
Abstract
Key message
The major quantitative trait loci associated with bunch weight and its component traits in the East African highland banana-breeding population are located on chromosome 3.
Abstract
Bunch weight increase is one of the major objectives of banana improvement programs, but little is known about the loci controlling bunch weight and its component traits. Here we report for the first time some genomic loci associated with bunch weight and its component traits in banana as revealed through a genome-wide association study. A banana-breeding population of 307 genotypes varying in ploidy was phenotyped in three locations under different environmental conditions, and data were collected on bunch weight, number of hands and fruits; fruit length and circumference; and diameter of both fruit and pulp for three crop cycles. The population was genotyped with genotyping by sequencing and 27,178 single nucleotide polymorphisms (SNPs) were generated. The association between SNPs and the best linear unbiased predictors of traits was performed with TASSEL v5 using a mixed linear model accounting for population structure and kinship. Using Bonferroni correction, false discovery rate, and long-range linkage disequilibrium (LD), 25 genomic loci were identified with significant SNPs and most were localized on chromosome 3. Most SNPs were located in genes encoding uncharacterized and hypothetical proteins, but some mapped to transcription factors and genes involved in cell cycle regulation. Inter-chromosomal LD of SNPs was present in the population, but none of the SNPs were significantly associated with the traits. The clustering of significant SNPs on chromosome 3 supported our hypothesis that fruit filling in this population was under control of a few quantitative trait loci with major effects.
Similar content being viewed by others


Introduction
The ability of banana to produce seedless fruits was the key to banana domestication along with other attributes such as plant vigour, which was a consequence of polyploidization (Simmonds 1962; Heslop-Harrison and Schwarzacher 2007; Cenci et al. 2019). Deciphering the genetic basis of expressed phenotypes is important for breeders to make useful gene introgression that lead to high-yielding and resilient crops. Yield in banana is evaluated by measuring bunch weight per unit area and over a defined time, often 1 year. The components of bunch weight include a number of traits such as number of hands and fruits; fruit length and circumference; and the diameter of both fruit and pulp (Nyine et al. 2017). Fruit filling refers to the sink capacity of the fruit that is directly proportional to the pulp content, or the edible part of banana. Fruit circumference and length indicate how well the banana fruits are filled with pulp. Fruit filling is one of the main traits that banana breeders use in the preliminary selection of hybrids because it contributes significantly to the bunch weight. Simmonds (1962) reported that parthenocarpy in banana was controlled by three genes designated as P1, P2 and P3, while Turner and Gibbs (2018) described the process of bunch formation and asserted that photosynthate availability plays an important role in regulating the number of fruits and hands on the developing banana inflorescence. There is still, however, a knowledge gap concerning the genetic factors regulating bunch component traits in this crop with important consequences to both food security and economic development in emerging markets.
In well-investigated monocots such as wheat, barley and rice, grain yield has been shown to be controlled by a network of genes involved in plant height determination, flowering regulation, floral architecture, and grain number, width and length (Nadolska-Orczyk et al. 2017; Dixon et al. 2018; Zhang et al. 2018). The available literature shows that in Musa acuminata, MCM1-AGAMOUS-DEFENSINS-SRF (MaMADS) genes, MYB and AP2/ERF transcription factors are involved in fruit architecture, development and ripening (D’Hont et al. 2012; Liu et al. 2017). In plantain hybrids, variation in fruit size and parthenocarpy were linked to the segregation of one dominant parthenocarpy gene P1 (Ortiz and Vuylsteke 1995). Moreover, in triploid banana clones (AAA genomes), three dominant P genes have been reported to control parthenocarpy. A parthenocarpic diploid clone Pisang Lilin (AA genome), was shown to be heterozygous for all the three P genes while variation in the number of dominant alleles has been reported in other diploids (Simmonds 1962). In spite of the available genomic resources, the location and mode of action of these P genes are not fully understood.
Wild diploid progenitors of cultivated banana are the main sources of host plant resistance to pathogens and pests. They have, however, inferior fruit characteristics that are often inherited together through linkage drag with the resistance genes during the breeding process. The consequence of this is that hybrids from breeding programs are sometimes less acceptable by the farmers due to substandard fruit qualities when compared to the existing cultivars (Ortiz et al. 1995). Production of seedless fruit by hybrids arising from non-parthenocarpic and parthenocarpic parents is not unusual (Swennen and Vuylsteke 1993; Vuylsteke et al. 1993a, b), but the majority contain very little, or no pulp. The lack of pulp (poor fruit filling), is one of the main reasons why approximately 90% of hybrids generated in crossbreeding programs are discarded by breeders in early evaluation trials. Early identification of hybrids with poor fruit filling characteristics will save considerable breeding resources. Research by Sardos et al. (2016) noted several loci associated with the seedless phenotype in a panel of 105 diploids, but the disconnection between seedlessness and fruit filling in triploid breeding populations raises an important question, whether the quantitative trait loci (QTL) controlling seedless fruit production are the same as those responsible for fruit filling. Simmonds (1953) pointed out that there was no absolute relationship between seed sterility and parthenocarpy in bananas. In rice, a large effect QTL on chromosome 7 was associated with grain width and length and was complemented by a QTL with smaller effect on chromosome 2 (Begum et al. 2015). Grain weight in wheat is controlled by the TaGW2 gene, an orthologue of the rice gene OsGW involved in grain development. It is known to be a negative regulator of grain weight by modulating cell number and length (Zhang et al. 2018). Low expression of this gene results in increased seed width and reduction in seed length, but generally increases the thousand kernel weight. Perhaps, similar mechanisms are involved in the control of banana fruit size, but such genome-wide association studies (GWAS) or association genetics research have not been attempted in banana.
GWAS relies on high-density molecular markers such as single nucleotide polymorphisms (SNPs) to detect historical and present recombination events mostly in diversity panels (Huang and Han 2014; Begum et al. 2015; Sardos et al. 2016). This makes GWAS a useful tool for identifying genomic loci that are associated with traits of interest (Platt et al. 2010). The number of potentially useful SNPs available for analysis in GWAS is dependent on the genotyping platform, and size and genetic diversity of the utilized population. SNPs can be increased by imputation from a densely genotyped reference panel into a low-density genotyped target population provided identity by descent exists between them (Browning and Browning 2016). SNPs arise from mutations that occur in the life history of a species and the persistence of a certain pattern in a population in disequilibrium to a given trait can be detected through GWAS. However, care must be taken to ensure that genetic heterogeneity due to population structure, relatedness, variation in phenotyping and genotype ascertainment bias do not confound the GWAS results if they are to be reproducible (McCarthy et al. 2008).
The power of GWAS depends on the ability to detect non-random association of SNP alleles linked to the genes that control the trait of interest. This is commonly referred to as linkage disequilibrium (LD). LD has been utilized in human GWAS to identify causal alleles for various diseases (Kerem et al. 1989; Corder et al. 1994) and in plants to identify QTL that control important agronomic traits including yield, or yield components (Buckler et al. 2009; Peiffer et al. 2014; Begum et al. 2015; Sardos et al. 2016). Changes in LD (LD decay) are influenced by the mating design in the population, the frequency of recombination, the distribution of recombination hotspots and population structure (Flint-Garcia et al. 2003; Dyer et al. 2007; Sabeti et al. 2007). LD decay has been shown to vary widely among species and even between subpopulations of the same species and can range from a few hundred base pairs to several hundred kilobase pairs (Flint-Garcia et al. 2003). In a diploid banana diversity panel, rapid LD decay was observed between 10 and 100 kb, but long-range LD was also noted (Sardos et al. 2016).
Inter-chromosomal LD also referred to as genome-wide LD is a phenomenon that reflects the complex control of traits through several regulatory pathways and epistasis. For this reason, Boyle et al. (2017) proposed an ‘omnigenic’ model to account for the effect of modifier genes outside the core pathways involved in the expression of complex phenotypes. Although the use of this concept of inter-chromosomal LD is not widespread in GWAS, careful examination of this concept could uncover the mystery of “missing heritability” in GWAS (Manolio et al. 2009). Indeed, some research articles indicate the existence of non-homologous LD in animals and plants (Farnir et al. 2000; Rostoks et al. 2006; Kulminski, 2011). Using long-range and inter-chromosomal LD, SNPs whose probability (P) value is below the threshold of significant association with the trait could be identified. In humans, this concept has been exploited to highlight the role of autophagy in Crohn’s disease, the role of adipocyte thermogenesis and central nervous genes in obesity (Jostins et al. 2012; Claussnitzer et al. 2015; Locke et al. 2015).
In this study, we sought to understand the genetics underlying bunch weight and its component traits in banana through GWAS. Following the high predictive ability of fruit filling traits (> 0.7), in the genomic selection training population (Nyine et al. 2018), we hypothesized that fruit filling was under control of a few QTL with major effects. The objectives were to identify the genomic loci associated with bunch weight and its component traits, the genes closest to, or containing significantly associated SNPs, the level of LD that exists between and within loci associated with the traits and how fast LD decays along the chromosomes having significant marker–trait associations in comparison to the rest of the chromosomes. We also wanted to find out if inter-chromosomal LD exists in banana and to what level could it be utilized to identify the QTL for complex traits such as bunch weight.
Materials and methods
Breeding population
The banana population used in this study was described in detail by Nyine et al. (2017) and Nyine et al. (2018). In brief, the population consisted of 307 genotypes from an East African highland banana (EAHB) breeding program of the International Institute of Tropical Agriculture (IITA, Namulonge, Uganda). The population was made up of 31 breeding clones (11 diploids, 12 triploids and 8 tetraploids), and 276 hybrid offspring most of which were triploids. Three fields were established using a completely randomized design with three replications per genotype. Two fields were established at Namulonge in central Uganda, at 0.53°N 32.58°E, while the third field was established in Western Uganda at the Mbarara Zonal Agricultural Research Development Institute, 0.60°S, 30.58°E. One field at Namulonge was maintained under low input management (no mulching and no application of synthetic fertilizer), while the second field and the one at Mbarara were maintained under high input management, which included mulching and addition of 480 g of nitrogen, phosphorus and potassium (NPK), fertilizer per mat per year mixed in the ratio of 25:5:5. Data for three crop cycles were collected from the three fields in the period between 2013 and 2018 because the field in Mbarara was established in 2015, and generally, bananas do not have synchronized flowering and harvesting period.
Phenotype data
Bunch weight component traits data used in the study included the number of hands and fruits; fruit length and circumference; and diameter of both fruit and pulp. Detailed description of these traits is documented by Nyine et al. (2017). A linear mixed-effect model with restricted maximum likelihood implemented in R-package lme4 (R Core Team 2019) was used to fit the phenotype data as follows,
In the equation, yijk is the phenotype of the ith genotype in jth field and kth crop cycle, μ is the intercept, Gi is the ith genotype, Ej is the jth field, Ck is the kth cycle, GECijk is the genotype by field by crop cycle interaction and εij is random residual. The effect of field was fixed, while the effects of cycle and genotype were random. The variance components from the mixed-effect linear model were used to calculate the broad sense heritability (H2) of the bunch weight and its component traits using the equation below.
where VG is the variance of the genotype, VC is the variance of the crop cycle, VGEC is the variance of the genotype interaction with the field and crop cycle and \( {V_\varepsilon } \) is the variance of random residual. Best linear unbiased predictors (BLUPs) were extracted from the model as random effects of the genotype and used as pseudo-phenotypes for GWAS.
Genotype data
The population was genotyped using the genotyping by sequencing (GBS) approach (Elshire et al. 2011) and ensuing data were processed as described by Nyine et al. (2018). Raw vcf files for the SNP data can be accessed from ftp://ftp.musabase.org/musaGBS/Nyines2018/. Raw SNPs with minor allele frequency (MAF) below 0.05 and maximum missingness above 75% were filtered out using vcf-filter tools, which resulted in 27,178 bi-allelic SNP sites based on double-haploid Pahang genome sequence assembly v2 (Martin et al. 2016). The missing SNPs were imputed using Beagle software program v5.0 (Browning and Browning 2016). A custom Perl script was used to convert the SNP vcf file to haplotype map (hmp) format required for downstream analysis based on IUPAC nucleotide nomenclature.
Population structure and kinship
The SNP hmp file was imported in TASSEL v5 (Bradbury et al. 2007) and the first three principal components (PCs) were calculated. Genotypes were plotted on the first two PCs in order to delineate the population structure using R-package ggplot2. Hybrid genotypes and the parental lines were identified using different colours on the plot and ellipses were used to identify individuals belonging to the same group by assuming a multivariate normal distribution. Similarly, the kinship coefficients were calculated from the SNP data and used together with the first three PCs to correct for any possible confounding of trait–marker association due to population structure and relatedness.
Linkage disequilibrium (LD)
In order to determine the number of possible loci that control bunch weight and its component traits in banana, pairwise LD between SNPs were determined with the squared coefficient of correlations between alleles (r2) and the standardized disequilibrium parameter (D′) calculated in TASSEL v5 using an LD sliding window size of 50. A threshold r2 value was set at 0.1, below which SNP pairs were considered to have weak LD. All SNPs that had a significant association with bunch weight and its component traits were compared with the rest of the SNPs. SNPs that had LD equal, or above 0.1 with the significant SNPs were considered to be associated with the trait even though their P values were above the statistically significant threshold. Intergenic SNPs within 1 to 10 kb window were considered to be on the same locus. LD decay plots of r2 against the physical distance in base pairs (bp) between SNPs from all chromosomes and from individual chromosomes were generated using R-package ggplot2. A smoothened LD curve generated from generalized additive model (GAM) implemented by geom_smooth option of ggplot2 was added to the LD plot for proper visualization of the LD decay trend. Inter-chromosomal LD was searched in the LD data to determine whether some SNPs had an r2 equal, or above 0.1 with SNPs significantly associated with the traits. The number of SNPs with r2 equal, or above 0.1 was recorded together with the D′ values for each chromosome pair.
Genome-wide association study
The mixed linear model (MLM) implemented in TASSEL v5 including both kinship matrix (K) and the first three PCs to account for population structure (PCA) was used to test for the marker–trait association. BLUPs were used as pseudo-phenotypes for bunch weight and its component traits. The quartile–quartile (Q–Q) plots for the negative log10-transformed expected and observed P values for the SNPs were generated for each trait using R-package ‘qqman’ (R Core Team 2019). These were used to visualize any deviance from the null hypothesis of no genetic association between SNPs and the trait. Manhattan plots for the negative log10-transformed raw P values and false discovery rate (FDR) adjusted P values against each chromosome were also generated for each trait using R-package qqman. Bonferroni correction was applied to the raw P values at 5% significance level (α = 0.05), and the threshold P value was 1.84e−6 (0.05 divided by 27,178 SNPs), for the SNPs that were significantly associated with the traits. The results from Bonferroni correction were compared to those of FDR at a threshold q value of 0.05. In order to eliminate any biases due to ploidy level effect, the diploids and tetraploids were excluded from the data set and GWAS was performed only on the triploids since they constituted more than two-thirds of the entire population and results were compared to those of the whole population based on FDR q values. Lastly, the level of stringency was reduced to a threshold q value of 0.1 so that more SNPs potentially associated with bunch weight and its component traits could be identified.
Linking significant SNP to putative genes
The gff3 file for double-haploid Pahang genome sequence assembly v2 (Martin et al. 2016) was downloaded from https://banana-genome-hub.southgreen.fr/. Using a custom Perl script, bed files were generated for the gene models from gff3 and the SNPs that were significantly associated with bunch weight and its component traits. The closest-features option from BEDOPS v2.4.35 tools (Neph et al. 2012), was used to determine the gene that contained, or was closest to the significant SNP. The annotation of the gene was obtained from the gff3 file using the gene identifier. To further gain a deeper understanding, the effects of all SNPs used in this study were annotated using SnpEff software (Cingolani et al. 2012). The database for double-haploid Pahang genome sequence assembly v2 was locally built in SnpEff software following the user manual before the annotation was done because the default settings could only allow access to the first version of the Musa genome present at Ensembl genomes website.
Results
Heritability and population structure
After filtering the data for missing entries and obvious scoring errors, 297 accessions were retained that had data from all the three experimental fields. These accessions were used to calculate the variance components of the variables, BLUPs and other downstream analyses (Supplementary material Table S1). The H2 for bunch weight was 0.92, number of hands was 0.88, number of fruits was 0.83, fruit length was 0.98, fruit circumference was 0.98, fruit diameter was 0.98 and pulp diameter was 0.97.
PCA of these accessions based on 27,178 SNPs did not reveal a clear distinct population structure amongst the hybrids assuming a multivariate normal distribution (Fig. 1). However, they showed a wide distribution on the two PCs, which was indicative of the high genetic variability despite some shared pedigree. Among the parents, the triploid EAHB landraces formed a unique cluster away from the rest of the population, while the tetraploids clustered together amongst the hybrids. The diploid parents had the widest spread on the two PCs (Fig. 1).

Distribution of genotypes in the study population on the first and second principal components. Where diploid (red), triploid (green) and tetraploid (magenta) are the parents that generated the hybrids (blue) (color figure online)
Linkage disequilibrium
Pairwise analysis of LD between 27,178 SNPs in TASSEL v5 produced 1,357,625 comparisons. The number of chromosome-specific pairwise SNP LD was 1,287,876 out of which only 4.6% (59,263) had r2 equal, or above 0.1. The remaining pairs were from inter-chromosomal LD and unanchored scaffolds (labelled as chromosome U). The average r2 and D′ for the 4.6% pairs were 0.29 ± 0.26 and 0.73 ± 0.22, respectively. Inter-chromosomal LD SNP pairs between the 11 chromosomes were 11,281 out of which only 0.5% (56) had r2 equal, or above 0.1. The average r2 and D′ for the 0.5% pairs were 0.15 ± 0.04 and 0.53 ± 0.14, respectively. The smoothened LD curve from GAM for all SNP pairs showed a very sharp drop in r2 within the first 10 kb followed by a slight increase before stabilizing (Fig. 2). The average r2 for the first 100 kb was 0.38 ± 0.31. However, long-range LD with r2 equal, or above 0.1 was observed across all chromosomes and it varied from 1.9 Mb on chromosome 11 to 7.5 Mb on chromosome 7 (Table 1). Only 0.72% of SNP pairs had long-range LD with r2 equal, or above 0.5 at a distance ranging between 0.9 Mb on chromosome 7 and 4.1 Mb on chromosome 5 (Table 1). Chromosome 3 had a unique trend in the LD smoothened curve because after 1.5 Mb, it started to show an increase in r2, while other chromosomes remained constant, or slightly reduced (Supplementary material Fig. S1). The average LD distance (and standard error) between SNPs significantly associated with bunch weight and its component traits based on the entire population and triploid subset was 0.97 ± 0.1 Mb (1 bp to 2.8 Mb), while the average r2 was 0.28 ± 0.03 (0.003 to 1) and D′ was 0.63 ± 0.03 (0.16 to 1).

Linkage disequilibrium plot of r2 against genomic distance between all SNP pairs. The smoothened curve (red) was generated using generalized additive model implemented in gglpot2 geom_smooth function (color figure online)
Marker–trait association
Mixed linear model accounting for both population structure and kinship, MLM (PCA + K) was used to identify the marker–trait association. The Q–Q plots showed deviation from the null hypothesis, thus indicating the presence of a significant association between SNP markers and bunch weight, and its component traits (Fig. 3a, Supplementary material Fig. S2). To identify the significant SNPs, we used both the Bonferroni correction at a threshold P value of 1.84e−6 and an FDR at a threshold q value of 0.05. All SNPs above the threshold line (red line) on the Manhattan plots were considered to be strongly associated with the traits (Fig. 3b, Supplementary material Fig. S3). Based on Bonferroni correction, 20 SNPs were associated with the number of hands, fruit length and circumference; and the diameter of both fruit and pulp (Table 2, Supplementary material Table S2), and no SNPs were associated with bunch weight and number of fruits. However, based on FDR, 32 SNPs were in strong association with bunch weight, number of hands; fruit length and circumference; and the diameter of both fruit and pulp (Table 2, Supplementary material Fig. S4). Two SNPs on chromosome 10 were weakly associated with the number of fruits based on FDR. Altogether, 34 out of the 27,178 SNPs used in the study, distributed across 20 loci located on five chromosomes and on unanchored scaffolds were significantly associated with bunch weight and its component traits. Twenty-six of the significant SNPs were located on chromosome 3. Chromosomes 6, 9 and 11 had one significant SNP each, while two SNPs were located on chromosome 10 and three SNPs were on unanchored scaffolds (U). The location of SNPs, the putative genes to which they mapped, or that was closest to them and the effect of the SNPs on the traits are summarized in (Supplementary material Table S3). Most of the SNP alleles had a negative effect on the traits.

Single nucleotide polymorphisms (SNPs) associated with fruit circumference. a Quartile–quartile (Q–Q) plot showing deviation from null hypothesis of no significant association, b Manhattan plot showing location of significant SNPs, the red line indicates the Bonferroni correction threshold at a P value of 1.84e−6 (color figure online)
Eight out of the 26 significantly associated SNPs on chromosome 3 are located in, or are near the genes that encode either hypothetical or uncharacterized proteins. Two SNPs mapped to MYB-related protein 308-like gene, which belongs to a family of transcription factors involved in various cellular processes including cell development. Three SNPs are located upstream of the transcription factor bHLH49 known to regulate cell elongation and expansion in Arabidopsis. Two SNPs mapped to proline-rich protein HaeIII known to be involved in stress response and cell wall development, one SNP mapped to kinesin-like protein while another one mapped to auxilin-like protein, both involved in intracellular transport. The remaining SNPs on chromosome 3 mapped to F-Box protein, pentatricopeptide repeat-containing protein, random slug protein, putative triactylglycerol lipase 2 and muscle M-line assembly protein unc-89-like. One of the three SNPs on unanchored scaffolds mapped to a gene encoding ribokinase, while the other two SNPs are located in a gene encoding 2-aminoethanethiol dioxygenase-like protein.
GWAS with triploid genotypes
When GWAS was performed with the triploid subset, most of the SNPs identified using the entire population were also identified in this subset at an FDR threshold q value of 0.05 (Supplementary material Tables S2, S3). However, some SNPs that showed association in the whole population (WP) were below the threshold yet, 14 new SNPs were identified to be significantly associated with fruit filling traits and these were all located on chromosome 3. One of the 14 SNPs (c3_17545341), however, was significantly associated with fruit circumference and pulp diameter in the WP but in the triploid subset, it was only significantly associated with fruit diameter (Supplementary material Table S2). These SNPs were distributed on six loci with half of them mapped to the genes such as UPF0481 protein At3g47200-like, enhancer of polycomb-like protein 1 and TIMELESS-interacting protein known to be involved in DNA replication and repair, and cell cycle regulation. The other half of the SNPs mapped upstream of ervatamin-B-like, hypothetical protein and membrane-anchored ubiquitin-fold protein 3-like genes (Supplementary material Table S3). Interestingly, some of the 14 SNPs had strong LD with SNPs that had a significant association with bunch weight and its component traits based on WP.
Inter-chromosomal and long-range LD SNPs
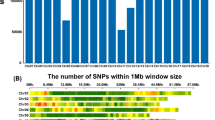
Only 56 SNP pairs had inter-chromosomal LD with r2 equal, or above 0.1 from all possible chromosome combinations and none of them was in LD with SNPs significantly associated with bunch weight and its component traits. However, 87 nonsignificant SNPs on chromosome 3 were in LD with SNPs significantly associated with bunch weight and its component traits at r2 equal, or above 0.1 (Supplementary material Table S4). Gene models closest to these SNPs were not different from those of the significant SNPs. The distance between significant SNPs on chromosome 3 varied from 1 bp to 2.9 Mb with some distant SNP pairs showing high LD. One interesting region was between 15,110,500 and 17,864,044 bp where the r2 values between the two SNP pairs; c3_15110553 and c3_17857961, c3_15110553 and c3_17857966 were 0.42 and 0.41, respectively, despite a 2.75 Mb distance between them. This explains the trend observed from the LD plot of this chromosome where the smoothened curve started to increase at around 15 Mb. SNP c3_15110553 is located in the promoter region of uncharacterized LOC103978321 gene (Ma03_g15320), while c3_17857961 and c3_17857966 are both located in the coding region of hypothetical protein ~ At3g50860 (Ma03_g16130). These three SNPs have two haplotypes TTA and CCG, which show positive and negative effect on fruit filling related traits, respectively (Supplementary material Table S3). When the interval was scanned, 82 genes were identified and a majority of them were either annotated as encoding uncharacterized, or hypothetical proteins (Supplementary material Table S5). Several genes encoding proteins such as AP2-like/ERF, transcription factor bHLH18-like and bHLH10-like, growth regulating factor 6, auxin-responsive protein IAA1, endoglucanase 24-like, CDPK-related kinase 3-like, ervatamin-B-like, E3 ubiquitin-protein ligase SINAT5-like and others were, however, present. Since most of these genes are involved in cell cycle regulation, cell wall development, cell elongation and expansion, signal transduction and transcriptional regulation of other genes, it is likely that differential expression of these genes affects different hybrid genotypes in diverse ways, resulting in the source to sink capacity variation. Using a 1 Mb sliding window, the SNP count in the interval was only 69 barely above the average of 66 SNPs per 1 Mb for the whole genome (Supplementary material Fig. S5). The region between 16 and 17 Mb on chromosome 3 had only 4 out of the 69 SNPs, which limited the possibility to detect any significant associations in the region. The presence of long-range LD suggested, therefore, that this interval bears genes that could affect bunch weight and its component traits.
Annotation of SNPs
After relaxing the level of stringency to a q value of 0.1, 19 new SNPs were identified that were significantly associated with bunch weight and its component traits. Thirteen of the SNPs were located on chromosome 3, while chromosomes 4, 6, 9 and 10 had one SNP each, and two SNPs were located on the unanchored scaffolds (Supplementary material Table S6). However, linking these SNPs to genes did not reveal any new genes but mapped to genes such as TIMELESS-interacting protein and others that had been already identified.
When the 27,178 SNPs were annotated using SnpEff software, 12,156 SNPs had functional effects (File S1). There were 53.37% (6488) missense SNPs, 45.76% (5562) silent SNPs and 0.87% (106) nonsense SNPs giving a ratio of 1.17, missense/silent mutations. However, when the base changes (SNPs) were analysed, the ratio of transitions/transversions (Ts/Tv) was 1.37 indicating the presence of a high proportion of SNPs that cause amino acid changes in the genes. At high stringency threshold P value and q value, we identified four missense SNPs on chromosome 3 (Supplementary material Table S6). Three of them (chr03_14800401, chr03_14800413 and chr03_14800497) were located in the TIMELESS-interacting protein (Ma03_g15020), while one (chr03_11198135) was present in MYB-related protein 308-like (Ma03_g14020). These two genes are known to play a role in cell cycle regulation.
Discussion
Population structure in breeding population
Association genetics should rely on high-density genetic markers evenly distributed across the genome to identify QTL associated with traits of interest. In this study, we used 27,178 SNP markers with a minimum minor allele frequency of 0.05 to find association with bunch weight and its component traits in a banana-breeding population derived from the triploid EAHB as grandparents. The average marker density was 66 SNPs per 1 Mb window across all 11 chromosomes, but variations along the chromosomes were noted with a reduction towards the centromeric region. We did not find a clear population structure in our population based on PCA especially amongst hybrids (Fig. 1). The triploid EAHBs formed, however, a unique cluster indicating low genetic diversity between them. Kitavi et al. (2016) and Němečková et al. (2018) showed that this subgroup of bananas is genetically uniform and postulated that it arose from single clone with M. acuminata ssp. zebrina and ssp. banksii being the most probable parents. The tetraploids that arose from them also clustered together indicating that they had a high genetic dosage from the EAHB parents (Batte et al. 2019; Brown et al. 2017; Nyine et al. 2017). The lack of a clear population structure within the hybrids can be attributed to their shared complex pedigree. And the wide distribution of hybrids on the two components reflects high diversity among the male parents.
The distribution and the frequency of marker alleles carried by different subpopulations in diversity panels commonly used in most association studies are influenced by evolutionary dynamics such as mutation rates, gene flow, recombination frequency and geographical isolation among others (Zhao et al. 2011). These evolutionary forces can lead to population structure characterized by genetic heterogeneity and cryptic relatedness that confound the marker-trait associations in such panels (Korte and Farlow, 2013). In breeding populations, however, genetic heterogeneity may not be strong due to the influence of the population structure, but relatedness between individuals is usually high, which can lead to false-positive association (Korte and Farlow, 2013). Mixed linear models accounting for both population structure and kinship have been used in GWAS to minimize false-positive association (Yu et al. 2006; Zhao et al. 2007; Kang et al. 2008; Zhang et al. 2010; Müller et al. 2019), thereby leading to identification of candidate genes and associated markers that can be used in marker-assisted selection for traits in both plant and animal breeding programs. This approach was used to identify the mutations in the dwarf8 gene that controls flowering time and plant height in maize (Thornsberry et al. 2001), and candidate genes that could be involved in seedless fruit production in bananas (Sardos et al. 2016). We used the first three PCA and kinship matrix derived from the SNP markers to correct for any spurious association followed by the stringent Bonferroni and a relatively relaxed FDR correction method. Hence, we expect that the associations detected in our study point to loci involved in the control of bunch weight and its component traits in banana. Further research including knockout or editing of genes having missense SNPs that are significantly associated with bunch weight and its component traits could lead to identifying genetic factors controlling yield in banana and the development of DNA markers that can be used routinely for selection of hybrids.
Linkage disequilibrium
Association genetic analysis on diversity panels usually captures linkage disequilibrium that has persisted through historical recombination (Flint-Garcia et al. 2003). In breeding population, both historical and present recombination events are captured. Hence, the results from such analysis are likely to have more immediate application in breeding programs than those from diversity panels that require validation (Begum et al. 2015). In this study, we used a breeding population that was composed of many half-sib families derived from related female parents (Kitavi et al. 2016; Němečková et al. 2018), but diverse male parents. We observed a rapid decay in LD within the first 10 kb on each chromosome, which was lower than that observed in the diploid diversity panel for seedless phenotype in banana (Sardos et al. 2016). The trend of LD decay based on the smoothened curves generated by GAM was comparable to that observed by Sardos et al. (2016), where rapid LD decay was followed by long-range LD. Tremendous differences in LD decay were reported in various species. In humans, LD decay was reported to vary from 60 to 500 kb, in other animals it varied from 1 kb to several centimorgans (Remington et al. 2001). In maize, LD reduced to less than 25% within the first 200 bp, yet in the diverse inbred lines of maize, rapid LD decay was observed at 1500 bp (Remington et al. 2001), while in Arabidopsis thaliana LD decayed within 250 kb to several megabases depending on the population and gene under selection (Flint-Garcia et al. 2003).
Long-range LD varied from 1.9 to 7.5 Mb in our population. The variation in LD has been attributed to mating designs in population such as outcrossing versus selfing, rate of recombination and the distribution of recombination hotspots, population bottlenecks during domestication, locked genomic portions due to presence of repetitive sequences and continuous selection of domestication genes (Labate et al. 2000; Jeffreys et al. 2001; Rafalski 2002; Weil, 2002; Whitt et al. 2002; Jordan et al. 2018). Unlike highly selfing species such as Arabidopsis and wheat, banana can maintain long-range LD blocks due to its vegetative propagation that limits the number of recombination events. Hence, the high LD observed in this study could be an indication of the selection pressure that banana underwent during the domestication process and has been maintained by vegetative propagation. The occurrence of large linkage blocks could be a problem for banana improvement if unfavourable alleles are present in such blocks, but if they contain only favourable alleles, fixation of the traits controlled by those loci in the hybrids becomes easy. The reduction in LD decay range compared to the diversity panel of Sardos et al. (2016), could be due to recent meiotic recombination following cross-pollinations. In Drosophila recens, genome-wide LD observed within the driving X chromosome (XD) was attributed to the presence of multiple inversions that suppressed recombination of XD with wild type thus generating non-random association between alleles on a 130 cM haplotype (Dyer et al. 2007).
Although we observed some inter-chromosomal LD between SNP markers, none of them had any significant association with bunch weight and its component traits, indicating that this phenomenon may not play a role in bunch weight and its component traits in banana. Kulminski (2011) observed, however, inter-chromosomal LD in both parental and offspring generation of Framingham heart study participants and concluded that it was caused by biogenetic mechanisms possibly associated with favourable, or unfavourable epigenetic evolution. Further research using other populations with different genetic background and traits will be therefore required to rule out the role of inter-chromosomal (genome-wide) LD in banana trait-marker associations.
Markers associated with bunch weight and its component traits
After accounting for population structure and kinship in the mixed linear model, we identified a total of 34 SNPs using the entire population and an additional 14 SNP using the triploid subset and when combined together, 47 unique SNPs were significantly associated with bunch weight and its component traits (Table 2). Altogether, they mapped to 25 genomic loci located on chromosomes 3, 6, 9, 10 and 11, and on unanchored scaffolds, the majority of which were located in protein-coding regions while a few were in the promoter, or 3′ UTR regions based on their physical distances from the closest putative gene. Most genes having significant SNPs encoded either uncharacterized, or hypothetical proteins based on the double-haploid Pahang reference genome annotation (D’Hont et al. 2012; Martin et al. 2016). Some of the SNPs were, however, located in genes with known functions and these included mostly transcription factors such as MYB-related protein gene, AP2-like/ERF, basic helix-loop-helix-like (bHLH-like) family of transcription factors that were found on chromosome 3. These transcription factors are well characterized in model plants such as Arabidopsis thaliana and other plants and were shown to be involved in diverse cellular developmental, differentiation and metabolic processes (Dubos et al. 2010; Feller et al. 2011; Ambawat et al. 2013; Pireyre and Burow, 2015).
Besides transcription factors, other genes that were identified on chromosome 3 through long-range LD SNP included genes that control DNA replication, gene transcription and cell cycle regulation such as enhancer of low casing for polycomb-like protein 1, TIMELESS-interacting protein, CDPK-related protein kinase 3-like, ervatamin-B-like and E3 ubiquitin-protein ligase SINAT5-like amongst others (Levine et al. 2004; Borde & Lichten 2014). Enhancer of polycomb proteins were shown to be involved in epigenetic maintenance of gene expression patterns and posttranscriptional modification in cells (Levine et al. 2004). In Nicotiana tabacum, gene NtCDPK1 was shown to be mainly expressed in proliferating root and shoot meristems as well as in developing floral buds. Inhibition of its expression was associated with abnormal cells and premature cell death, which confirmed its role in cell division, differentiation and death (Lee et al. 2013). E3 ubiquitin-protein ligase SINAT5-like regulates auxin signals in plant cells thereby controlling plant development (Xie et al. 2002). The TIMELESS-interacting protein was shown to be involved in both the circadian rhythm and cell cycle checkpoint regulation (Yoshizawa-Sugata and Masai, 2007; Engelen et al. 2013). Knockdown of TIMELESS-interacting protein or the presence of mutations in the gene that lead to modification in the protein were associated with inefficient progression of S phase and DNA synthesis during the cell cycle (Yoshizawa-Sugata and Masai 2007). We can therefore hypothesize that the presence of missense mutations in the TIMELESS-interacting protein and MYB-related protein 308-like genes that are involved in cell cycle regulation possibly contribute to the variation in fruit filling in banana.
Our results suggest that continued cell division and expansion in the female inflorescence are key processes in determining the sink capacity of banana fruits. The actual mechanisms and gene pathways involved in the control of bunch weight and its component traits remain to be investigated. We can only hypothesize that genotypes with low, or abolished expression of genes involved in floral cell division, differentiation and expansion are likely to have a smaller number of cells in fruits to store the photosynthetic products, which results in poor fruit filling and low bunch weight. In crops such as wheat and rice, grain yield was reported to be controlled by grain weight gene OsGW (rice) and TaGW2 (wheat), which determine seed size by modulating cell number and length (Zhang et al. 2018). In tomato, fruit weight was correlated with the number of cells due to cell division, cell expansion and endopolyploidization (Cheniclet et al. 2005).
The clustering of transcription factors and cell cycle regulation genes on chromosome 3 region that had SNPs associated with bunch weight and its component trait supported our hypothesis that fruit filling in this population was under control of a few QTL with major effects. This could also explain the high broad sense heritability of these traits, which were above 95%. However, given the size of our population, the resolution could have been limited. A future research should use a larger population to capture more recombination events on chromosome 3 so as to underpin the causal loci and unveil other loci that could have been missed out. Further research should also target populations with different genetic background such as Mchare, or plantain to verify if the loci controlling bunch weight and its components traits are conserved across the groups of Musa spp.. Martin et al. (2017) showed that heterozygous reciprocal translocations between chromosomes 1 and 4 resulted in segregation distortion, reduced recombination and linkage in the distal segments of the two chromosomes in the progeny of Musa acuminata ssp. malaccensis. A recent research by Dupouy et al. (2019) also identified two large reciprocal translocations in Musa acuminata ssp. burmanica involving chromosomes 2 and 8, and chromosomes 1 and 9. Studies comparing structural rearrangements and selection sweeps in the banana genome are required to shed more light on possible selection pressure that some regions underwent during domestication and their effects on traits such as yield. This could help to explain why most of the SNPs significantly associated with bunch weight and its component traits were clustered on chromosome 3. We expect that these results will stimulate further research in banana-breeding populations and diversity panels to underpin causal mutations that underlie the variations in bunch weight and its components in addition to other traits. More importantly, research geared towards the development of molecular markers in regions with significant SNPs could facilitate marker-assisted breeding and increase breeding speed to achieve high genetic gains.
References
Ambawat S, Sharma P, Yadav NR, Yadav RC (2013) MYB transcription factor genes as regulators for plant responses: an overview. Physiol Mol Biol Plants 19:307–321
Batte M, Swennen R, Uwimana B, Akech V, Brown A, Tumuhimbise R, Hovmalm HP, Geleta M, Ortiz R (2019) Crossbreeding East African highland bananas: lessons learnt relevant to the botany of the crop after 21 years of genetic enhancement. Front Plant Sci 10:1–9
Begum H, Spindel JE, Lalusin A, Borromeo T, Gregorio G, Hernandez J, Virk P, Collard B, McCouch SR (2015) Genome-wide association mapping for yield and other agronomic traits in an elite breeding population of tropical rice (Oryza sativa). PLoS ONE 10:e0119873
Borde V, Lichten M (2014) A timeless but timely connection between replication and recombination. Cell 158:267–268
Boyle EA, Li YI, Pritchard JK (2017) An expanded view of complex traits: from polygenic to omnigenic. Cell 169:1177–1186
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brown A, Tumuhimbise R, Amah D, Uwimana B, Nyine M, Mduma H, Talengera D, Karamura D, Kuriba J, Swennen R (2017) Banana and plantains (Musa spp.). In: Campos H, Caligari PDS (eds) Genetic improvement of tropical crops. Springer, New York, pp 219–240
Browning BL, Browning SR (2016) Genotype imputation with millions of reference samples. Am J Hum Genet 98:116–126
Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S, Garcia A, Glaubitz JC et al (2009) The genetic architecture of maize flowering time. Science 9:714–718
Cenci A, Hueber Y, Zorrilla-Fontanesi Y, van Wesemael J, Kissel E, Gislard M, Sardos J, Swennen R, Roux N, Carpentier SC et al (2019) Effect of paleopolyploidy and allopolyploidy on gene expression in banana. BMC Genom 20:1–12
Cheniclet C, Rong WY, Causse M, Frangne N, Bolling L, Carde J-P, Renaudin J-P (2005) Cell expansion and endoreduplication show a large genetic variability in pericarp and contribute strongly to tomato fruit growth. Plant Physiol 139:1984–1994
Cingolani P, Platts A, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6:80–92
Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V et al (2015) FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med 373:895–907
Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE et al (1994) Protective effect of apolipoprotein-E type2 allele for late-onset Alzheimer-disease. Nat Genet 7:180–184
D’Hont A, Denoeud F, Aury J-M, Baurens F-C, Carreel F, Garsmeur O, Noel B, Bocs S, Droc G, Rouard M et al (2012) The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488:213–219
Dixon LE, Greenwood JR, Bencivenga S, Zhang P, Cockram J, Mellers G, Ramm K, Cavanagh C, Swain SM, Boden SA (2018) TEOSINTE BRANCHED1 regulates inflorescence architecture and development in bread wheat (Triticum aestivum). Plant Cell 30:563–581
Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L (2010) MYB transcription factors in Arabidopsis. Trends Plant Sci 15:573–581
Dupouy M, Baurens F-C, Derouault P, Hervouet C, Cardi C, Cruaud C, Istace B, Labadie K, Guiougou C, Toubi L et al (2019) Two large reciprocal translocations characterized in the disease resistance-rich burmannica genetic group of Musa acuminata. Ann Bot XX:1–11. https://doi.org/10.1093/aob/mcz078
Dyer KA, Charlesworth B, Jaenik J (2007) Chromosome-wide linkage disequilibrium as a consequence of meiotic drive. Proc Natl Acad Sci USA 104:1587–1592
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) Arobust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379
Engelen E, Janssens RC, Yagita K, Smits VAJ, van der Horst GTJ, Tamanini F (2013) Mammalian TIMELESS is involved in period determination and DNA damage-dependent phase advancing of the circadian clock. PLoS ONE 8:e56623
Farnir F, Coppieters W, Arranz JJ, Berzi P, Cambisano N, Grisart B, Karim L, Marcq F, Moreau L, Mni M et al (2000) Extensive genome-wide linkage disequilibrium in cattle. Genome Res 10:220–227
Feller A, Machemer K, Braun EL, Grotewold E (2011) Evolution and comparative analysis of MYB and bHLH plant transcription factors. Plant J 66:94–116
Flint-Garcia SA, Thornsberry JM, Buckler ES IV (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374
Heslop-Harrison JS, Schwarzacher T (2007) Domestication, genomics and the future for banana. Ann Bot 100:1073–1084
Huang X, Han B (2014) Natural variations and genome-wide association studies in crop plants. Annu Rev Plant Biol 65:531–551
Jeffreys AJ, Kauppi L, Neumann R (2001) Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat Genet 29:217–222
Jordan KW, Wang S, He F, Chao S, Lun Y, Paux E, Sourdille P, Sherman J, Akhunova A, Blake NK et al (2018) The genetic architecture of genome-wide recombination rate variation in allopolyploid wheat revealed by nested association mapping. Plant J 95:1039–1054
Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA et al (2012) International IBD Genetics Consortium (IIBDGC) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491:119–124
Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E (2008) Efficient control of population structure in model organism association mapping. Genetics 178:1709–1723
Kerem BS, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245:1073–1080
Kitavi M, Downing T, Lorenzen J, Karamura D, Onyango M, Nyine M, Ferguson M, Spillane S (2016) The triploid East African Highland Banana (EAHB) genepool is genetically uniform arising from a single ancestral clone that underwent population expansion by vegetative propagation. Theor Appl Genet 129:547–561
Korte A, Farlow A (2013) The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 9:1–9
Kulminski AM (2011) Complex phenotypes and phenomenon of genome-wide inter-chromosomal linkage disequilibrium in the human genome. Exp Gerontol 46:979–986
Labate JA, Lamkey KR, Lee M, Woodman W (2000) Hardy-Weinberg and linkage equilibrium estimates in the BSSS and BSCB1 random mated populations. Maydica 45:243–255
Lee SS, Cho HS, Yoon GM, Ahn J-W, Kim H-H, Pai H-S (2013) Interaction of NtCDPK1 calcium-dependent protein kinase with NtRpn3 regulatory subunit of the 26S proteasome in Nicotiana tabacum. Plant J 33:825–840
Levine SS, King IFG, Kingston RE (2004) Division of labor in polycomb group repression. Trends Biochem Sci 29:478–485
Liu J, Zhang J, Zhang J, Miao H, Wang J, Gao P, Hu W, Jia C, Wang Z, Xu B, Jin Z (2017) Genome-wide analysis of banana MADS-box family closely related to fruit development and ripening. Sci Rep 7:1–13
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, Lifelines Cohort Study, ADIPOGen Consortium, AGEN, BMI Working Group, CARDIOGRAMplusC4D Consortium, CKDGen Consortium, GLGC, ICBP, MAGIC Investigators, MuTHER Consortium, MIGen Consortium, PAGE Consortium, ReproGen Consortium, GENIE Consortium, International Endogene Consortium et al (2015) Genetic studies of body mass index yield new insights for obesity biology. Nature 518:197–206
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A et al (2009) Finding the missing heritability of complex diseases. Nature 461:747–753
Martin G, Baurens FC, Droc G, Rouard M, Cenci A, Kilian A, Hastie A, Doležel J, Aury JM, Alberti A et al (2016) Improvement of the banana “Musa acuminata” reference sequence using NGS data and semi-automated bioinformatics methods. BMC Genom 17:1–12
Martin G, Carreel F, Coriton O, Hervouet C, Cardi C, Derouault P, Roques D, Salmon F, Rouard M, Sardos J et al (2017) Evolution of the banana genome (Musa acuminata) is impacted by large chromosomal translocations. Mol Biol Evolut 34:2140–2152
McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JPA, Hirschhorn JN (2008) Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9:356–369
Müller BSF, Filho JEA, Lima BM, Garcia CC, Missiaggia A, Aguiar AM, Takahashi E, Kirst M, Gezan SA, Silva-junior OB et al (2019) Independent and Joint-GWAS for growth traits in Eucalyptus by assembling genome-wide data for 3373 individuals across four breeding populations. New Phytol 221:818–833
Nadolska-Orczyk A, Rajchel IK, Orczyk W, Gasparis S (2017) Major genes determining yield-related traits in wheat and barley. Theor Appl Genet 130:1081–1098
Němečková A, Christelová P, Čížková J, Nyine M, Van den Houwe I, Svačina R, Uwimana B, Swennen R, Doležel J, Hřibová E (2018) Molecular and cytogenetic study of East African highland banana. Front Plant Sci. https://doi.org/10.3389/fpls.2018.01371
Neph S, Kuehn MS, Reynolds AP, Haugen E, Thurman RE, Johnson AK, Rynes E, Maurano MT, Vierstra J, Thomas S et al (2012) BEDOPS: high-performance genomic feature operations. Bioinformatics 28:1919–1920
Nyine M, Uwimana B, Swennen R, Batte M, Brown A, Christelová P, Hřibová E, Lorenzen J, Doležel J (2017) Trait variation and genetic diversity in a banana genomic selection training population. PLoS ONE 12:e0178734
Nyine M, Uwimana B, Blavet N, Hřibová E, Vanrespaille H, Batte M, Akech V, Brown A, Lorenzen J, Swennen R, Doležel J (2018) Genomic prediction in a multiploid crop: genotype by environment interaction and allele dosage effects on predictive ability in banana. Plant Genome. https://doi.org/10.3835/plantgenome2017.10.0090
Ortiz R, Vuylsteke D (1995) Effect of the parthenocarpy gene P 1 and ploidy on fruit and bunch traits of plantain banana hybrids. Heredity 75:460–465
Ortiz R, Ferris RSB, Vuylsteke DR (1995) Banana and plantain breeding. In: Gowen S (ed) Banana and plantain. Chapman & Hall, London, pp 110–146
Peiffer JA, Romay MC, Gore MA, Flint-Garcia SA, Zhang Z, Millard MJ, Gardner CA, McMullen MD, Holland JB, Bradbury PJ, Buckler ES (2014) The genetic architecture of maize height. Genetics 196:1337–1356
Pireyre M, Burow M (2015) Regulation of MYB and bHLH transcription factors: a glance at the protein level. Mol Plant 8:378–388
Platt A, Viljálmsson BJ, Nordborg M (2010) Conditions under which genome-wide association studies will be positively misleading. Genetics 186:1045–1052
R Core Team (2019) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rafalski A (2002) Applications of single nucleotide polymorphisms in crop genetics. Curr Opin Plant Biol 5:94–100
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES IV (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA 98:11479–11484
Rostoks N, Ramsay L, MacKenzie K, Cardle L, Bhat PR, Roose ML, Svensson JT, Stein N, Varshney RK, Marshall DF et al (2006) Recent history of artificial outcrossing facilitates whole-genome association mapping in elite inbred crop varieties. Proc Natl Acad Sci USA 103:18656–18661
Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R et al (2007) Genome-wide detection and characterization of positive selection in human populations. Nature 449:913–918
Sardos J, Rouard M, Hueber Y, Cenci A, Hyma KE, van den Houwe I, Hřibová E, Courtois B, Roux N (2016) A Genome-wide association study on the seedless phenotype in banana (Musa spp.) reveals the potential of a selected panel to detect candidate genes in a vegetatively propagated crop. PLoS ONE 11:e0154448
Simmonds NW (1953) Segregation in some diploid bananas. J Genet 51:458–469
Simmonds NW (1962) Evolution of the bananas. Longmans, Green & Co., London
Swennen R, Vuylsteke D (1993) Breeding black sigatoka resistant plantains with a wild banana. Trop Agric 70:74–77
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28:286–289
Turner DW, Gibbs DJ (2018) A functional approach to bunch formation in banana. In: Kema GHJ, Drenth A (eds) Achieving sustainable cultivation of bananas: volume 1—cultivation techniques. Burleigh Dodds Science Publishing, Cambridge
Vuylsteke D, Swennen R, Ortiz R (1993a) Registration of 14 improved tropical Musa plantain hybrids with Black Sigatoka resistance. Hortic Sci 28:957–959
Vuylsteke D, Swennen R, Ortiz R (1993b) Development and performance of Black Sigatoka-resistant tetraploid hybrids of plantain (Musa spp. AAB group). Euphytica 65:33–42
Weil CF (2002) Finding the crosswalks on DNA. Proc Natl Acad Sci USA 99:5763–5765
Whitt SR, Wilson LM, Tenaillon MI, Gaut BS, Buckler ES (2002) Genetic diversity and selection in the maize starch pathway. Proc Natl Acad Sci USA 99:12959–12962
Xie Q, Guo H-S, Dallman G, Fang S, Weissman AM, Chua N-H (2002) SINAT5 promotes ubiquitin-related degradation of NAC1 to attenuate auxin signals. Nature 419:167–170
Yoshizawa-Sugata N, Masai H (2007) Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J Biol Chem 282:2729–2740
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB et al (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhang Z, Ersoz E, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA, Bradbury PJ, Yu J, Arnett DK, Ordovas JM et al (2010) Mixed linear model approach adapted for genome-wide association studies. Nat Genet 42:355–360
Zhang Y, Li D, Zhang D, Zhao X, Cao X, Dong L, Liu J, Chen K, Zhang H, Gao C, Wang D (2018) Analysis of the functions of TaGW2 homoeologs in wheat grain weight and protein content traits. Plant J 94:857–866
Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C, Tang C, Toomajian C, Zheng H, Dean C, Marjoram P et al (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet 3:71–82
Zhao K, Tung C-W, Eizenga GC, Wright MH, Ali ML, Price AH, Norton GJ, Islam MR, Reynolds A, Mezey J et al (2011) Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat Commun 2:467
Acknowledgements
This work was supported with funding from Bill and Melinda Gates Foundation, investment ID: OPP1093845 through the IITA coordinated Project “Improvement of Banana for Smallholder Farmers in the Great Lakes Region of Africa.” This work was also supported by the ERDF project “Plants as a tool for sustainable global development” (No. CZ.02.1.01/0.0/0.0/16_019/0000827). The authors are also grateful to all donors who supported this work through their contributions to the CGIAR Fund (https://www.cgiar.org/funders/) and in particular to the CGIAR Research Program for Roots, Tubers and Bananas (CRP-RTB).
Author information
Authors and Affiliations
Contributions
JL, MN and JD conceived the idea; MN, BU, JL, RO and RS developed the methodology; JL, RS, JD, BU and AB sourced the funding; MN and VA carried out data curation; MN and BU analysed the data; MN drafted the manuscript; BU, VA, AB, RO, JD, JL and RS revised and gave further inputs to this manuscript
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Communicated by P. Heslop-Harrison.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nyine, M., Uwimana, B., Akech, V. et al. Association genetics of bunch weight and its component traits in East African highland banana (Musa spp. AAA group). Theor Appl Genet 132, 3295–3308 (2019). https://doi.org/10.1007/s00122-019-03425-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-019-03425-x