% normal serum (ZYMED) from the species in which the secondary antibody was raised, followed by incubation of primary antibodies for 1 h at room temperature. Biotinylated goat anti-rabbit and biotinylated rabbit anti-mouse IgGs (ZYMED, 1:200) were used as secondary antibodies. Peroxidase-conjugated streptavidin (ZYMED, 1:200) was used by developing the substrate of 3-amino-9-ethylcarbazole. Light counterstaining was performed with haematoxylin. Microwave treatment in citrate buffer was necessary to retrieve the antigenicity of BrdU, whereas 0.1% pepsin–0.1 mol/l HCl was optimal for cytokeratin 19 (CK19) and hormone retrieving in paraffin sections. For double staining of BrdU and Chro A, the Vectastain ABC-kit (Vector, Burlingame, CA, USA) was used. Double immunofluorescent staining was performed to check for co-localisation of endocrine markers, N-CAM, and CK19, using conjugated secondary antibodies as follows: fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit, FITC-conjugated donkey anti-guinea-pig, tetrarhodamine isothiocyanate (TRITC)-conjugated donkey anti-mouse, and TRITC-conjugated donkey anti-guinea-pig IgGs (DAKO, 1:50).
Insulin and DNA measurement
For the analysis of insulin and DNA content, the cells in one well of six-well plates were collected and washed twice in PBS, resuspended in 300 μl distilled cold water, and homogenised by sonication on ice. Aliquots of the homogenates in duplicate were either analysed fluorometrically for their DNA content [20] or extracted with acid ethanol overnight and measured for insulin content using a solid-phase RIA kit (DPC, Los Angeles, CA, USA).
Cell separation
The initial cell clusters were dissociated with 0.16 mg/ml trypsin and 0.1 mmol/l EDTA [21] and filtered through a 30-μm nylon mesh (Miltenyi Biotec, Bergisch Gladbach, Germany) to remove residual cell clumps. The single cell suspension was incubated for 15 min at 6 °C with microbeads conjugated to a monoclonal antibody against N-CAM (Miltenyi Biotec) diluted 1:5. Cell separation was carried out on a MiniMACS magnetic cell separation system according to the manufacturer’s instructions (Miltenyi Biotec). The N-CAM-negative cells were purified by passage through two consecutive MS columns while N-CAM-positive cells were retained in the columns. The separated cells were plated on gelatin-coated 12-well plates and cultured in CMRL-1066 medium (Invitrogen) supplemented with 10% fetal calf serum (PromoCell). In some experiments, 15 ng/ml hepatocyte growth factor (HGF) was used to promote maximal expansion of the monolayers. Differentiation of the expanded cells was induced by Matrigel overlay and serum-free DMEM/F12 differentiation medium as before. In addition, several other protocols were applied to induce differentiation in these cells, including transfer of the cells after the expansion phase into high-density culture either in conical culture wells or on Matrigel in the serum-free medium.
Results
In the first part of this study, we used islet isolates with variable purity from five donors to study proliferation of endocrine cells and discover evidence of de-differentiation of pre-existing islet cells during monolayer culture.
Proliferation
The vast majority of the cells in the monolayer cultures maintained a round epithelial phenotype. When the cell aggregates first attached and spread out, the epithelial cell population was quite homogeneous (Fig. 1a). During the following days, some of these epithelial cells proliferated actively and became larger. All of the large proliferating epithelial cells were positive for the duct cell marker CK19 (Fig. 1b). Based on double immunocytochemistry for BrdU and Chro A, a low level of endocrine cell proliferation could be detected. This peaked at day 2, but the maximal labelling index was only 0.7% in a high-purity islet preparation (Fig. 2). Both proliferative and non-proliferative endocrine cells were small compared with the majority of ductal cells (Fig. 1c, Fig. 3b). Within the CK19-positive ductal population, more than 95% of BrdU labelling appeared in the large cells among a population of non-proliferative smaller ductal cells (Fig. 1b). Since only a few endocrine cells proliferated in the early monolayer cultures, we chose 24-h BrdU labelling to estimate their replication rate. However, it has to be recognised that during this long labelling period de-differentiation of endocrine cells may have occurred. Therefore, the proliferation rate of endocrine cells might have been slightly underestimated.

Immunocytochemical staining of cytocentrifuge preparations of monolayer cells during the early phase of culture. a Homogeneous cell populations at day 1 in culture; most of the epithelial cells are CK19-positive (red) ductal cells. b From day 3, cells become heterogeneous with more proliferation (BrdU-positive, blue) and larger ductal (CK19-positive, red) cells (arrows). c One proliferating (BrdU-positive, blue) endocrine (Chro A-positive, red) cell (arrow) at day 3. Original magnification ×20

Proliferation rates of pre-existing islet cells (percentage of BrdU-positive cells in total Chro A-positive cells) during monolayer culture of four islet preparations of variable purity. Open circles, Hu57 (76.5% beta cells); filled circles, Hu58 (24.7% beta cells); triangles, Hu59 (4.4% beta cells); and diamonds, Hu60 (27.6% beta cells)

Double-immunofluorescence staining of Chro A (green) and CK19 (red) in fresh tissues before culture (a), in early monolayers (b), and at the end of the differentiation phase (c). Transitional cells only appear in monolayers (b, arrows and insets), but not in fresh tissues (a) or in the differentiated cysts/CHIBs (c). Nuclei are counterstained by DAPI (blue). Original magnification ×20. d Quantitative analysis of the proportion of transitional cells during the first five days of culture. Data are the means±SE of five separate experiments with cells from different donors
De-differentiation
Transitional cells coexpressing Chro A and CK19 were detected in cultures derived from all five donor tissues. These transitional cells were relatively small with a weaker staining intensity than non-transitional endocrine or ductal cells. They were frequently identified during the expansion phase but never in the fresh fractions at the onset or in the differentiated CHIBs (Fig. 3a–c). As shown in Fig. 3d, more than 10% of all endocrine cells coexpressed Chro A and CK19 after the first 24 h of culture. During the following 4 days, this proportion gradually diminished after the initial peak. Further double immunostaining of insulin, glucagon, somatostatin and pancreatic polypeptide together with CK19 showed that transition occurred in all four endocrine cell types (Fig. 4).

Double-immunofluorescence staining of cytocentrifuge preparations of day 5 monolayers. Co-staining (arrows) of insulin (ins), glucagon (glu), somatostatin (soma), and pancreatic polypeptide (pp) (all green) with CK19 (red) suggests that de-differentiation occurs in all four types of endocrine cells. Nuclei are counterstained by DAPI. Original magnification ×40
Endocrine differentiation
Consistent with our previous results [13], increases were observed in the percentage of insulin-positive cells as well as of glucagon-positive cells and also in the insulin per DNA content (Fig. 5) after Matrigel overlay and serum-free culture. In this study, we made calculations on the absolute numbers of insulin- and glucagon-positive cells at the stage of final differentiation (day 28) as compared with a similar sized sample of cells at monolayer culture (day 5). The absolute cell number was calculated by multiplying the percentage of insulin- or glucagon-positive cells by the total DNA content and then dividing by 7.0 pg of a single human diploid nucleus [12]. Using this approach, we found that the absolute number of insulin-positive cells increased 10-fold and that of glucagon-positive cells increased 15-fold during the culture of low-purity islets from one pancreas (Hu 59, Fig. 5b). However, both beta and alpha cell numbers increased two- to four-fold, even in cultures of purer islets from three other pancreases (Fig. 5b). The endocrine cells in CHIBs were only partially differentiated when comparing the insulin content and insulin mRNA level with those in fresh human islets. The insulin/DNA content of CHIBs (8.6±1.7 pmol/μg) was 5% of that found in pure human islets (160±29 pmol/μg) [12], and the insulin mRNA expression level in CHIBs was 4–5% of that found in human islets [13].

Ratios of insulin per DNA content (a) and ratios of absolute numbers (b) and absolute numbers of insulin- (c) and glucagon-positive (d) cells at days 5 (d5) and 28 (d28), based on immunostaining, insulin and DNA quantification of aliquots of cells (one culture well in a six-well plate). Data are presented separately for four different islet preparations of variable purity (Hu57, 77%; Hu58, 25%; Hu59, 4%; and Hu60, 28% beta cells)
Cell sorting experiments
We next performed magnetic cell separation experiments with cells from five donors based on the cell surface expression of N-CAM to clarify whether the de-differentiated endocrine cells and neogenic islets in our culture model originated from the pre-existing ductal or islet cell compartments. It is known that within the rat pancreas N-CAM is expressed specifically in endocrine islet cells [21]. Using MACS, we could indeed effectively eliminate the endocrine cells from the N-CAM-negative fraction (remaining Chro A positivity 0.3±0.1%, Table 1). However, the N-CAM-positive fraction was not purely endocrine because in addition to the Chro A-positive cells it also contained a significant number of CK19-positive cells. Only a few mesenchymal (vimentin-positive) cells were present (Table 1). By double immunofluorescent staining of freshly isolated and dispersed human adult pancreatic cells, besides the N-CAM expression in endocrine cells (Fig. 6a,b), a minority of the CK19-positive cells were found to also express N-CAM (Fig. 6c). The total percentage of different types of cells in the sorted fractions was not 100% (Table 1). This is probably because 20–30% of dead cells (trypan blue staining) were not excluded. When the separated cells were put in the same culture conditions that were used for the unseparated cultures, they attached but only formed small patches of confluent monolayers. Various modifications of the culture conditions were tested. When gelatinised culture plates were used and the medium was supplemented with 15 ng/ml HGF, the purified non-endocrine cells could form large confluent monolayers (Fig. 6d). Immunostaining for CK19 showed that most of these cells were ductal (Fig. 6e). Matrigel coating and serum-free differentiation medium induced the formation of three-dimensional ductal cysts with small islet buds (Fig. 6f) from the N-CAM-negative cell population. However, as compared with cultures of unseparated cells the number of Chro A-positive cells was very small and there was no increase in the cellular insulin per DNA content after differentiation induction (1.6±0.2 pmol/μg before culture versus 1.0±0.2 pmol/μg after culture). The outcome was similar in differentiation experiments on the more patchy monolayers expanded in the absence of gelatin and HGF.

Double-immunofluorescence staining in fresh isolated human pancreatic cells for (a) N-CAM (green) and synaptophysin (red); (b) N-CAM (green) and insulin (red); and (c) N-CAM (green) and CK19 (red), showing that in addition to the endocrine cells, also a subpopulation of CK19-positive cells express N-CAM. (d) MACS-purified N-CAM-negative cells form confluent monolayers on gelatinised plates when cultured in the presence of HGF. Most of the cells are positive for CK19 (red, e). Three-dimensional cysts develop from these cells (f) during the differentiation phase in serum-free medium on Matrigel. Very few individual Chro A-positive cells are found within these cysts (red, f). Original magnifications ×40 (a–c) and × 20 (d–f)
Discussion
In this study, we again show that human islet-cell neogenesis can be induced during the culture of mixed human pancreatic cells originating from islet isolation fractions of low purity. We also show that a similar neogenic process is induced from very pure islet fractions. These experiments are characterised by an initial phase of proliferation, associated with a decrease in endocrine cell number, and a later differentiation phase, which results in a net increase of endocrine islet cells. In our previous study [13] we found that the neogenic islet cells were derived from CK19-positive ductal epithelium-like cells which proliferated during the initial days of culture. This time we focused on the role of the pre-existing differentiated islet cells, which are present at the onset of culture. Our results show that a significant proportion of islet cells start to express CK19 soon after plating, thus attaining a transitional phenotype. Although it is theoretically possible that these transitional cells could represent pre-existing ductal cells that start to express endocrine proteins, this is unlikely because these cells are encountered at an early stage of culture, characterised by decreasing numbers of endocrine cells, and a decrease in the cellular insulin content by 50% [13]. The transitional cells were only observed for a limited time, suggesting that they stopped expressing markers of fully differentiated islet cells (such as Chro A). Significant epithelial cell proliferation was only observed in CK19-positive cells, but not in Chro A-positive cells. Consequently, it is possible that the de-differentiated islet cells started to proliferate after losing their endocrine phenotype, and may have acted as precursors of newly differentiated islet cells during the differentiation phase. We tested this hypothesis by eliminating the pre-existing islet cells before starting the cultures.
The role of beta cell proliferation has been emphasised based on cell lineage experiments performed in mice [1]. However, many studies show that there are obvious differences between species in the capacity of beta cells to replicate. Proliferation of human beta cells in vivo appears to be extremely low at all ages [2, 6, 22], and this is true also for isolated islets even after transplantation to nude mice [3, 7, 23]. However, when placed in suitable culture conditions, human beta cells may transiently proliferate but this is invariably associated with loss of differentiated function, at least in monolayer cultures [15, 24]. In this study, we also observed a low-level transient stimulation of beta cell replication at the time when transitional cell types emerged, consistent with the association of proliferation and de-differentiation.
All experimental studies published so far on human islet cell neogenesis have been hampered by ‘contamination’ through pre-existing islet cells in the starting material [7, 12, 13, 25]. This is a difficult problem because it appears to be impossible to identify any fractions from dispersed human pancreatic cells processed for islet isolation that would be completely devoid of islet endocrine cells. We developed a method for the elimination of these endocrine cells based on their expression of N-CAM [21]. Magnetic microbeads coated with N-CAM antibody effectively bound to the islet cells, allowing us to generate 99.7% endocrine-cell-free human pancreatic cells. When plated in culture, most of these cells expressed CK19 and proliferated. However, the differentiation phase was dramatically altered. The formation of three-dimensional cystic structures with small islet buds, which we have regularly seen in 50 consecutive experiments, did not occur when the Matrigel overlay and serum-free culture medium were applied for the expanded N-CAM-negative cell population. This was not the result of poor viability of the cells, because their rate of proliferation was not clearly different from that of the unseparated cell population. However, because of the relatively low numbers of cells that could be processed with the MACS system, and the inevitable loss of cells during separation, we were not able to achieve equally large confluent monolayers as with the unseparated cell mass. Using a smaller scale, we applied various alternative protocols to induce differentiation. The only positive results were observed when the expanded cells were transferred in relatively high density to culture wells coated with Matrigel. During 2–3 weeks in serum-free differentiation medium, these cultures formed a number of cysts resembling the original ‘CHIBs’ (Fig. 6f). However, there were practically no dense cell buds growing out of these cysts, and although a few Chro A-positive cells could be detected by immunocytochemistry, there were very few insulin-positive cells and also the insulin content of the extracted cultures remained low.
The specific expression of N-CAM in adult pancreatic islets of rodents has been reported [21, 26], but in the human pancreas, N-CAM may be found also in ducts [27, 28]. We performed the MACS experiments to deplete the pre-existing islets. However, our results show that a cell population coexpressing N-CAM and CK19 was also depleted from the N-CAM-negative fraction. These cells were also detected by double immunocytochemistry in the uncultured fresh material. It is possible that the N-CAM/CK19 double-positive cells could represent a pool of islet cell progenitors or partially de-differentiated islet cells. This interesting pool of cells needs to be characterised in more detail in further studies.
The plasticity of differentiated pancreatic cells may be an important mechanism behind islet-cell expansion, as has been demonstrated by several studies in rodents. Transdifferentiation of acinar cells to ductal cells and further differentiation of these cells into islets has recently been demonstrated in vivo [29] and in vitro [30]. There are fewer observations in human pancreatic cells but clear evidence exists, particularly for the islet/ductal transitional cells both in vivo [6] and in vitro [14, 15, 31]. However, only a recent report [31] and our study have observed a relatively large number of transitional cells within all four types of endocrine cells and presented the quantitative results while only occasional co-staining of insulin and ductal markers is found by others. Our demonstration of endocrine–ductal transitional cells and the fact that depletion of endocrine cells from the starting material severely reduces the endocrine differentiation of the expanded CK19-positive cells promotes the hypothesis that this particular form of plasticity could be important for human islet expansion. This would imply that instead of direct replication, the human islet cells could de-differentiate into a duct-like phenotype that could then re-differentiate into islet cells after a phase of proliferation. The data presented here suggest that the endocrine precursors within the ductal cell compartment might actually belong to this pool of committed cells circulating between the endocrine and ductal phenotypes. More direct evidence for this might be obtained if the freshly isolated endocrine and ductal cells could be separately labelled prior to the in vitro culture experiments.
Recently, the process of de-differentiation and re-differentiation in cultured human islets cells has also been reported by Gershengorn et al. [17]. However, in contrast to the islet–ductal transition found in our study, they showed a reversible epithelial-to-mesenchymal transition. The transitional vimentin-positive cells were positive for proinsulin mRNA until passage 10, suggesting that de-differentiated beta cells were proliferative. The majority of proliferating vimentin-positive cells were also positive for nestin, which makes it possible that these cells were identical to the populations reported by Zulewski et al. [9]. However, the re-differentiated epithelial cells had a very low insulin expression level of less than 0.02% of that seen in human islets. In comparison, the CHIBs were much more differentiated, with 5% of the insulin level found in pure human islets. However, it is clear that the expansion of the de-differentiated epithelial cells in our study was very limited in comparison with the long-term proliferation reported for the mesenchymal cells [17].
Taken together, we can conclude that the islet cell neogenesis in our culture system at least partly represents the de-differentiation of non-proliferative islet cells into a proliferative duct-cell-like phenotype with further re-differentiation in appropriate conditions. Further studies are needed to establish whether all ductal cells, and also acinar cells, participate in the pool of endocrine precursors, or whether only a pool of cells committed for endocrine differentiation circulate between endocrine and ductal phenotypes. This concept is also compatible with the notion strongly suggested by mouse experiments [1] that self-duplication rather than stem cell differentiation is responsible for the expansion of the adult beta cell mass.
Abbreviations
- BrdU:
-
bromodeoxyuridine
- CHIB:
-
cultivated human islet bud
- CK19:
-
cytokeratin 19
- Chro A:
-
chromogranin A
- HGF:
-
hepatocyte growth factor
- MACS:
-
magnetic cell sorting
- N-CAM:
-
neural cell adhesion molecule
- PFA:
-
paraformaldehyde
- TRITC:
-
tetrarhodamine isothiocyanate
References
Dor Y, Brown J, Martinez OI, Melton DA (2004) Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429:41–46
Bouwens L, Lu WG, Dekrijger RR (1997) Proliferation and differentiation in the human fetal endocrine pancreas. Diabetologia 40:398–404
Tyrberg B, Eizirik DL, Hellerström C, Pipeleers DG, Andersson A (1996) Human pancreatic beta-cell deoxyribonucleic acid-synthesis in islet grafts decreases with increasing organ donor age but increases in response to glucose stimulation in vitro. Endocrinology 137:5694–5699
Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE (1993) A second pathway for regeneration of adult exocrine and endocrine pancreas: a possible recapitulation of embryonic development. Diabetes 42:1715–1720
Bouwens L (1998) Transdifferentiation versus stem cell hypothesis for the regeneration of islet beta-cells in the pancreas. Microsc Res Tech 43:332–336
Bouwens L, Pipeleers DG (1998) Extra-insular beta cells associated with ductules are frequent in adult human pancreas. Diabetologia 41:629–633
Bogdani M, Lefebvre V, Buelens N et al (2003) Formation of insulin-positive cells in implants of human pancreatic duct cell preparations from young donors. Diabetologia 46:830–838
Street CN, Lakey JR, Shapiro AM et al (2004) Islet graft assessment in the Edmonton Protocol: implications for predicting long-term clinical outcome. Diabetes 53:3107–3114
Zulewski H, Abraham EJ, Gerlach MJ et al (2001) Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 50:521–533
Suzuki A, Nakauchi H, Taniguchi H (2004) Prospective isolation of multipotent pancreatic progenitors using flow-cytometric cell sorting. Diabetes 53:2143–2152
Ramiya VK, Maraist M, Arfors KE, Schatz DA, Peck AB, Cornelius JG (2000) Reversal of insulin-dependent diabetes using islets generated in vitro from pancreatic stem cells. Nat Med 6:278–282
Bonner-Weir S, Taneja M, Weir GC et al (2000) In vitro cultivation of human islets from expanded ductal tissue. Proc Natl Acad Sci U S A 97:7999–8004
Gao R, Ustinov J, Pulkkinen MA, Lundin K, Korsgren O, Otonkoski T (2003) Characterization of endocrine progenitor cells and critical factors for their differentiation in human adult pancreatic cell culture. Diabetes 52:2007–2015
Schmied BM, Ulrich A, Matsuzaki H et al (2001) Transdifferentiation of human islet cells in a long-term culture. Pancreas 23:157–171
Beattie GM, Itkin-Ansari P, Cirulli V et al (1999) Sustained proliferation of pdx-1(+) cells derived from human islets. Diabetes 48:1013–1019
Yuan S, Rosenberg L, Paraskevas S, Agapitos D, Duguid WP (1996) Transdifferentiation of human islets to pancreatic ductal cells in collagen matrix culture. Differentiation 61:67–75
Gershengorn MC, Hardikar AA, Wei C, Geras-Raaka E, Marcus-Samuels B, Raaka BM (2004) Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells. Science 306:2261–2264
Ricordi C, Lacy PE, Finke EH, Olack BJ, Scharp DW (1988) Automated method for isolation of human pancreatic islets. Diabetes 37:413–420
Ozmen L, Ekdahl KN, Elgue G, Larsson R, Korsgren O, Nilsson B (2002) Inhibition of thrombin abrogates the instant blood-mediated inflammatory reaction triggered by isolated human islets: possible application of the thrombin inhibitor melagatran in clinical islet transplantation. Diabetes 51:1779–1784
Hinegardner RT (1971) An improved fluorometric assay for DNA. Anal Biochem 39:197–201
Cirulli V, Baetens D, Rutishauser U, Halban PA, Orci L, Rouiller DG (1994) Expression of neural cell adhesion molecule (N-CAM) in rat islets and its role in islet cell type segregation. J Cell Sci 107:1429–1436
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
Tyrberg B, Ustinov J, Otonkoski T, Andersson A (2001) Stimulated endocrine cell proliferation and differentiation in transplanted human pancreatic islets: effects of the ob gene and compensatory growth of the implantation organ. Diabetes 50:301–307
Beattie GM, Rubin JS, Mally MI, Otonkoski T, Hayek A (1996) Regulation of proliferation and differentiation of human fetal pancreatic islet cells by extracellular matrix, hepatocyte growth factor and cell–cell contact. Diabetes 45:1223–1228
Lechner A, Nolan AL, Blacken RA, Habener JF (2005) Redifferentiation of insulin-secreting cells after in vitro expansion of adult human pancreatic islet tissue. Biochem Biophys Res Commun 327:581–588
Langley OK, Aletsee-Ufrecht MC, Grant NJ, Gratzl M (1989) Expression of the neural cell adhesion molecule NCAM in endocrine cells. J Histochem Cytochem 37:781–791
Eidelman S, Damsky CH, Wheelock MJ, Damjanov I (1989) Expression of the cell–cell adhesion glycoprotein cell-CAM 120/80 in normal human tissues and tumors. Am J Pathol 135:101–110
Fujisawa M, Notohara K, Tsukayama C, Mizuno R, Okada S (2003) CD56-positive cells with or without synaptophysin expression are recognized in the pancreatic duct epithelium: a study with adult and fetal tissues and specimens from chronic pancreatitis. Acta Med Okayama 57:279–284
Rooman I, Lardon J, Bouwens L (2002) Gastrin stimulates beta-cell neogenesis and increases islet mass from transdifferentiated but not from normal exocrine pancreas tissue. Diabetes 51:686–690
Baeyens L, De Breuck S, Lardon J, Mfopou JK, Rooman I, Bouwens L (2005) In vitro generation of insulin-producing beta cells from adult exocrine pancreatic cells. Diabetologia 48:49–57
Suarez-Pinzon WL, Lakey JR, Brand SJ, Rabinovitch A (2005) Combination therapy with epidermal growth factor and gastrin induces neogenesis of human islet β-cells from pancreatic duct cells and an increase in functional β-cell mass. J Clin Endocrinol Metab 90:3401–3409
Acknowledgements
This work was supported by grants from the Juvenile Diabetes Research Foundation, the Sigrid Juselius Foundation, the Finnish Diabetes Research Foundation, the Academy of Finland and the Helsinki University Hospital Research Funds (EVO). The authors thank Dr K. Salmela and the Nordic Network for Clinical Islet Transplantation for the procurement of human pancreatic cells. Ms P. Narinen is thanked for her skilled technical assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gao, R., Ustinov, J., Korsgren, O. et al. In vitro neogenesis of human islets reflects the plasticity of differentiated human pancreatic cells. Diabetologia 48, 2296–2304 (2005). https://doi.org/10.1007/s00125-005-1935-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-1935-8