
Abstract
Aims/hypothesis
The cyclooxygenase-2 (PTGS2, previously known as COX2) enzyme and its products, such as prostaglandin E2 (PGE2), have been implicated in the pathogenesis of several inflammatory diseases including islet dysfunction under diabetic conditions. In this study we evaluated whether diabetic conditions in vitro, such as high-glucose (HG) culture or AGE, or in vivo in animal models of diabetes can induce PTGS2 expression and activity in pancreatic islets.
Materials and methods
Isolated human pancreatic islets were treated for 24 h with HG (25 mmol/l) or with S100b (5 mg/l), a specific ligand for the AGE-specific receptor. PTGS2 and cyclooxygenase-1 (PTGS1, previously known as COX1) mRNA, protein expression and product PGE2 were analysed by RT-PCR, Western blots and specific enzyme immunoassay respectively. Islet PTGS2 production in animal models was assessed by immunofluorescence.
Results
Treatment of human pancreatic islets with HG and S100b led to a three–five-fold induction of PTGS2 mRNA (p<0.001). PTGS2 protein and its product PGE2 (351.4±13.05 fg/ml vs control 39.4±0.11 fg/ml) were also increased (p<0.001). Pretreatment with specific inhibitors demonstrated the involvement of protein kinase C and oxidant stress in S100b- and HG-induced PTGS2 expression. However, insulin secretion was not significantly altered by S100b. Double immunofluorescent staining showed increased PTGS2 production in pancreatic islets from diabetic mice relative to corresponding controls.
Conclusion/interpretation
These results show for the first time that diabetes as well as diabetic conditions such as AGE and HG in vitro can directly upregulate the expression of the inflammatory PTGS2 gene in pancreatic islets. This might contribute to the pathogenesis of islet dysfunction in diabetes.
Similar content being viewed by others


Introduction
Cyclooxygenase (PTGS, previously known as COX) enzymes catalyse the rate-limiting step in the conversion of arachidonic acid to prostaglandins [1–5], which are important mediators of acute and chronic inflammation, development and immune functions [6]. The constitutively expressed cyclooxygenase-1 (PTGS1, previously known as COX1) isoform appears to regulate many normal physiological functions in several cell types, whereas the inducible isoform, cyclooxygenase-2 (PTGS2, previously COX2), is usually expressed at low levels in most tissues and cells, but is significantly induced by a wide range of inflammatory stimuli such as lipopolysaccharide, cytokines and chemicals [1, 2]. PTGS2 overexpression has been demonstrated in several human inflammatory diseases [2]. In the pancreatic islet PTGS2 is constitutively and dominantly expressed [7, 8] and here PTGS2 products such as prostaglandin E2 (PGE2) are believed to play a role in inflammation, islet destruction and inhibition of insulin secretion [7–9]. A recent report showed that PTGS2 mRNA, however, was present in lower abundance than PTGS1 in islets [10].
The diabetic state impairs beta cell function in animals and humans. Type 1 diabetes mellitus is caused by autoimmune and inflammatory processes in the pancreas, leading to selective destruction of the beta cell [11, 12]. Evidence indicates that both hyperglycaemia and AGE are important mediators of beta cell dysfunction [13]. Exposure of human islets to a high glucose (HG) concentration resulted in increased apoptosis of beta cells and this was attributed to the IL1B-nuclear factor-kappa B (NF-κB) pathway [14]. Furthermore, inflammatory cytokines released by infiltrating macrophages have been implicated as effector molecules that participate in both islet inflammation and beta cell destruction during the development of diabetes [8, 15]. For instance, it was shown that when isolated rat islets are incubated with IL1B, IFNG and TNF they synergistically lead to the impairment of beta cell function and cause beta cell death; this appeared to be due the induction of PTGS2, oxidant stress and inducible nitric oxide synthase [8]. The molecular mechanisms responsible for the cytotoxic effect of these cytokines, particularly under diabetic conditions, remains to be elucidated. HG could induce IL1B in human pancreatic islets [14], and very recently Persaud et al. [10] showed that HG can induce PTGS2 in islets. However, it is not clear whether under diabetic conditions in vivo or in vitro, treatment with AGE can directly induce PTGS2 expression in mouse and human islets.
Chronic hyperglycaemia-mediated diabetic complications are, in part, related to non-enzymic glycation of proteins and lipids, and its sequelae. Early glycated molecules can be further modified by the formation of AGE that act via receptors such as the AGE-specific receptor (AGER, previously known as RAGE) and lead to cellular damage underlying the microvascular and macrovascular complications of diabetes [16]. However, it not known to what extent AGE directly influence pancreatic beta cell function.
In this study we show for the first time that a specific inflammatory ligand of the AGER, S100b, can induce PTGS2 expression and activity in isolated human pancreatic islets. This was similar to the effects of HG. We have also shown that Ptgs2 expression is increased in vivo in the islets of mouse models of type 1 and type 2 diabetes mellitus. These studies are significant because early cellular dysfunction and inflammation induced by HG and AGE in islets may play major roles in the pathogenesis of diabetes and its complications.
Materials and methods
Materials
RT-PCR reagents were from Applied Biosystems (Foster City, CA, USA) while Quantum RNA 18S Internal Standards were from Ambion (Austin, TX, USA). PGE2 enzyme immunoassay (EIA) kit was from Cayman Chemical (Ann Arbor, MI, USA). Anti-PTGS2 antibody was from BD Transduction Laboratories (Palo Alto, CA, USA) or from Cayman Chemical, and anti-insulin antibody was from Linco Immunoresearch (St Charles, MO, USA).
Cell culture and animal treatments
Normal human pancreatic islets were obtained from the Southern California Islet Consortium (City of Hope Medical Centre, Duarte, CA, USA) and cultured in RPMI-1640 medium supplemented with 10% FCS, glutamine, HEPES, streptomycin (100 μg/ml)-penicillin (100 U/ml) and d-glucose (3.5 mmol/l) (normal glucose, NG) in a 5% CO2 incubator at 37°C. For experiments, islets were treated with or without d-glucose (25 mmol/l) (HG) or S100b (5 mg/l) (Calbiochem, San Diego, CA, USA).
We purchased db/db mice and their genetic control db/m mice, and C57BL/6J mice from Jackson Laboratory (Bar Harbor, ME, USA). In addition, diabetes was induced in 11-week-old C57BL/6J mice by injection of 50 mg streptozotocin (STZ)/kg body weight (concentration 7.5 mg/ml) on five consecutive days. Streptozotocin (Sigma Chemical, St Louis, MO, USA) was dissolved in a sodium citrate solution (10 mmol/l) containing 0.9% NaCl, pH 4.5, immediately before administration. Diabetes induction was assessed by measuring glycaemia levels using Accu-chek Advantage (Roche Diagnostics, Indianapolis, IN, USA). STZ-treated mice were killed 4 days after the last injection when blood glucose levels reached >20 mmol/l. db/db and db/m mice were killed at about 11 weeks of age. Pancreases from all mice were dissected out and fixed in 10% formalin solution. All experimental procedures involving mice were performed in accordance with protocols approved by the City of Hope/Beckman Research Institute Research Animal Care Committee.
RNA preparation and relative RT-PCR
Human islets (500 islet equivalents per sample) in 3 ml medium containing 3.5 mmol/l (NG) alone or treated either with S100b (5 mg/l) or 25 mmol glucose/l (HG) or 21.5 mmol mannitol/l (osmolarity control) were cultured in duplicate in six-well dishes for 24 h. Total RNA was isolated by RNA-STAT-60 method [17, 18] and 1 μg used for the RT reaction using Gene Amp RNA PCR kit. Complementary DNA corresponding to 0.05 μg RNA was then used in multiplex PCR reactions as described in [17, 18]. Results are expressed as fold stimulation over NG after normalising with paired 18S RNA levels.
Western blot analysis and PGE2 EIA
Western blot analysis and PGE2 EIA were performed using lysed human islets and medium supernatant fractions respectively as described previously [17, 18]
In vitro insulin release assays
To assess the insulin release function of the islets, a static incubation assay was performed. The static incubation assays were performed on islets pre-treated overnight with S100b, alone or in combination with the inhibitors of oxidant stress (N-acetyl cysteine, NAC), mitochondrial complex II inhibitor (thenoyltrifluoroacetone, TTFA) and protein kinase C (bisindolylmaleimide, GFX) in RPMI medium containing low glucose (3.3 mmol/l). Before the static incubation assay, the islets were washed five times with low-glucose medium (RPMI containing 3.3 mmol glucose/l and 2% FCS) and then incubated in low-glucose medium. After 30 min, the medium was collected for insulin assay (first low-glucose sample) and then replaced by HG stimulation medium (RPMI 1640 containing 19.4 mmol glucose/l and 2% FCS). After incubation for another 30 min, supernatant fractions (HG samples) were collected, islets were washed five times with basal medium and further incubated with basal medium for 30 min to obtain a second low-glucose sample. Collected samples were frozen and stored until measurement of human insulin using an EIA kit (Alpco, Windham, NH, USA). The stimulation index was calculated as insulin released into the HG medium divided by insulin released into the low-glucose medium.
Analysis of PTGS2 expression by immunofluorescence microscopy
Formalin-fixed pancreas tissues from mice were processed, and paraffin sections were treated with anti-PTGS2 and anti-insulin antibodies for immunofluorescence analyses. Isolated human islets were treated with S100b (5 mg/l) or HG (25 mmol/l) for 24 h, fixed with formalin and embedded in paraffin. Paraffin-embedded samples were deparaffinised/rehydrated, treated with antigen retrieval solution (Vector Laboratories, Burlingame, CA, USA), according to the manufacturer’s protocols, and then processed for double immunofluorescence [19] using rabbit or mouse anti-PTGS2 antibodies (Becton Dickinson, San Jose, CA, USA) and guinea pig anti-insulin (Linco Immunoresearch) followed by anti-mouse, anti-rabbit or anti-guinea pig Ig antibodies conjugated with fluorescein isothiocyanate or Texas Red (Jackson Immunoresearch, West Grove, PA, USA). After intensive washing the samples were counterstained for DNA with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma Chemical.), embedded in Vectashield (Vector Laboratories, Burlingame, CA, USA), visualised and documented with an Olympus BX51 microscope (Olympus America, Melville, NY, USA) equipped with a Pixera 600 cooled charged coupled device camera (Pixera, Los Gatos, CA, USA). Fluorescent images were analysed using the ImagePro Plus image analysis software (Media Cybernetics, Silver Spring, MD, USA). The relative fluorescence intensity of the islet areas corresponding to PTGS2 staining was measured. The relative fluorescence was generated as mean and SD for each islet area. These were averaged and provided an estimate of relative fluorescence.
Data analyses
Data are expressed as mean ± SEM of multiple experiments. Paired Student’s t tests were used to compare two groups and ANOVA for multiple comparisons. Values of p<0.05 were considered statistically significant.
Results
HG and ligation of AGER with S100b increase the expression of PTGS2 but not PTGS1 isoform in human pancreatic islets
Human islets were treated for 24 h either with HG or S100b (5 mg/l), a specific AGER ligand. RNA isolated from these samples was analysed for PTGS2 mRNA expression by relative RT-PCR with 18S as internal control for normalisation. HG treatment significantly induced a two–three-fold increase, while S100b treatment led to a five–six-fold significant increase in PTGS2 mRNA expression in human islets compared with NG-grown islets (p<0.001; Fig. 1a,b). Both freshly isolated and frozen stored samples of human islets showed similar increases in PTGS2 mRNA expression (results not shown). In contrast, neither HG nor S100b had any effect on PTGS1 mRNA expression (Fig. 1c). Furthermore, Fig. 1d shows that PTGS2 mRNA was increased only by HG and not by equimolar concentrations of mannitol (osmolality control), indicating the specificity of HG effects. These results suggest that HG and S100b specifically upregulate the inducible inflammatory isoform encoded by PTGS2, but do not upregulate PTGS1 mRNA.

Ethidium bromide-stained agarose gels of RT-PCR products and bar graph showing that treatment of normal human donor islets with HG and S100b leads to increased PTGS2 mRNA expression. Relative RT-PCR with gene-specific primers were performed with total RNA isolated from normal human pancreatic islets treated with HG or S100b for 24 h. 18S RNA primers were included in each PCR reaction as internal control. PCR products were analysed on agarose (2.5%) gels. a RT-PCR for PTGS2; b Results quantitated from six separate experiments (n=6, ***p<0.001 vs NG); c PTGS1 mRNA; results representative of four similar experiments. d PTGS2 mRNA in cells treated with HG or mannitol (MN); results representative of four similar experiments
In the next step, we examined whether S100b-induced PTGS2 mRNA expression occurs via the AGER receptor in islets. Islets were pre-treated with a specific anti-AGER antibody for 1 h before S100b treatment. RT-PCR analyses of RNA from these islets showed complete blockade of the S100b induction of PTGS2 mRNA levels in antibody-treated islets (Fig. 2), thereby confirming that induction of PTGS2 mRNA by S100b is via AGER activation.

S100b-induced PTGS2 mRNA expression is via AGER. Normal human islets were pre-treated with anti-AGER antibody (70 μg/ml) for 1 h followed by S100b (5 mg/l) for 24 h. PTGS2 mRNA levels were analysed by RT-PCR using specific primers for PTGS2 and 18S internal control. Results are representative of four similar experiments
HG and AGER ligand increase the production of PTGS2 protein in human pancreatic islets
Our observations that islet PTGS2 mRNA levels were upregulated by simulated diabetic conditions in culture, such as HG and AGER ligation, prompted us to examine whether PTGS2 protein levels were also altered. Western blot analysis with a specific PTGS2 antibody was carried out using total protein prepared from human islets cultured with NG, HG or S100b. Treatment (24 h) of both HG and S100b showed a clear increase in PTGS2 protein levels (upper panel of Fig. 3). Equal loading of protein in each lane was confirmed by probing with an anti-actin antibody as internal control (Fig. 3).

Western blot analysis of PTGS2 protein in HG- and S100b-treated human islets. Total protein isolated from control untreated human islets or islets treated with HG or S100b for 24 h were resolved by SDS-PAGE, transferred onto Immobilon membranes, and probed with an anti-PTGS2 polyclonal antibody. As a control for protein loading, the PTGS2 immunoblot was probed with actin
We also examined PTGS2 protein production by immunofluorescence microscopy using specific anti-PTGS2 antibodies. Sections from untreated human islets or those stimulated with either HG or S100b were treated with anti-PTGS2 and anti-insulin antibodies. Fig. 4 shows results of PTGS2 and insulin immunofluorescence analyses. HG- and S100b-treated islets (Fig. 4d–i) showed clear increases in PTGS2 expression relative to islets cultured in NG (Fig. 4a–c). Increased PTGS2 production was detected in both insulin-positive as well as in insulin-negative cells (Fig. 4c,f and i). Similarly, immunofluorescence intensity analyses (Fig. 5) showed a significant increase in PTGS2 expression in isolated human islets that were treated with HG and S100b, when compared with control (p<0.05).

PTGS2 protein expression in S100b- and HG-treated human pancreatic islet sections as evaluated by immunofluorescence microscopy. PTGS2 and insulin shown in green and red, respectively. PTGS2, insulin and merged staining respectively in: a–c control human islets; d–f islets treated with HG; and g–i islets treated with S100b. After fixation, human islet sections were treated with anti-PTGS2 and anti-insulin antibodies, followed by secondary antibodies conjugated with fluorescein isothiocyanate and Texas Red respectively, counterstained for DNA (blue) and examined in a fluorescent microscope

Quantitative analysis of PTGS2 immunofluorescence data. The relative fluorescence intensity of PTGS2 staining in the islet areas was quantified using ImagePro Plus software (Media Cybernetics). This was performed in four random islet areas for pancreatic tissue sections from both mouse groups, and from human islets treated with or without HG or S100b. All values are expressed as mean±SEM (n=3–6). *p<0.05 (untreated control vs S100b, ***p<0.001 (control db/m vs db/db), †††p<0.001 (untreated control vs HG)
S100b and HG increase PTGS2 enzyme activity in human pancreatic islets
To determine whether the induction of PTGS2 mRNA and protein levels were also associated with increase in PTGS2 enzyme activity, we next examined the levels of the PTGS2 product PGE2 in HG and S100b-treated vs NG-grown normal human islets in vitro. PGE2 released into the culture supernatant fractions was measured by a specific EIA. As shown in Fig. 6, S100b or HG treatment for 24 h significantly increased PGE2 levels (351.4±13.05 fg/ml, n=4, p<0.001 for S100b; 355 fg/ml, n=2 for HG) compared with NG (39.44±0.1,135 fg/ml, n=4). Overall, these results show that diabetic islets have high levels of PTGS2 mRNA and protein expression and its product PGE2, and this might contribute to several adverse events.

PGE2 levels in HG- or S100b-treated islets relative to control NG-cultured islets. PGE2 was measured by EIA in culture supernatant fractions of human islets treated with or without HG and S100b for 24 h. Values are mean±SEM of three independent experiments (n=4). ***p<0.001 vs S100b. For HG-treated islets the value shown is the mean of two experiments
Signal transduction mechanisms involved in S100b and HG-induced PTGS2 mRNA expression
In order to determine the key signal transduction pathways involved in HG-induced PTGS2 mRNA in islets, we evaluated the effects of inhibitors of pathways known to be activated by HG and S100b, including signalling kinases and oxidant stress. Islets were pre-incubated with either an antioxidant (NAC), a mitochondrial complex-II inhibitor to block mitochondrial superoxide production (TTFA) or protein kinase C inhibitor (GFX); PTGS2 mRNA induction by HG or S100b was determined by RT-PCR. The representative RT-PCR blot in Fig. 7 shows that HG- and S100b-induced PTGS2 mRNA expression was blocked by protein kinase C inhibitor (GFX) and antioxidant (NAC) in islets. However, TTFA had no effect on HG-induced PTGS2 mRNA expression. These results implicate the involvement of multiple pathways including protein kinase C and oxidant stress in HG- and S100b-induced PTGS2 mRNA expression in islets.

Effects of antioxidant, superoxide and protein kinase C inhibitors on PTGS2 mRNA expression. Human islets were pre-treated with various inhibitors (protein kinase C inhibitor, GFX; mitochondrial complex II inhibitor, TTFA; antioxidant, NAC) for 1 h before HG or S100b stimulation. Total RNA was isolated post HG and S100b stimulation and PTGS2 mRNA levels were analysed by RT-PCR as described in Fig. 1. − absence, + presence of S100b or HG, respectively
In parallel experiments, the functionality of islets was analysed by examining glucose-induced insulin secretion after consecutive incubations in low glucose (3.3 mmol/l) and HG (19.4 mmol/l) glucose. The stimulation index was calculated as the ratio between the insulin secreted at HG and insulin secreted at low glucose. This was tested in islets pre-treated with S100b alone and in combination with the protein kinase C inhibitor, GFX or TTFA. Results shown in Table 1 show that the insulin stimulation index in islets treated with S100b alone or in the presence of inhibitors was slightly decreased relative to control, but this effect was not statistically significant, thus indicating that S100b had no significant effect on insulin secretion.
PTGS2 expression is increased in vivo in pancreatic islets of mouse models of type 1 and type 2 diabetes mellitus
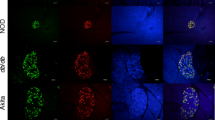
To further analyse the in vivo relevance of elevated PTGS2 expression, pancreatic tissue sections from STZ-induced diabetic mice (model of type 1 diabetes mellitus), from db/db diabetic mice (model of type 2 diabetes mellitus) and from their respective control groups were studied. PTGS2 protein production was analysed by immunofluorescent staining using specific anti-PTGS2 and anti-insulin antibodies to identify insulin-producing beta cells. PTGS2 staining was detected in both insulin-positive (beta) cells, as well as in insulin-negative cells. Our results in Fig. 8 show that there was a slight increase in PTGS2 protein production in the islets of STZ-induced diabetic mice (Fig. 8d–f) compared with saline-injected control C57BL/6 mice (Fig. 8a–c). However, quantitative image analysis (Fig. 5) shows that this was not statistically significant. On the other hand, PTGS2 protein production was greater in islets of diabetic db/db mice (Fig. 9d–f) compared with their genetic control, db/m mice (Fig. 9a–c) and this was statistically significant (p<0.001, Fig. 5).

Immunofluorescence analyses of PTGS2 expression in pancreatic sections from control and STZ-treated diabetic mice injected with citrate buffer (Control) or treated with STZ. Blood glucose levels were >20 mmol/l in STZ-treated mice. Pancreatic sections were treated with (a, d) anti-PTGS2 (green), (b, e) and anti-insulin (red). (c, f) Merged stains. The staining was performed as in Fig. 4 (see Materials and methods)

Immunofluorescence analyses of PTGS2 expression in pancreas tissue from control (db/m) and db/db mice. Pancreatic tissue was fixed and treated with anti-PTGS2 (green) and anti-insulin antibody (red) as in Fig. 4 (see Materials and methods). (a–c) Immunofluorescent staining for PTGS2, insulin and merged staining, respectively in control mice, and (d–f) in db/db mice
Discussion
In this study we have demonstrated that treatment of normal human donor pancreatic islets with diabetic stimuli such as AGER ligands or HG can lead to a significant increase in PTGS2 mRNA and protein expression and product formation. In contrast, HG or S100b did not alter the expression of PTGS1 mRNA. Furthermore, human islets maintained at the equimolar concentration of mannitol did not induce PTGS2 mRNA, thereby suggesting that increased PTGS2 mRNA is specific to HG. The effects of the AGER ligand, S100b, on PTGS2 mRNA induction was blocked by an anti-AGER antibody, thereby demonstrating the involvement of AGER in PTGS2 induction.
PTGS2 protein expression was also increased in vitro in normal human islets treated with HG or S100b. Furthermore, we also noted increased PTGS2 in vivo in islets of mouse models of type 1 and type 2 diabetes mellitus. Increased PTGS2 expression/staining was present in beta cells (insulin-positive cells) as well as in non-beta cells (insulin-negative cells). We noted similar increases in PTGS2 immunostaining compared with controls in the islets of diabetic db/db mice and STZ-induced diabetic mice, and with the same mosaic pattern in beta and non-beta cells. Expression levels of PTGS2 in beta cells were greater in db/db mice relative to STZ-injected mice. This might be a reflection of the fact that the STZ-injected mice have greatly reduced numbers of islets and extensive beta cell destruction. Interestingly, in a preliminary unpublished study we similarly noted a marked increase in PTGS2 expression (by RT-PCR and immunofluorescence) in the islets of a human donor with type 2 diabetes relative to normal non-diabetic donors (results not shown).
Although PTGS2 and its products have been implicated in beta cell destruction and islet dysfunction, the exact source of PTGS2 in the islet was not verified in these earlier studies. Cytokines produced by infiltrating macrophages have been shown to clearly mediate beta cell apoptosis and dysfunction. Our study suggests that, in addition, diabetic conditions can also directly increase PTGS2 expression in islet cells, including beta cells, and this may also contribute to islet dysfunction. Increased PTGS2 expression could be either direct or via induction of cytokines, such as IL1B, that are potent inducers of PTGS2. Furthermore, PTGS2 expression is known to be regulated by both transcriptional and post-transcriptional mechanisms [20] and it is possible that both are in operation in islets as demonstrated in monocytes [17]. While our demonstration of increased PTGS2 expression in vitro and in vivo under diabetic conditions does not directly implicate PTGS2 in the pathogenesis of diabetes, it is highly likely-given the fact that PTGS2 is induced by inflammatory cytokines and that PTGS2 products have inflammatory properties—that increased PTGS2 expression can mediate islet dysfunction and the development or progression of diabetes. This is supported by studies showing the beneficial effects of PTGS2 inhibitors in a model of diabetes [21].
Our studies with inhibitors showed that an oxidant NAC and a protein kinase C inhibitor GFX blocked HG- or S100b-induced PTGS2 expression in islets. In contrast, TTFA, which inhibits mitochondrial superoxide production, did not have any effect. These results suggest that multiple signalling pathways including oxidant stress and protein kinase C are involved in HG- or S100b-induced PTGS2 expression in islets. Furthermore, treatment of islets with S100b alone or in the presence of these inhibitors did not significantly affect glucose-induced insulin secretion. These results are consistent with published results showing that cytokine-induced PTGS2 did not affect insulin secretion in islets [22, 23] and further support observations that PTGS2 inhibitors failed to prevent cytokine-induced beta cell dysfunction [23]. In addition, a very recent study [24] showed that PTGS2 inhibition enhances anti-tumour immunity. These authors suggest that PTGS2 and its product PGE2 can modulate immune responses by upregulating the activity of T-lymphocytes. In the context of islets and our results, it is possible that S100b-, HG- and diabetes-induced PTGS2 expression may modulate autoimmunity and thereby cause islet dysfunction. More extensive studies are needed, including the evaluation of PTGS2-deficient mice, specific PTGS2 inhibitors or similar anti-inflammatory agents, to fully determine the role of PTGS2 in the pathogenesis of diabetes and its complications.
Abbreviations
- AGER:
-
AGE-specific receptor
- DAPI:
-
4′,6-diamidino-2-phenylindole
- EIA:
-
enzyme immunoassay
- GFX:
-
bisindolylmaleimide
- HG:
-
high glucose
- NF-κB:
-
nuclear factor-kappa B
- NAC:
-
N-acetyl cysteine
- NG:
-
normal glucose
- PGE2 :
-
prostagladin E2
- PTGS:
-
cyclooxygenase
- PTGS1:
-
cyclooxygenase-1
- PTGS2:
-
cyclooxygenase-2
- STZ:
-
streptozotocin
- TTFA:
-
thenolytrofluoroacetone
References
Smith WL, Dewitt DL (1996) Prostaglandin endoperoxide H synthases-1 and -2 In: Dixon FJ (eds) Advances in Immunology, Vol 62, Academic, Orlando, FL, pp. 167–215
Hla T, Ristimaki A, Appleby SB, Barriocanal JG (1993) Cyclooxygenase gene expression in inflammation and angiogenesis. Ann N Y Acad Sci 696:197–204
Fu J, Masferrer J, Seibert K, Raz A, Needleman P (1990) The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J Biol Chem 265:16737–16740
Herschman HR (1996) Prostaglandin synthase 2. Biochim Biophys Acta 1299:125–140
Vane, JR, Bakhle, YS, Botting, RM (1998) Cyclooxygenases 1 and 2. Ann Rev Pharmacol Toxicol 38:97–120
Williams CS, Mann M, DuBois RN (1999) The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 18:7908–7916
Robertson RP (1998) Dominance of cyclooxygenase-2 in the regulation of pancreatic islet prostaglandin synthesis. Diabetes 47:1379–1383
McDaniel ML, Kwon G, Hill JR, Marshall CA, Corbett JA (1996) Cytokines and nitric oxide in islet inflammation and diabetes. Proc Soc Exp Biol Med 211:24–32
Tran PO, Gleason CE, Poitout V, Robertson RP (1999) Prostaglandin E (2) mediates inhibition of insulin secretion by interleukin-1beta. J Biol Chem 274:31245–31248
Persaud SJ, Burns CJ, Belin VD, Jones PM (2004) Glucose-induced regulation of COX-2 expression in human islets of Langerhans. Diabetes 53 [Suppl 1]:S190–S192
Gepts W (1965) Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 14:619–633
Donath MY, Storling J, Maedler K, Mandrup-Poulsen T (2003) Inflammatory mediators and islet beta cell failure: a link between type 1 and type 2 diabetes. J Mol Med 81:455–470
Tajiri Y, Moller C, Grill V (1997) Long term effects of aminoguanidine on insulin release and biosynthesis: evidence that the formation of advanced glycosylation end products inhibits B cell function. Endocrinology 138:272–280
Maedler K, Sergeev P, Ris F, et al. (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110:851–860
Hohmeier HE, Tran VV, Chen G, Gasa R, Newgard CB (2003) Inflammatory mechanisms in diabetes: lessons from the beta cell. Int J Obes Relat Metab Disord 27 [Suppl 3]:S12–S16
Schmidt A, Hori O, Brett J, Yan SD, Wautier JL, Stern D (1994) Cellular receptors for advanced glycation end products. Implications for induction of oxidant stress and cellular dysfunction in the pathogenesis of vascular lesions. Arterioscler Thromb 14:1521–1528
Shanmugam N, Kim, YS, Lanting L, Natarajan R (2003) Regulation of cyclooxygenase-2 expression in monocytes by ligation of the receptor for advanced glycation end products. J Biol Chem 278:34834–34844
Shanmugam N, Gaw Gonzalo IT, Natarajan R (2004) Molecular mechanisms of high glucose-induced cyclooxygenase-2 expression in monocytes. Diabetes 53:794–802
Todorov IT, Attaran A, Kearsey SE (1995) BM28, a human member of the MCM2-3-5 family, is displaced from chromatin during DNA replication. J Cell Biol 129:1433–1445
Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM (2000) Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3′-untranslated region. J Biol Chem 275:11750–11757
Tabatabaie T, Waldon AM, Jacob JM, Floyd RA, Kotake Y (2000) COX-2 inhibition prevents insulin-dependent diabetes in low-dose streptozotocin-treated mice. Biochem Biophys Res Commun 273:699–704
Heitmeier MR, Kelly CB, Ensor NJ, et al. (2004) Role of cyclooxygenase-2 in cytokine-induced beta-cell dysfunction and damage by isolated rat and human islets. J Biol Chem 279:53145–53151
Hughes JH, Easom RA, Wolf BA, Turk J, McDaniel ML (1989) Interleukin 1-induced prostaglandin E2 accumulation by isolated pancreatic islets. Diabetes:38:1251–1257
Sherven Sharma, Li Zhu, Seok Chul Yang, et al. (2005) Cyclooxygenase 2 inhibition promotes IFN-γ-dependent enhancement of antitumor responses. J Immunol 2005 175: 813–819
Acknowledgements
The authors would like to thank the Southern California Islet Cell Resource Centre at City of Hope for the human islet samples, and L. Lanting, L. Valiente and S. Loera (City of Hope Anatomic Pathology Laboratory) for their excellent technical assistance. We thank D. Stern and A.-M. Schmidt for the generous gift of the anti-AGER antibody. These studies were supported by grants from the National Institutes of Health (NIH RO1 DK065073, NIH IU42RR16607), the Juvenile Diabetes Research Foundation, SC-ICR and also in part by a GCRC grant from NCRR (MO1RR00043 awarded to the City of Hope).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shanmugam, N., Todorov, I.T., Nair, I. et al. Increased expression of cyclooxygenase-2 in human pancreatic islets treated with high glucose or ligands of the advanced glycation endproduct-specific receptor (AGER), and in islets from diabetic mice. Diabetologia 49, 100–107 (2006). https://doi.org/10.1007/s00125-005-0065-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-0065-7