
Abstract
Aims/hypothesis
Sodium tungstate has recently emerged as an effective oral treatment for diabetes. We examined the effects of tungstate administration in the beta-cell mass of the pancreas as well as its therapeutic potential.
Methods
Sodium tungstate was administered via drinking water to healthy and neonatal streptozotocin (nSTZ)-diabetic rats for one month. The pancreas from each rat was removed and morphometric and immunocytochemical studies were carried out. The molecular mechanism of tungstate’s action was also studied.
Results
In nSTZ rats administration of this compound normalised glycaemia, and increased insulinaemia and islet insulin content. Blood glucose concentrations were normalised as early as on day 4 of treatment, and tungstate treatment produced a partial recovery of beta-cell mass. The rats remained normoglycaemic after tungstate withdrawal. Morphometric studies showed that the increase in beta-cell mass was not due to beta-cell hypertrophy but to hyperplasia, with an increase in islet density in treated diabetic rats. Tungstate treatment increased extra-islet beta-cell replication without modifying intra-islet beta-cell replication rates. Moreover, the treatment induced increases in insulin-positive cells located close to ducts; and in PDX-1 positive cells scattered in the exocrine tissue, suggesting active neogenesis. In islets from treated diabetic rats, tungstate is able to increase the phosphorylation state of PDX-1 through the activation of p38.
Conclusion/interpretation
These observations indicate that tungstate treatment is able to regenerate a stable, functional pancreatic beta-cell population which leads to and maintains normoglycaemia.
Similar content being viewed by others

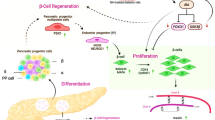

Diabetes is a major public health problem. Its impact on society is huge [1] and its chronic complications cause enormous human suffering [2]. The development of new therapies that are able to improve glycaemia management and even to cure diabetes is of great interest. The idea of inducing the formation of new beta cells is a novel approach for managing diabetes. In recent years, major advances in our understanding of pancreas embryogenesis have led to the identification of several transcription factors that are involved in this process [3]. Among these, PDX-1 plays a critical role in the development of the pancreas, since mutations in this gene result in pancreatic agenesis in both mice and humans [4, 5]. Moreover, studies on the dynamics of the adult pancreas have shown that adult beta cells adjust their mass to distinct metabolic states that occur during the life span, such as pregnancy [6] or obesity [7, 8]. The mechanisms involved in this dynamic state shape a delicate balance between cell growth, proliferation, and destruction [9]. The formation of new beta cells arising from the ductal epithelium has been shown in the adult pancreas, although the precise progenitor cell population remains a matter of debate. In this regard, two hypotheses have been proposed: a transdifferentiation of beta cells derived from ductal or acinar cells or differentiation from stem cells located in and around the ductal epithelium [10].
Previous studies have shown sodium tungstate to be an effective oral anti-diabetic agent, in both short- and long-term treatments, in several animal diabetes mellitus models [11, 12, 13, 14]. This compound reduces and, in most cases, normalises glycaemia, and also decreases hypertriglyceridaemia [13]. Tungstate treatment normalises glucose metabolism in the liver [11, 14]. We found that in treated nSTZ rats, a Type 2 diabetes model [15], the reversion of the diabetic phenotype observed is due to an increase in the beta-cell mass through neogenesis. Moreover, this beta-cell population is functional and stable even when the treatment is withdrawn.
Materials and methods
Animals and experimental design
Principles of laboratory animal care were followed (European and local government guidelines) and protocols were approved by the Animal Research Committee of the University of Barcelona. Newborn Wistar rats (Charles River Laboratories, Wilmington, Mass., USA) were divided randomly into two groups, diabetic (nSTZ) and healthy rats. Diabetes was induced as described previously [15]. All the rats used were 9-weeks-old.
Both the diabetic and healthy rats were further divided into two groups: untreated and tungstate-treated. The treatment was carried out as described previously [12]. Fluid and food intake, body weight, and blood glucose (Glucometer Elite, Química Farmaceutica Bayer, Barcelona, Spain) were measured every 2 days (10:00–12:00 am). At the end of the treatments, the rats were decapitated (10:00–12:00 am) and the blood was immediately collected. Plasma insulin was measured by radioimmunoassay (CIS, Gif-Sur-Yvette, France). Pancreata were excised, weighed and immediately frozen in liquid N2 or alternatively fixed in 4% paraformaldehyde and embedded in paraffin.
Islet isolation and insulin content
Pancreata were digested with collagenase (Roche Diagnostics, Basel, Switzerland) followed by islet separation in a density gradient using Histopaque (Sigma Diagnostics, St Louis, Mo., USA) as described elsewhere [16]. Islets were hand-picked under a stereomicroscope and separated in batches of ten to determine insulin content by radioimmunoassay (CIS, France).
Morphometric analysis
Pancreatic serial cryosections were immunostained for insulin or glucagon using indirect immunofluorescence techniques. Anti-insulin and anti-glucagon (ICN Pharmaceuticals, Costa Mesa, Calif., USA) were used as primary antibodies. To measure the total islet area toluidine-blue pancreatic sections were used. Morphometry was done with a manual optical image analyser (MOP-01, Olympus, Tokyo, Japan). For PDX-1 detection, paraffin sections were treated with 10 mmol/l sodium citrate buffer (pH 6.0). Simultaneous incubation with anti-PDX-1 antibody (kindly provided by J.F. Habener) and anti-insulin antibody or anti-somatostatin antibody (ICN Pharmaceuticals) was carried out.
Beta-cell volume determination
Beta-cell size was measured in insulin-stained sections co-stained with propidium iodide at 10 µg/ml (Sigma) with RNAse 100 µg/ml (Sigma) at 37°C. Beta-cell nuclei on a random section were counted, and the area of beta-cell tissue in that section was measured by planimetry as described above. Individual beta-cell volume was determined by dividing the beta-cell area by the number of nuclei. However, this method can lead to the overestimation of the size of the beta cells since not all are sectioned across their nuclei. A minimum of 500 beta-cell nuclei were counted.
Beta-cell replication studies
Five hours before they were killed, the rats were injected intraperitoneally with 5-bromo-2-deoxyuridine (BrdU) (Sigma) at a concentration of 50 mg per kg body weight. Frozen pancreatic sections were used to determine replication rates by an anti-BrdU antibody (Sigma). To calculate the BrdU labelling index, the number of positive cells for insulin or glucagon and BrdU was counted. At least 1000 beta-cell nuclei were counted per pancreas. Based on the labelling index measured and an estimated S-phase (Ts) duration for beta cells of 6.4 h [17] the cell birth rate (the production of new cells per 24 h) can be calculated from the equation: Cell birth rate = (Labeling Index/Ts)×24 h.
Confocal microscopy
Fluorescence was monitored with a Leica TCS NT microscope with excitation from the 480 nm line of an argon/krypton filter centred at 530 nm for FITC and above 590 nm for TRITC.
1D and 2D western blotting
Rats were fasted overnight before being killed. Pancreatic islets were prepared by collagenase digestion as described [16], snap frozen in liquid nitrogen and kept at −80°C until use. Proteins were extracted during 30 min at 4°C in a buffer containing Tris (50 mmol/l, pH 7.5), EDTA (5 mmol/l), NaCl (150 mmol/l), Triton X100 (1%), iodoacetate (5 mmol/l), sodium phosphate (10 mmol/l), sodium fluoride (10 mmol/l), sodium vanadate (1 mmol/l) and protease inhibitors (Sigma cocktail). Proteins were separated by SDS-PAGE and transferred to PVDF membranes by standard protocols. Phosphorylation of p38 was assessed by Western blotting using specific antibody against phospho-p38 MAP Kinase (Thr180/Tyr182; Cell signalling). Membranes were then stripped and incubated with anti-p38 (Cell signalling). Phosphorylation of PDX-1 was determined by two-dimensional electrophoresis (2D-PAGE) as described [18]. Briefly, islet lysates were re-suspended in a buffer containing urea (7 mol/l), thiourea (2 mol/l), CHAPS (2% w/v), DTT (65 mmol/l), ampholines (0.5% v/v), orthovanadate (10 mmol/l) and protease inhibitors and were separated by 2D-PAGE using immobilised gels of pH 3–10 (IPG strips, Biorad, Hercules, Calif., USA) as the first dimension and home made 10% acrylamide gels as the second dimension. Proteins were then transferred to PVFD membranes and incubated with antibody against PDX-1 (Chemicon, Temecula, Calif., USA).
Northern blot analysis
Total islet mRNA was obtained by modifying the Chomczynski method [19] (RNAzol B, TEL-TEST, USA) and analysed by northern blot. Briefly, aliquots of 20 µg RNA were electrophorised on a 1% agarose-5% formaldehyde gel and transferred to a nylon membrane (Hybond N, Amersham, Uppsala, Sweden). Membranes were hybridised with [32P] labelled specific cDNA probe for insulin (a gift from G. Bell) and quantified using a Personal FX Phosphoimager and QuantityOne software (BioRad). The values were normalised with a 28S ribosomal cDNA probe.
Statistical analysis
Data are expressed as means ± SEM. Differences between the experimental groups were evaluated using the unpaired Student’s t test for several independent observations. A p value of less than 0.05 was considered significant.
Results
Tungstate treatment normalises blood glucose and insulin plasma concentrations
nSTZ rats showed mild hyperglycaemia (8.4±0.7 mmol/l, Fig. 1). After 4 days of treatment, blood glucose concentrations were already normalised to 6.0±0.3 mmol/l, and these were maintained throughout the treatment (28 days) without any hypoglycaemic episode. In contrast, tungstate administration did not affect blood glucose values in healthy rats. The normalisation of glycaemia in treated diabetic rats was accompanied by an increase in plasma insulin concentrations. Untreated diabetic rats showed lower insulinaemia concentrations than healthy ones (27±4 µU/ml vs 41±4 µU/ml; p<0.05), and this parameter was restored after tungstate treatment (57±5 µU/ml on day 4 and 49±3 µU/ml at the end of treatment). Treatment did not affect insulinaemia in healthy rats (46±4 µU/ml).

Effects of tungstate administration on glycaemia of untreated healthy (open circles), treated healthy (filled circles), untreated diabetic (open squares) and treated diabetic (filled squares) rats. Values are expressed as means ±S EM (n=12 rats/group). *p<0.05, and ** p<0.001 compared to untreated diabetic rats
Treated diabetic rats showed a decrease in food intake (65±2 vs 87±1 g·kg−1·day−1, p<0.05). To determine whether this diminished intake was responsible for the decrease in glycaemia, we did additional pair-feeding studies on diabetic rats. One group of diabetic rats was fed for 15 days with the same amount of chow diet as their treated counterparts. Only tungstate-treated diabetic rats reached normoglycaemic status while their pair-fed untreated counterparts remained hyperglycaemic (Table 1). Concomitantly, only the former showed improved plasma insulin concentrations. These experiments thus ruled out the possibility that tungstate treatment normalised blood glucose concentrations by reducing food intake.
Morphological and functional analysis of pancreatic islets
Tungstate-treated diabetic rats showed higher islet density than their untreated counterparts (27±3 vs 16±2 islets/area; p<0.05). Moreover, pancreatic sections from tungstate-treated diabetic rats showed a recovery of beta-cell mass to exocrine-tissue ratio at the end of treatment (Table 2) to values similar to those found in healthy rats. The beta-cell mass to exocrine-tissue ratio was already partially recovered on day 4 of treatment (1.48±0.09 vs 0.77±0.07%, treated vs untreated diabetic rats; p<0.001). Beta-cell hypertrophy was not observed in either treated-diabetic or treated-healthy rats (Table 2). Moreover, pancreatic weight was not affected by tungstate treatment.
The replication rate of intra-islet beta cells in untreated diabetic rats was higher than in the healthy ones (1.29±0.30 vs 0.76±0.20%; p<0.05). Tungstate treatment did not modify this parameter in either diabetic (1.20±0.17%) or healthy rats (0.77±0.14%). Interestingly, extra-islet beta-cell replication was higher in treated diabetic rats, than in their untreated counterparts (0.71±0.09 vs 0.24±0.06%; p<0.001). Similarly, when the production of new beta cells per day was calculated, it showed that tungstate treatment affected only diabetic rats and that the effects were restricted to the extra-islet beta-cell population (Fig. 2). No differences in α-cell replication were observed in any experimental group.

Effects of tungstate administration on beta-cell cell birth rate. Intra-islet (white columns) and extra-islet (black columns) cell birth rate (CBR) was calculated as described in the methods section. Values are expressed as means ± SEM (n=8 rats/group) from HU (healthy untreated), HT (healthy treated), DU (diabetic untreated), and DT (diabetic treated) rats. *p<0.05 untreated and treated diabetic vs healthy rats. **p<0.001 treated diabetic vs all groups
The increase in beta-cell mass observed in the treated-diabetic rats was mainly due to an increase of cells located outside rather than within well-formed islets. These extra-islet beta cells, individually or in small clusters, were often found close to ducts, indicating an activation of neogenic pathways (Fig. 3a, b). Afterwards, PDX-1 expression was studied in the pancreas of the different experimental groups. On the one hand, we found double positive PDX-1/insulin cells located outside islets in diabetic rats on day 4 of treatment, thereby reinforcing our hypothesis of tungstate-induced neogenesis. On the other hand, single PDX-1 positive cells were also found scattered in the exocrine tissue on day 4 of treatment but were absent on day 15 (Fig. 3e). The initial presence of PDX-1 positive cells, which are negative for other endocrine hormones, together with the subsequent increase in beta-cell density, indicate differentiation from a precursor cell to a mature insulin-producing cell. No co-staining with somatostatin was detected in these PDX-1 positive single cells.

Insulin and PDX-1 expression in pancreatic sections. a Representative confocal image of a propidium iodide (red) and insulin (green) staining of a pancreatic section from a tungstate-treated diabetic rat. This section illustrates the emergence of clusters from a duct (×400). Arrowheads, pancreatic duct; asterisks, clusters of cells. b Same as before, in this image single insulin-positive cells emerging from the duct. Arrowheads, pancreatic duct; asterisks, single cells. c–f PDX-1 expression in pancreatic sections from untreated healthy (c) and untreated diabetic rats (d), and diabetic rats treated for 4 days (e) and 15 days (f). Confocal images of PDX-1 (green) and insulin (red) immunostaining. Arrows indicate extra-islet PDX-1 positive cells (×630). The images are representative from three different rats
Tungstate mechanism of action on islets
PDX-1 protein concentrations were not increased in treated versus untreated rats. The activity of PDX-1 can be regulated by phosphorylation and this can be assessed by two-dimensional electrophoresis (2D) as a decrease in the pI. PDX-1 2D-western blots from isolated islets from healthy rats showed the presence of two different forms of PDX-1 with similar molecular weight but different pI (Fig. 4a, 55±8%). In vitro treatment with tungstate, as well as with glucose or insulin which are known to activate and phosphorylate PDX-1, resulted in a complete shift of PDX-1 towards the most acidic form of PDX-1 (Fig. 4b,c; glucose 88±12, insulin 78±10, and tungstate 94±14%, p<0.05). In islets from diabetic treated rats, tungstate also induced a shift of PDX-1 from basic to more acidic forms (Fig. 5a). Tungstate treatment doubled the amount of PDX-1 in the more acidic forms, compared to PDX-1 forms in the untreated diabetic rats (2.08±0.14, p<0.05), thus suggesting an increase in PDX-1 phosphorylation. Tungstate administration also increased the phosphorylation state of p38 without changing the p38 total concentration (Fig. 5b, ratio phosphorylated p38/ total p38 of 0.11±0.04 in untreated diabetic rats vs 0.41±0.12 in treated diabetic rats, p<0.01).

Effects of tungstate on the in vitro phosphorylation of PDX-1 in islets analysed by 2D western blots. Pancreatic islets were isolated from healthy rats, and were cultured in RPMI medium containing 11.1 mmol/l glucose and 10% FCS during 36 h, followed by 5 h incubation in 1.8 mmol/l glucose and 0.1% FCS and treated with different stimuli for 30 min. The figure is representative of three independent experiments

The 2D western blots were done from isolated islets of 4-day treated rats. a Tungstate decreases PDX-1 pI. b Activation of p38 by tungstate. Controls positive (C+) and negative (C−) were obtained from an insulin cell line (βTC3); the cells were serum and glucose starved and incubated with either media alone (C−) or supplemented with insulin (100 nmol/l, 5 min; C+). c Effect of tungstate treatment on islet insulin mRNA expression in diabetic treated and untreated rats. All figures are representative of three independent experiments
In addition to its involvement in neogenesis, PDX-1 also regulates the expression of several beta-cell genes including insulin. Indeed, we observed an increase in islet insulin content after tungstate treatment. This parameter, which was lower in diabetic rats than in healthy ones (284±22 vs 1102±36 µU/islet; p<0.001), was normalised in treated diabetic rats (918±47 µU/islet). The increase observed in the islet insulin content correlated with a twofold increase in insulin mRNA (Fig. 5c) expression as measured by Northern blot.
Tungstate effects persist after withdrawal of treatment
To study whether the effects induced by tungstate in beta-cell mass were maintained in the long-term, we withdrew treatment after 28 days and did a follow-up study for 15 additional days. Consistent with the half-life of tungstate (approximately 1.7 h in the rat), 36 h after the last administration tungstemia is undetectable. After withdrawing the treatment, previously treated diabetic rats remained normoglycaemic for the following 15 days (Fig. 6). Moreover, at the end of this 15-day period, their plasma insulin concentrations were similar to those of healthy rats (50.0±6.8 µU/ml). Morphometric analyses showed that the pancreas from these rats maintained the same values of beta-cell to exocrine-tissue ratio reached on day 28 of treatment (1.56±0.18%). Therefore, the new beta-cell population induced by tungstate is fully functional and able to maintain glucose homeostasis.

Glycaemia of rats after treatment withdrawal. Blood glucose concentrations were measured in untreated (open squares) and treated (filled squares) diabetic rats for 15 days after treatment. Values are expressed as means ± SEM (n=12 rats/group). *p<0.001 treated diabetic vs untreated diabetic rats
Discussion
In recent years, a line of diabetes research has focused on the development of new strategies addressed to recover beta-cell function and mass arising from our increasing knowledge on adult pancreas plasticity. The dynamics of beta-cell mass involves a tight regulation between cell formation and destruction to obtain the population required to cope successfully with a range of metabolic situations [20]. It has been reported that the nSTZ rat model (the model used in the present paper), partially regenerates beta-cell mass (approximately 40%) during the weaning period, mainly through differentiation from precursor ductal cells (neogenesis) [21]. Our data show that the normoglycaemic effects of tungstate in this model are mediated through a process of pancreatic regeneration leading to total beta-cell mass restoration. This new cell population is functional, allowing the decrease of glycaemia in diabetic rats, and maintaining normoglycaemia even after treatment is withdrawn. These results suggest that tungstate acts by switching on stable beta-cell regeneration.
Since no pancreatic sections from treated rats presented cell hypertrophy, we studied the possible involvement of two processes of beta-cell regeneration: replication and neogenesis [9, 20]. As described before [22], an increase in intra-islet beta-cell replication in diabetic rats was observed, which was not changed by tungstate administration. Since the results clearly ruled out the replication of intra-islet beta cells as the mechanism responsible for the restoration of the beta-cell mass, we studied the involvement of neogenesis. The current criteria to identify this process include the presence of single beta cells or small clusters associated to ducts and/or scattered within the exocrine pancreas, which is what we observed in the treated diabetic rats. In addition, a molecular approach based on the expression of certain transcription factors involved in pancreas development has also been used to study neogenesis [3]. Among these, PDX-1 plays a key role in the recapitulation of this process in adult animals [23]. We present evidence of the involvement of neogenesis as the cause of the cell formation induced by tungstate. The observation of single insulin positive cells or clusters in close contact with ducts suggests an active process of differentiation from precursor cells. Therefore, the initial presence of PDX-1 positive cells, which are negative for other endocrine hormones, together with the subsequent increase in beta-cell density, indicates differentiation from a precursor cell to a mature insulin-producing cell.
The nSTZ model shows active beta-cell mass regeneration during the neonatal period [21]; a regeneration that continues till week 6 of life [22], coinciding with a deterioration in cell function. So it is reasonable to assume that the described effects of tungstate treatment, which started at week 9 of life, could be related to an enhancement of neogenesis, thus lengthening the regenerative period. Thus, assuming that tungstate effects are due to an enhancement of beta-cell neogenesis, our results support the idea of an activation of this pathway. Treatment of nSTZ rats with either phlorizin or vanadate, an insulin-like agent similar to tungstate, decreases hyperglycaemia but fails to normalise beta-cell mass and glucose-induced insulin secretion [24]. Therefore, we can affirm that the effects observed on beta-cell mass in tungstate-treated diabetic rats are due to the direct effects of this compound observed on the pancreas and not to the reduction of glucotoxicity.
Tungstate is a well-known inhibitor of phosphoprotein tyrosine phosphatases (PTP) [25], and the effects described could be mediated by this inhibitory activity. We assessed the molecular mechanism of tungstate action in islets cultured in vitro with tungstate, and also in islets from treated diabetic rats. Activation of PDX-1 involves phosphorylation and this event is dependent on p38 protein [26] and ERK1/2 [27], although some disagreements with the role of PDX-1 phosphorylation have arisen [28]. The control PDX-1 activity may integrate signals from multiple pathways: for example, glucose–stimulated PDX-1 transactivation is ERK1/2 dependent and it has been suggested that p38 cascade could affect PDX-1 in response to other agents [27]. Our data prove that tungstate treatment in diabetic rats is able to activate the islet p38 protein by increasing its phosphorylation state, without changing total p38 concentrations. The 2D experiments show that tungstate decreased the pI of PDX-1 protein both in islets from treated diabetic rats and in cultured islets. The decrease in the pI is probably due to an increase in the phosphorylation state of PDX-1, which decreases the overall charge of the protein. However, we can not discard other post-translational modifications.
Although the results obtained in the diabetic rats treated with tungstate were similar to those found with GLP-1 [29], no differences were observed in the concentrations of GLP-1 between all the rats either treated or untreated. Therefore, it is reasonable to assume that, as with GLP-1, tungstate induces the activation of PDX-1, which is the main regulator of insulin expression, albeit using a different mechanism.
In conclusion, our data demonstrate that the anti-diabetic effect of tungstate in nSTZ rats is mediated through pancreatic regeneration, which leads to a restoration of beta-cell mass. These results suggest that tungstate acts probably by increasing PDX-1 phosphorylation; and, therefore, activity in the islets of treated rats. Our results indicate that sodium tungstate could help us to understand the molecular mechanisms underlying the regulation of glucose homeostasis and endocrine plasticity. The understanding of beta-cell regeneration may point the way towards new strategies for the treatment of diabetic patients with a decreased or impaired beta-cell mass.
Abbreviations
- nSTZ:
-
Neonatal streptozotocin
- PDX-1:
-
pancreatic duodenal homeobox-1
- BrdU:
-
5-bromo-2′-deoxyuridine
References
King H, Aubert RE, Herman WH (1998) Global burden of diabetes, 1995–2025: prevalence, numerical estimates, and projections. Diabetes Care 21:1414–1431
Bloomgarden ZT (2001) European Association for the Study of Diabetes Annual Meeting, 2000: pathogenesis of type 2 diabetes, vascular disease, and neuropathy. Diabetes Care 24:1115–1119
Schwitzgebel VM (2001) Programming of the pancreas. Mol Cell Endocrinol 185:99–108
Jonsson J, Carlssson L, Edlund T, Edlund H (1994) Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 371:606–609
Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF (1997) Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet 15:106–110
Sorenson RL, Brelje TC (1997) Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res 29:301–307
Klöppel G, Löh M, Habich K, Oberholzer M, Heitz P (1985) Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Path Res 4:110–125
Milburn JL Jr, Hirose H, Lee YH et al. (1995) Pancreatic beta-cells in obesity. Evidence for induction of functional, morphologic, and metabolic abnormalities by increased long chain fatty acids. J Biol Chem 270:1295–1299
Bonner-Weir S (2001) Beta-cell turnover: its assessment and implications. Diabetes 50 [Suppl 1]:S20–S24
Zulewski H, Abraham EJ, Gerlach MJ et al. (2001) Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 50:521–533
Barberà A, Rodriguez-Gil JE, Guinovart JJ (1994) Insulin-like actions of tungstate in diabetic rats. Normalization of hepatic glucose metabolism. J Biol Chem 269:20047–20057
Barberà A, Fernandez-Alvarez J, Truc A, Gomis R, Guinovart JJ (1997) Effects of tungstate in neonatally streptozotocin-induced diabetic rats: mechanism leading to normalization of glycaemia. Diabetologia 40:143–149
Muñoz MC, Barberà A, Dominguez J, Fernandez-Alvarez J, Gomis R, Guinovart JJ (2001) Effects of tungstate, a new potential oral antidiabetic agent, in Zucker diabetic fatty rats. Diabetes 50:131–138
Barbera A, Gomis RR, Prats N et al. (2001) Tungstate is an effective antidiabetic agent in streptozotocin-induced diabetic rats: a long-term study. Diabetologia 44:507–513
Portha B, Levacher C, Picon L, Rosselin G (1974) Diabetogenic effect of streptozotocin in rat during the perinatal period. Diabetes 23:889–895
Guest P, Rhodes CJ, Hutton JC (1989) Regulation of the biosynthesis of insulin-secretory-granule proteins. Co-ordinate translational control is exerted on some, but not all, granule matrix constituents. Biochem J 257:431–437
Swenne I (1982) The role of glucose in the in vitro regulation of cell cycle kinetics and proliferation of fetal pancreatic B-cells. Diabetes 31:754–760
Barceló-Batllori S, André M, Servis C et al. (2002) Proteomic analysis of cytokine induced proteins in human intestinal epithelial cells: implications for inflammatory bowel diseases. Proteomics 2:551–560
Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159
Bernard-Kargar C, Ktorza A (2001) Endocrine pancreas plasticity under physiological and pathological conditions. Diabetes 50 [Suppl 1]:S30–S35
Wang RN, Bouwens L, Klöppel G (1994) Beta-cell proliferation in normal and streptozotocin-treated newborn rats: site, dynamics and capacity. Diabetologia 37:1088–1096
Wang RN, Bouwens L, Klöppel G (1996) Beta-cell growth in adolescent and adult rats treated with streptozotocin during the neonatal period. Diabetologia 39:548–557
Sharma A, Zangen DH, Reitz P et al. (1999) The homeodomain protein IDX-1 increases after an early burst of proliferation during pancreatic regeneration. Diabetes 48:507–513
Serradas P, Bailbe D, Blondel O, Portha B (1991) Abnormal B-cell function in rats with non-insulin-dependent diabetes induced by neonatal streptozotocin: effect of in vivo insulin, phlorizin, or vanadate treatments. Pancreas 6:54–62
Stankiewicz PJ, Gresser MJ (1988) Inhibition of phosphatase and sulfatase by transition-state analogues. Biochemistry 27:206–212
Elrick LJ, Dockerty K (2001) Phosphorylation-dependent nucleocytoplasmic shuttling of pancreatic duodenal homeobox-1. Diabetes 50:2244–2252
Khoo S, Griffer SC, Xia Y, Baer RJ, German MS, Cobb MH (2003) Regulation of insulin gene transcription by ERK1 and ERK2 in pancreatic β cells. J Biol Chem 278:32969–32977
Moede T, Leibiger B, Pour HG, Berggren PO, Leibiger IB (1999) Identification of a nuclear localization signal, RRMKWKK, in the homeodomain transcription factor PDX-1. FEBS Lett 461:229–234
Perfetti R, Zhou J, Doyle ME, Egan JM (2000) Glucagon-like peptide-1 induces cell proliferation and pancreatic-duodenum homeobox-1 expression and increases endocrine cell mass in the pancreas of old, glucose-intolerant rats. Endocrinology 141:4600–4605
Acknowledgements
The authors thank Dr. A. Bosch (Serveis Científico-Tècnics, Universitat de Barcelona) for technical assistance in confocal microscopy, A. Truc and M. Julià for their technical assistance, Dr. J.F. Habener and Dr. G. Bell for generously providing PDX-1 antibody and insulin cDNA probe respectively, Dr. J. Ferrer for helpful criticism of the results and M. Maudsley and T. Yates for correcting the manuscript. This study was supported by grants from Ministerio de Ciencia y Tecnología SAF 2000/0053, Marató TV3, and FIS 02/0483, Red de Centros C03/08, and RGDM G03/212 from Ministerio de Sanidad y Consumo (Spain). J. Fernandez-Alvarez was a recipient of a grant from the Fundació Biomedica Clínic, B. Nadal was a recipient of a grant from the Generalitat de Catalunya and M. Claret was a recipient of a grant from the Ministerio de Ciencia y Tecnologia of the Spanish government.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fernández-Alvarez, J., Barberà, A., Nadal, B. et al. Stable and functional regeneration of pancreatic beta-cell population in nSTZ-rats treated with tungstate. Diabetologia 47, 470–477 (2004). https://doi.org/10.1007/s00125-004-1332-8
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1332-8