
Abstract
Key message
Current development of advanced biotechnology tools allows us to characterize the role of key genes in plant productivity. The implementation of this knowledge in breeding strategies might accelerate the progress in obtaining high-yielding cultivars.
Abstract
The achievements of the Green Revolution were based on a specific plant ideotype, determined by a single gene involved in gibberellin signaling or metabolism. Compared with the 1950s, an enormous increase in our knowledge about the biological basis of plant productivity has opened new avenues for novel breeding strategies. The large and complex genomes of diploid barley and hexaploid wheat represent a great challenge, but they also offer a large reservoir of genes that can be targeted for breeding. We summarize examples of productivity-related genes/mutants in wheat and barley, identified or characterized by means of modern biology. The genes are classified functionally into several groups, including the following: (1) transcription factors, regulating spike development, which mainly affect grain number; (2) genes involved in metabolism or signaling of growth regulators—cytokinins, gibberellins, and brassinosteroids—which control plant architecture and in consequence stem hardiness and grain yield; (3) genes determining cell division and proliferation mainly impacting grain size; (4) floral regulators influencing inflorescence architecture and in consequence seed number; and (5) genes involved in carbohydrate metabolism having an impact on plant architecture and grain yield. The implementation of selected genes in breeding programs is discussed, considering specific genotypes, agronomic and climate conditions, and taking into account that many of the genes are members of multigene families.
Similar content being viewed by others


Introduction
Allohexaploid bread wheat (Triticum aestivum L., 2n = 6x = 42, AABBDD) and diploid barley (Hordeum vulgare) are very important crops in global agriculture. Wheat ranks third after maize and rice, and barley is the fourth (Schulte et al. 2009; Tester and Langridge 2010). As temperate crops, their production in EU countries is especially high.
The harvested production of wheat and barley in 2014 was 149.7 and 60.8 million metric tons, which makes up, respectively, 44.8 and 18.2% of the total cereal production in the EU (http://ec.europa.eu/eurostat/statistics-explained/index.php/Agricultural_production_-_crops). The species are major crops in the Triticeae tribe. Because of their close relationship, they share common traits, which are distinct from rice and maize. The Triticeae grown worldwide are well adapted to a temperate climate, have winter and spring types, and have similar architecture of inflorescences and spikes. Winter varieties, in contrast to spring type, require vernalization to promote flowering in response to long-day (LD) conditions.
The Green Revolution (GR) period, which was initiated in the 1940s and continued until the late 1960s, resulted in a significant increase in cereal production mainly due to higher yields. It was achieved by combining natural semi-dwarf mutants with specifically changed plant architecture and improved agrotechnology including irrigation and fertilization. Further research revealed that the most important traits were caused by single gene mutations: Reduced height-1 (Rht1) in wheat and semidwarf1 (sdw1) in barley (reviewed in Hedden 2003). The genes and the mutations affecting gibberellin signaling and metabolism led to pleiotropic changes which were beneficial for crop production (Börner et al. 1993; Chandler and Harding 2013; Franckowiak and Lundqvist 2012; Peng et al. 1999; Saville et al. 2012; Wen et al. 2013).
Currently, bred varieties are still characterized by higher yields, but the increase has never been as spectacular as that observed during the GR. An important limitation is related to the large and complex genomes of both crops, which makes selection of crucial traits particularly difficult. However, continuing from the 1970s, a significant increase of knowledge on cereal genomes and gene functions as well as development of new biotechnological tools has opened new possibilities to repeat the success of the GR. The knowledge on a particular gene’s function in relation to grain yield is especially important for efficient selection of new haplotypes in classical breeding. This can be a new driver of wheat and barley breeding.
Several reviews on genetic factors influencing various aspects of cereal yield have already been published. There are articles on: genetic factors of floral development and inflorescence architecture in cereals (Sreenivasulu and Schnurbusch 2012), control of flowering time (Cockram et al. 2007), aspects of senescence, nutrient remobilization and yield in wheat and barley (Distelfeld et al. 2014), traits associated with phenology, photosynthesis, assimilate partitioning, and lodging resistance in rice and wheat (Valluru et al. 2014), genetic factors improving barley culm robustness (reviewed by Dockter and Hansson 2015), and genes regulating cytokinin levels in various species (reviewed by Jameson and Song 2015).
In our review, we focus on the most important and newly identified genes determining yield-related traits in the Triticeae species wheat and barley. The selected genes represent different groups with various biological functions. We start with those that were so successfully used during the GR but thoroughly characterized in the last decade, and then continue with the newly identified genes. We also indicate the sources and the types of gene modifications (mutants, transgenics, positional cloning, etc) and discuss how the changes affect yield components.
Plant architecture and grain yield traits in Triticeae
Barley and wheat belong to the Triticeae species possessing similar plant architecture with an unbranched inflorescence known as the spike. The architecture as well as their climatic and agronomic requirements differ from the model species rice. Spikes of wheat and barley most likely evolved from an ancestral compound inflorescence, producing branches (Endress 2010; Kellogg et al. 2013; Remizowa et al. 2013). Archetypal inflorescences and spikes bear three single-flowered spikelets per rachis node. The spikelets are arranged in two opposite rows along the main axis. In barley, if the two outer lateral spikelets at each node are sterile, the spikes are two-rowed; when all three are fertile, the spikes are six-rowed (Ramsay et al. 2011). Final grain yield depends on grain number and grain weight (reviewed by Sreenivasulu and Schnurbusch 2012; Kesavan et al. 2013; Distelfeld et al. 2014). The yield components influencing grain number include number of tillers bearing fertile spikes, extension of vegetative as well as reproductive growth and differentiation phase, inflorescence architecture, culm hardiness, spike initiation, elongation and branching as well as spikelet formation. Grain weight is affected by grain cell number (Brocklehurst 1977) and sink capacity (Millet and Pinthus 1984).
The genomes of barley and wheat
The size and the complexity of barley and bread wheat genomes are very important obstacles in developing different strategies of selection and breeding. The size of the barley (Hordeum vulgare) genome is 5.1 Gbp (Doležel et al. 1989), and that of wheat (Triticum aestivum) is over three times bigger, 17 Gbp (Bennett and Smith 1976; The International Wheat Genome Sequencing 2014). The two genomes also differ considerably with respect to their complexity. Barley is diploid, containing seven chromosome pairs (2n = 2x = 14). Wheat is allohexaploid (AABBDD), with three diploid genomes originating from different species (2n = 6x = 42). More than 124,000 gene loci were annotated by The International Wheat Genome Sequencing Consortium (2014). The genes are distributed in gene-rich regions at the telomeres across the homoeologous chromosomes and subgenomes (Gill et al. 2006a, b). The data gathered by the consortium evidenced dynamic genome change including gene gain, loss, and duplication since the divergence of wheat lineages.
Due to the allohexaploidy, the majority of wheat genes are represented by three homologs in each of the three A, B, and D genomes. However, considering their effect on phenotype, the most important question is whether they are expressed and, if so, what their expression pattern is. Data collected so far indicate that only about 20% of genes were always expressed from all three genomes (Mochida et al. 2003). The remaining ones showed preferential expression from certain genomes, which could vary between tissues and organs (Appleford et al. 2006; Mochida et al. 2003). Analysis of the wheat grain transcriptome revealed cell type and stage-dependent genome dominance and asymmetric expression for some groups of genes (Pfeifer et al. 2014). There are only a few examples of wheat genes that are located exclusively in one genome, such as Pin genes, which determine grain hardness (Nadolska-Orczyk et al. 2009).
The genes of the Green Revolution
The Green Revolution (GR) and its founder Norman Borlaug are widely recognized iconic symbols of the 20th century agriculture. The novel approach to breeding crop varieties and a significant increase in productivity crucial to meet food demand in hunger-threatened regions were recognized by awarding the 1970 Nobel Peace Prize to Norman Borlaug. The goal was achieved by the development of high-yielding varieties together with the application of modern agrotechnologies, such as irrigation, synthetic fertilizers and pesticides (reviewed by Hedden 2003).
These high-yielding varieties, which were the key to success, were developed by the introduction of single major genes into cereals. The new varieties were shorter, with improved resistance to stem lodging and greater ability to tolerate nitrogen-based fertilizers (Gale and Youssefian 1985). Moreover, due to photoperiod insensitivity and rust resistance, the cultivars were adapted to a wide range of agricultural environments (Borlaug 1983).
Semi-dwarf phenotype is the most important feature of GR cultivars, and in wheat, it depends on the presence of the Reduced height-1 (Rht1) gene. The gene was identified in Japan after crossing a Japanese semidwarf wheat cultivar with American high-yielding varieties to produce Norin 10 (reviewed by Hedden 2003). Two alleles of the gene are located at one of two loci on genome B and genome D of hexaploid wheat and are named, respectively, Rht-B1 and Rht-D1 (Börner et al. 1996). They have been found in the majority of varieties grown worldwide (Evans 1998). The alleles confer a limited response to the growth phytohormone gibberellin (GA) (Gale and Marshall 1973). Further research revealed that the Rht alleles encode DELLA proteins (Peng et al. 1999), which are transcriptional regulators that act to repress GA signaling (Pearce et al. 2011). The mutated alleles contain single nucleotide substitutions that determine premature stop codons in the N-terminal coding region (Peng et al. 1999). Rht-B1 and Rht-D1 are orthologs of the Arabidopsis Gibberellin Insensitive (GAI) gene and maize dwarf-8 (d8) (Chandler et al. 2002; Peng et al. 1999) as well as barley Slender1 (HvSln1) (Wen et al. 2013). The direct effect of the mutation is reduced plant height, but there is also an important pleiotropic effect causing increased assimilate partitioning to developing ears and an increased number of grains per spike (Börner et al. 1993; Flintham et al. 1997; Youssefian et al. 1992). In addition, selected alleles of the Rht gene play a role in disease resistance (Saville et al. 2012; Srinivasachary et al. 2009).
In barley, the Green Revolution genes semidwarf1 (sdw1) located at the sdw1/denso locus (Druka et al. 2011; Franckowiak and Lundqvist 2012) and uzu1.a (Chono et al. 2003; Dockter et al. 2014) are involved, respectively, in the metabolism of the GA and brassinosteroid hormones. The genes determine shorter and stronger culms supporting spikes and preventing lodging. Since both phytohormones control many processes in different plant tissues, their pleiotropic effects also cause a number of unwanted agronomic traits, such as reduction of grain size. Moreover, sdw1 confers late flowering and might be associated with lowered malt quality, while uzu1 determines temperature sensitivity, and therefore, there were attempts to replace them with alleles of alternative genes (Dockter and Hansson 2015; Wang et al. 2014a).
The pleiotropic effect of another GR dwarfing gene of barley, Sln1 mentioned above, include, similar to their orthologous Rht genes of wheat, disease resistance (Saville et al. 2012), which should be appreciated in plant breeding.
Approaches for the identification of major yield determining genes
Natural or induced mutations were the only source of agricultural important traits until the end of the 1970s. Diploid barley has been, since the early 20th century, a model for obtaining induced mutants and exploring the potential of mutation breeding in crop improvement (Druka et al. 2011). The large mutant resources are collected in the NordGen genebank (http://www.nordgen.org/), and are proposed to be explored using the tools of modern genetics. Barley mutants have been used to characterize and/or utilize genes of nitrate reductase (Somers et al. 1983), the row-type gene, SIX-ROWED SPIKE 1 (Vrs1; Komatsuda et al. 2007), short culm UZU DWARF (Uzu1; Chono et al. 2003), and semi-dwarf, slender-type plant architecture SLENDER1 (Sln1; Chandler et al. 2002). The nature of some morphological mutants of barley was proposed to be explained by a new model of barley phytomer (Forster et al. 2007). Selection of such mutants is especially difficult in the complex, polyploid wheat genome.
Since the 1980s, more sophisticated approaches to acquire new phenotypes for research and breeding have been developed. The methods include genetic transformation leading to transgene or native gene overexpression or native gene silencing. RNAi-based silencing of a selected gene is currently the most powerful technology among genetic modification methods, especially useful for analysis of gene function in species with large and complex genomes including wheat (reviewed in Fu et al. 2007; Gasparis et al. 2011, 2013; Lawrence and Pikaard 2003; Travella et al. 2006).
The availability of molecular marker-based genetic maps allowed detection of hundreds of yield-related quantitative trait loci (QTL), and, in species with smaller genomes, e.g., rice, multiple QTL have been cloned (Mayer et al. 2011). Direct cloning of yield-related genes using strategies of gene high-resolution mapping and map-based cloning in wheat and barley is still very difficult due to the complexity of the genomes. However, availability of the sequenced genomes and transcriptomes as well as the development of comparative genomics creates new possibilities of gene identification and gene function analysis (Christiansen et al. 2016; Hou et al. 2014; Ma et al. 2015a; Poursarebani et al. 2015; Qu et al. 2015; Vu et al. 2010a, b).
The most impressive progress has been made for rice, a species with a relatively small, fully sequenced and annotated genome, which has become a model cereal species. The Triticeae species differ from rice with respect to their plant architecture including inflorescence as well as their climatic and agronomic requirements. It is worth noting that phenotype differences found for rice and Triticeae correspond to differences and relationships between groups of major genes. However, despite the differences, some yield-related genes in wheat and barley have been identified using comparative genomics with rice (Distelfeld et al. 2012; Hanif et al. 2016; Houston et al. 2013; Hu et al. 2016; Jiang et al. 2011; Ma et al. 2015a, Su et al. 2011; Zheng et al. 2014) or other species including the model plant Arabidopsis (Jost et al. 2016). Typically, the identified orthologs retained the same biological function, although in certain cases, they might determine different phenotypes (Distelfeld et al. 2012; Gasparis et al. 2013). This shows that a particular function of a gene of interest should be established in a target species. Some information on expected function might be derived from the temporal and spatial expression profile of selected genes (Christiansen et al. 2011; Ma et al. 2015b; Song et al. 2012; Zalewski et al. 2014).
Currently, genome editing is expected to be the most powerful biotechnology tool to perform specific changes through targeted mutagenesis, precise gene editing, multigene transformation, and gene stacking. The technology will allow for gene inactivation or generation of a new functional allele. There are already the first papers on induction of targeted, heritable mutations in barley, and Brassica oleracea (Lawrenson et al. 2015) and gene replacement in barley (Watanabe et al. 2015). Moreover, a group of researchers from the Chinese Academy of Sciences showed that TALEN and CRISPR-Cas9 technologies have been efficient and specific in allopolyploid species such as common wheat (T. aestivum), with triplicate homoeologous genes of the three genomes (Shan et al. 2014; Wang et al. 2014a, b). Application of the strategies to yield-related traits will require further technical improvement, adaptation to different genotypes and documentation of heritability. The gene silencing approach by RNAi-based technology might remain the method of choice in polyploid species, although the potential big advantage of genome editing tools is the possibility to obtain plants that might be excluded from GMO legislation.
The major genes of the second GR
Barley (Hordeum vulgare) genes and wheat (Triticum aestivum, T. durum) orthologs
Barley, with its smaller and less complex, compared to wheat, diploid genome, is more suitable for identification of natural or induced mutants, which result in higher yield. Among the 20 genes listed in Table S1, 18 are natural or induced mutants, and 2 genes were modified by RNAi-based silencing. The genes are classified functionally into three general groups (Table S1). The first one (I), associated with higher grain number, includes transcription factors and other regulators affecting spikelet development, inflorescence architecture and growth. Group II, controlling plant architecture, contains genes that directly affect metabolic processes or signaling of growth regulators. The genes changing cytokinin metabolism have an important influence on grain number; the genes modifying gibberellin and brassinosteroid metabolism mainly influence culm (stem) length and robustness/hardiness. Other mutants, frequently of unknown function, control spike and spikelet characteristics, awn and culm length and robustness (reviewed by Dockter and Hansson 2015). For some of the barley genes listed in Table S1, orthologs in wheat (Table S2) were found, and in such a situation, the genes of both species are described together. A list of barley and wheat yield-determining genes with known orthologs in other cereals/species is presented in Table 1.
Transcription factors
A group of transcription factors regulate inflorescence architecture and spikelet development. Although mutants of some of them were identified decades ago, the detailed molecular analysis of selected alleles has been carried out during the last few years.
The main genes modifying spikelet development by regulation of lateral spikelet fertility, SIX-ROWED SPIKE 1 (VRS1) (Komatsuda et al. 2007; Lundqvist et al. 1997; Lundqvist and Lundqvist 1988) and INTERMEDIUM-C (INT-C) (Lundqvist and Lundqvist 1988; Ramsay et al. 2011), were found as natural mutants. Modifications of the genes in barley have resulted in two different cultivation types, two-rowed and six-rowed forms. Both forms are known and their origin is dated to the domestication period (Ramsay et al. 2011). A schematic diagram of relationship between these and other major genes determining plant architecture in barley and wheat is presented in Fig. 1.

Major genes determining plant architecture in barley (left) and wheat (right). Vertical arrows indicate down or up enzyme regulation/gene expression; horizontal arrows indicate direction of gene co-regulation; bold main/key allele; ellipsis multiple alleles
vrs1 is a loss-of-function allele, derived from the wild two-rowed ancestor. The wild-type allele Vrs1.b encodes the homeodomain-leucine zipper I-class (HD-Zip I) type transcriptional repressor that inhibits the development of lateral spikelet fertility (Komatsuda et al. 2007). The protein controls cell division and development specifically in the lateral spikelets. The vrs1 loss-of-function allele determines the development of six-row spikes. The pleiotropic effect of the allele is the reduction in the number of tillers per plant, and thus spikes per plant, that is compensated for by the increase in number of grains per spike (Lundqvist et al. 1997). Vrs1 orthologs of wheat, Brachypodium, rice, sorghum, and maize were not found, indicating that this transcription factor is specific to barley (Sakuma et al. 2010).
Phenotypes determined by Vrs1 alleles are modified by alleles at the INTERMEDIUM-C (INT-C) locus (Ramsay et al. 2011). Lundqvist and Lundqvist (1988) found up to ten INT alleles. Seventeen additional barley mutations of the INT-C coding region correlating with lateral spikelet fertility were identified (Ramsay et al. 2011). The alleles influence variations in male fertility and grain development from two-rowed to six-rowed spikes. INT-C is an ortholog of the maize domestication gene TEOSINTE BRANCHED 1 (TB1). Besides Vrs1 and Int-C, Six-rowed spike4 (Vrs4) is the third locus found to control row type in barley (Koppolu et al. 2013). Vrs4 is an ortholog of the maize inflorescence architecture gene RAMOSA2 (RA2), which encodes a lateral organ boundaries (Lima Neto et al. 2014) domain transcription factor. Eighteen coding mutations of the gene were associated with lateral spikelet fertility and loss of spikelet determinacy (Koppolu et al. 2013).
Photoperiod-1 (Ppd-H1) is the major determinant of barley photoperiod response (Turner et al. 2005). The ppd1-H1 late-flowering mutant belongs to a class of genes involved in circadian clock function (Turner et al. 2005). The allele shows a reduced response to photoperiod, which is explained by altered circadian expression of the photoperiod pathway gene CONSTANS (CO) and reduced expression of its down-stream target FLOWERING LOCUS (FT), a key regulator of flowering (Turner et al. 2005). Both FT1 and Ppd-H1 have not been implicated in the control of two- or six-rowed spikes. However, the genes regulate optimum flowering time, specific for geographical regions, and essential for enhanced barley productivity. The latest research on CONSTANS genes in barley, HvCO1 and HvCO2, showed that the genes control floral repression by upregulating VERNALIZATION 2 (VRN-H2) (Mulki and Korff 2016).
Comparative mapping shows Ppd-H1 of barley to be collinear with a wheat member of the pseudo-response regulator (PRR) gene family, Ppd-D1 located on chromosome 2D (Beales et al. 2007). The Ppd-1 gene in wheat, like in barley, is a key regulator of inflorescence architecture and paired spikelet development (Boden et al. 2015). The gene controls photoperiod-dependent floral induction by regulating expression of FT. Loss-of-function alleles promote a constitutive long-day (LD) response (Beales et al. 2007; Shaw et al. 2012, 2013; Turner et al. 2005). Among five Ppd-D1 mutants found by Beales et al. (2007), only the Ppd-D1a allele was associated with photoperiod insensitivity. This loss-of-function allele has a 2 kb deletion upstream of the coding region and determines early flowering on short or long days. According to the authors (Beales et al. 2007), this type of photoperiod-insensitive wheat mutant allowed adaptation of cultivars obtained during the GR to broad range of environments. The Ppd-1 photoperiod insensitive mutations were found in A, B, and D genomes, but there was no genome specificity associated with these mutants (Shaw et al. 2012). The flowering-affecting alleles act mostly by upregulation of TaFT1 and suppression of CONSTANS TaCO1.
Most of the genes described above have already been used in breeding. The breeding target proposed by Boden et al. (2015) will be the modulation of the expression of the two genes Ppd-1 and FT to obtain wheat inflorescence showing better arrangements and an increased number of grains in spikelets. Moreover, reduced photoperiod responsiveness of the ppd-H1 mutant of barley is considered to be advantageous in spring varieties (Turner et al. 2005). Regulation of interactions between HvCO and PpdH1 with VRN-H2 is proposed for manipulation of the photoperiod response together with the vernalization pathway in barley (Mulki and Korff 2016). A diagram of the potential relationship between the genes controlling the photoperiod in barley and wheat is presented in Fig. 2. Precise models for the coregulation of these genes in the SD or LD light (Cockram et al. 2007) and before and after vernalization were recently proposed by Mulki and von Korff (2016).

Major genes controlling photoperiod in barley and wheat. For the precise model of coregulation of these genes before and after vernalization, see Mulki and von Korff (2016)
An induced mutant of inflorescence architecture named ‘Compositum-Barley’ displays noncanonical spike branching (Poursarebani et al. 2015). The spikelets are replaced by lateral branch-like structures forming small secondary spikes. The phenotype is determined by the compositum 2 (com2) locus. COM2 was found to be orthologous to the branched head t (bh t) locus regulating spike branching in tetraploid ‘Miracle-Wheat’, and also orthologous to maize BRANCHED SILKLESS (BD1), to rice FRIZZY PANICLE/BRANCHED FLORETLESS 1 (FZP/BFL1) (Poursarebani et al. 2015) and to WFZP (W from wheat) in hexaploid wheat (Dobrovolskaya et al. 2015). FRIZZY PANICLE (FZP) is the main gene acting in spikelet development during the floret meristem transition phase. The orthologs encode a number of the APETALA2/Ethylene Response Factor transcription factor family members. The COM2 expression in barley was found to be regulated by Six-rowed spike 4 influencing branch repression in barley, while HvIDS1 (an ortholog of maize INDETERMINATE SPIKELET 1) is a putative down-stream target of COM2. Wheat mutants developing supernumerary spikelets (SSs) are a recessive type of FZP/BFL1, and three WFZP homoeologous genes were found. The most severe effect was observed in genotypes sharing WFZP-D lesions and WFZP-A frameshift. The final result of noncanonical branch formation in tetra- or hexaploid wheat was production of significantly more grains per spike and higher yield. Therefore, supernumerary spikelet (SS) mutants are proposed to be included in wheat breeding to increase grain number (Dobrovolskaya et al. 2015; Poursarebani et al. 2015). Moreover, spike branching is expected to enhance sink capacity in wheat spikes (Lawlor and Paul 2014), which might further increase yield. Otherwise, the same mutation in branch repressor genes of barley and other diploid grass species did not influence grain number (Poursarebani et al. 2015).
A genome-wide analysis of common wheat identified seven QTL regulating supernumerary spikelet SS formation located on five chromosomes (2D, 5B, 6A, 6B, and 7B) (Echeverry-Solarte et al. 2014). Sequence analysis of the bh t locus in a collection of mutant and wild-type tetraploid wheat accessions revealed that a single amino acid substitution in the DNA-binding domain of the encoded protein gave rise to ‘Miracle-Wheat’ (Poursarebani et al. 2015).
The family of NAC transcription factors is known to be involved in regulation of important agronomic traits in plants. Comparison of selected barley NAC genes with closely related NAC genes from other species suggests their conserved role in cell wall biosynthesis, leaf senescence, root development, seed development, nutrient remobilization, and hormone-regulated stress responses (Christiansen et al. 2011). However, the functions for some NAC orthologs in wheat and rice were divergent (Distelfeld et al. 2012), which underlines the importance of separate functional characterization for each gene in a target species. Analyzed by comparative genomics, barley HvNAC005 was shown to be a strong positive regulator of senescence (Christiansen et al. 2016). The authors indicate the gene as a target for tuning the gene expression to improve nutrient remobilization related to senescence in barley.
In wheat, the most prominent example of NAC is NAM-B1 located at the Gpc-B1 locus (Uauy et al. 2006a). The wild allele encodes a NAC transcription factor that accelerates senescence and improves grain protein, zinc, and iron content. Contrary silencing of expression of multiple NAM homologs by RNAi delayed senescence and significantly reduced protein, zinc, and iron in the grains. Gpc-B1 is considered as a domestication gene affecting grain size (Dubcovsky and Dvorak 2007; Uauy et al. 2006b). Modern wheat varieties carry a nonfunctional GPC-B1. The authors suggest that this is a result of selection by breeders of larger seeds, which are the result of non-accelerated grain maturity.
Another nitrate-signaling and cereal-specific NAC transcription factor, TaNAC2-5A, was isolated and re-introduced into wheat to study nitrate-dependent signaling (He et al. 2015). Overexpression of the gene enhanced root growth and the nitrate influx rate, and as a consequence increased the root’s ability to acquire nitrate from the soil. Transgenic wheat lines revealed higher grain yield and higher nitrogen accumulation in aerial parts, which was allocated to grains. TaNAC2-5A was found to be involved in nitrate signaling and is proposed as a gene resource for breeding crops with more efficient use of fertilizers (He et al. 2015).
Barley spike density has been shown to be controlled by several loci: erectoides-a (ert-a), mapped to the centromere of chromosome 7H dense spike (dsp) (Shahinnia et al. 2012), and ZEOCRITON1 Zeo1, Zeo2, Zeo3 (Druka et al. 2011), with the most important alleles of HvAPETALA2 (HvAP2) interacting with microRNA172 (miR172) (Houston et al. 2013). The function of the first two loci has not been characterized. The Zeo loci were genetically mapped to the main arm of chromosome 2H. High-resolution genetic mapping of the Zeo1.b region assigned it to MLOC_81350 and MLOC_43830 (Houston et al. 2013). The loci encode a transcription factor containing an APETALA2 DNA-binding domain and miR172-binding site in the last exon of MLOC_43830. In mutants of HvAP2 internode, elongation was reduced in both the culm and the spike, which was the effect of perturbed interaction between miR172 and its corresponding binding site in the mRNA of AP2-like transcription factor (Houston et al. 2013). Orthologs reported for rice (Qi et al. 2011) and maize (Jiang et al. 2012) affected internode length as well. The HvAP2 and dsp alleles of barley were included in the discussed group of genes, because they might increase grain yield by controlling spike density, as suggested (Houston et al. 2013; Shahinnia et al. 2012).
Molecular characterization of the Q locus in wheat revealed that it encodes an AP2-like transcription factor (Simons et al. 2006). The gene mainly confers the free-threshing character, and is considered as another main domestication gene of polyploid wheat (Dubcovsky and Dvorak 2007).
Genes modulating hormone activity
This is another group of genes with a potentially large impact on yield and function in cytokinin and brassinosteroid metabolism and signaling pathways. The genes seem to be of special importance for plant breeding, because both groups of hormones regulate developmental processes and traits directly related to productivity, such as shoot branching, seed number, seed size, root system, and leaf inclination angle.
Cytokinins
Cytokinins are crucial in promoting cell division, cell growth and differentiation, axillary bud growth, and in delaying leaf senescence (Jameson and Song 2015; Mok and Mok 2001). This group of plant hormones participates in local and long-distance signaling, interacting with auxins and other substances.
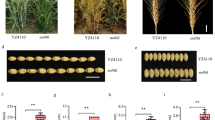
Regulation dependent on cytokinins contributes to crop productivity by controlling cell division and lateral meristem activity (reviewed in Mok and Mok 2001). This growth regulator was shown to be a limiting factor in flower and pod development or during seed setting (Jameson and Song 2015). An important part of the cytokinin regulatory mechanism relies on spatial and temporal changes in cytokinin level. It is precisely controlled in plant tissues and organs. The level of active cytokinins depends on balanced regulation of synthesis [isopentenyl transferase (IPT)], activation [Lonely Guy (LOG)], inactivation (O-glucosyl transferase), re-activation (β-glucosidase), and degradation [cytokinin dehydrogenase (CKX)] (Jameson and Song 2015; Sakakibara 2006). The enzyme cytokinin dehydrogenase (CKX) catalyzes cytokinin degradation and is encoded by the family of CKX genes (Werner et al. 2006). The expression of individual CKX genes, each showing a diverse pattern of spatial and developmental regulation, leads to the desired pattern of cytokinin accumulation in various organs. The significantly higher number of grains observed in an OsCKX2 knock-out rice mutant was explained to be a result of lower activity of CKX enzyme catalyzing cytokinin degradation and consequently high cytokinin accumulation in the inflorescence meristem (Ashikari et al. 2005). The same effect of increased seed number after RNAi-based silencing of two HvCKX genes was reported in barley (Zalewski et al. 2010, 2012, 2014). Decreased expression of HvCKX1 and HvCKX9 led to a higher number of seeds and spikes. We proposed that the effect of higher plant productivity is related to the silencing of these HvCKX genes, showing high original expression in developing kernels of wild-type plants. The phenotype was significantly stronger in HvCKX1-silenced plants compared to HvCKX9-silenced ones. This correlated well with higher expression of HvCKX1 vs. HvCKX9 in developing kernels of wild-type plants.
In wheat, five haplotype variants of TaCKX6-D1 (a–e) were evaluated. Haplotype TaCKX6-D1a associated with higher grain weight showed decreased expression relative to haplotype TaCKX6-D1b in eight DAP seeds (Zhang et al. 2012). Recently, a novel allele of TaCKX6, TaCKX6a02, was identified by a PCR-based approach with primers designed for expressed sequence tags (ESTs), which were homoeologous to the TaCKX gene family. The allele was associated with grain size, filling rate, and weight (Lu et al. 2015). All these results agree with other data indicating that cytokinins are a key driver of seed yield in various species, as described in the recent review by Jameson and Song (2015).
Unexpectedly, an opposite effect was reported for TaCKX4 variants of recombinant inbred lines (RIL). Grain weight and chlorophyll content in the flag leaf were significantly higher in lines with a suggested higher copy number of the gene (Chang et al. 2015). The authors did not explain how this higher copy number of TaCKX4 could influence higher grain weight and moreover whether it was correlated with higher expression of the gene. These and others major genes determining grain size in barley and wheat are listed in Fig. 3.

Major genes determining grain size in barley (left) and wheat (right)
Brassinosteroids
Brassinosteroids affect a wide range of physiological processes including cell elongation, cell division and differentiation, reproductive biology (flowering, ovule and seed number, seed size), senescence, root growth and development, activation of photosynthesis, and the antioxidant system (Cao et al. 2005; Che et al. 2015; Houimli et al. 2008; Shahbaz et al. 2008; Wu et al. 2008). Many of the regulated processes are directly associated with crop yield. It is well documented that application of exogenous BR improves plant tolerance for environmental stresses (reviewed in Khripach et al. 2000). Genetic modification of selected BR-related genes led to a crop yield increase by 20–60% (reviewed in Divi and Krishna 2009). Sakamoto et al. (Sakamoto et al. 2006) reported higher grain yield of the rice brassinosteroid-deficient osdwarf4-1 mutant, characterized by erect leaf phenotype. Increase in the yield, compared with wild-type plants, was evident under conditions of dense planting. According to the authors, the phenotype of more erect leaves improved not only light distribution and photosynthesis but also the size and use of nitrogen reservoirs (Sakamoto et al. 2006).
uzu1 and brh are natural mutants of barley deficient in brassinosteroid biosynthesis. The first one known from the GR period and a big group of brh mutants determine reduced stem length. The wild-type Uzu1 encodes the BR receptor HvBRI1 (BRASSINOSTEROID-INSENSITIVE1; MLOC_5176). The GR mutant allele uzu1.a is a monogenic recessive mutant of the brassinosteroid hormone receptor, insensitive to exogenously applied BR. The mutant has been introduced in Japanese hull-less barley cultivars. The Uzu1 gene is an ortholog of the rice D61 gene (Chono et al. 2003; Dockter et al. 2014).
Members of another large group of brh (brachytic) mutants including 27 semidwarf mutants in 18 independent genetic loci (Dahleen et al. 2005) are deficient in brassinosteroid biosynthesis. Their phenotype is very similar to brassinosteroid-deficient mutants. The brh1, bhr4.j, and bhr8.ad genes have shorter kernels with lower weight compensated by an increased number of kernels per inflorescence and upright plant architecture (Dahleen et al. 2005; Franckowiak and Lundqvist 2012).
Yield-related orthologs of Uzu1 and brh genes in wheat have not been found.
The GL2 locus in rice, identified as one of the grain-length associated QTLs, has the potential to improve grain weight by up to 27%. It is allelic to OsGRF4, which is a component of the BR signaling pathway. Mutation of the gene at the miR369 targeting site results in activation of BR responses and mediates specific regulation of grain length by this group of hormones (Che et al. 2015). Organ specific expression of genes encoding C-22 hydroxylases the enzymes controlling BR levels in rice resulted in plants producing more tillers and more seeds compared with the WT control. The changes led to up to a 44% increase in grain yield per plant (Wu et al. 2008). These two examples of rice genes involved in brassinosteroid signaling or metabolism are included here to indicate the possible huge impact of such genes on yield-related traits. We have suggested searching for their orthologs in wheat and barley.
Gibberellins
The GA signaling and metabolism pathways have been prime targets for manipulation for further improvements in crop yield (reviewed in Hedden 2003). The specific genes affecting GA biosynthesis or signaling and used in breeding during GR have been reviewed in the first section. The genes determine the typical phenotype, which is semi-dwarf, with reduced culm internode length and increased lodging resistance.
The widely used semidominant GA insensitive wheat mutant Rht-1 is an example of a GA signaling mutation. The most important alleles are Reduced height-1 (Rht1-B1b, Rht1-D1b), which in wheat are located in B and D genomes. Additional Rht-1 dwarfing mutations in B and D genomes were characterized at the molecular level. No semi-dwarfing alleles of Rht-A1 were found (Pearce et al. 2011). Another Rht-B1c allele was partially dominant or co-dominant for plant height, and differed from Rht-B1a by a single 2 kb Veju retrotransposon insertion, several SNPs and one 197 bp insertion (Wen et al. 2013). Haplotype investigations indicated that retrotransposon insertion was responsible for the extreme dwarfing effect. RhtB1c caused reduction of total Rht-1 transcript levels and up-regulation of GATA-like transcription factors (Wen et al. 2013). The pleiotropic effect of the gene is in this case beneficial for other agricultural traits. It resulted in increased tiller and root number, a higher photosynthetic rate, and a higher chlorophyll content (Zhang et al. 1995; Li et al. 2006). In addition, Rht-B1c determines inhibition of α-amylase synthesis in ripening grains, and enhanced seed dormancy (Flintham and Gale 1982; Flintham et al. 1997). Such a pleiotropic effect, although not always positively associated with higher yield, is typical for the genes involved in plant hormone signaling or metabolism.
Numerous sdw-type mutants of barley (Börner et al. 1996; Druka et al. 2011; Franckowiak and Lundqvist 2012; Vu et al. 2010a, b) as well as HvSLN1 (Wen et al. 2013) represent GA-insensitive phenotype and function in the GA signaling pathway. HvSLN1 is an ortholog of Rht1 in wheat and GAI in Arabidopsis.
Another set of GA-responsive barley mutations function in GA biosynthesis. The wild Grd5 (GIBBERELLIN-RESPONSIVE DWARF5) encodes CYPSSA (cytochrome P450 ent-kaurenoic acid oxidase), the enzyme that catalyzes conversion of ent-kaurenoic acid to GA12, which is the first step of the gibberellin biosynthesis pathway. The three independently obtained grd5 mutants, defective in CYPSSA enzyme, accumulate ent-kaurenoic acid in developing grains and determine the dwarf phenotype (Helliwell et al. 2001).
Breviaristatum (ari) represents another group of mutations characterized by semidwarf phenotype. There are 32 ari mutant alleles represented in a set of Bowman near-isogenic lines of barley and several hundred at the Nordic Genetic Resource Centre (Dockter and Hansson 2015; Druka et al. 2011; Franckowiak and Lundqvist 2012). The old, widespread cultivated barley cv. Golden Promise carries ari-e GP (erectoides) recessive mutation (Liu et al. 2014).
Identification and characterization of barley Polycomb group (PcG) homologs FERTILIZATION INDEPENDENT SEED (HvFIE), ENHANCER OF ZESTE E(Z) [HvE(Z)], SUPPRESSOR OF ZESTE [HvSu(z)12a], and HvSu(z)12b revealed that expression of the four genes was significantly different among tissues, developmental stages of seed and barley cultivars. The differences were found to be associated with seed size. Significant differences in HvFIE and HvE(Z) gene expression affected the level of the abiotic stress-related hormone abscisic acid (ABA), known to be involved in seed maturation, dormancy, and germination (Kapazoglou et al. 2010).
Other yield-related genes of wheat without known orthologs in barley
Out of 20 yield-related genes characterized in wheat, for only three have the orthologs been reported in barley. There was one gene modifying GA signaling and two representing a group of transcription factors. In addition, two genes encoding enzymes of cytokinin metabolism represent the same family of CKX genes that have already been characterized. The 15 other genes with an impact on plant architecture and grain yield (Table S2) include the following: transcription regulators affecting tillering and grain development, and in consequence grain number, regulators of cell divisions mainly affecting grain size, and genes regulating plant architecture by sucrose and starch metabolism.
Transcription regulators
In the group of transcription regulators, Ppd-1 in hexaploid wheat, which is an ortholog of Ppd-H1 in barley, and WFZP in hexaploid wheat as well as bh t in tetraploid wheat, which are orthologs of COM2 in barley, have already been characterized. The transcript elongation factor TaTEF-7A is a newly selected gene that represents a multigene family in wheat having a strong impact on grain number (Zheng et al. 2014). The rice ortholog OsTEF1 was proved to regulate the tillering process by activating expression of cytochrome P450 and regulating expression of another set of over 100 genes (Paul et al. 2012). The wheat TaTEF-7A allele, found to increase grain number per spike, was identified as a result of analysis of haplotypes, which showed the highest expression in young spikes and developing seeds. The allele was found to influence grain number per spike. Moreover, ectopic overexpression of TaTEF-7A in A. thaliana caused a pleiotropic effect on vegetative and reproductive development visible as enhanced grain length, silique number, and silique length (Zheng et al. 2014).
Plant growth and development and consequently grain yield are to a large extent dependent on nitrogen and phosphorus uptake. One component of the uptake system, the trimeric complex Nuclear Factor Y (NF-Y), binds to the CCAAT box, which is a universal element of the eukaryotic promoter. Each of three NF-Y subunits (NF-YA, NF-YB, and NF-YC) is encoded by multiple genes expressed either constitutively or in an organ-specific manner (Stephenson et al. 2007). Many of the genes are involved in the stress response of different species, including cereals (Nelson et al. 2007; Stephenson et al. 2007; Wei et al. 2010). TaNF-YC11 (Stephenson et al. 2007) and TaNF-YB3 (Stephenson et al. 2011), two of the light-upregulated NF-YC members in wheat, are involved in the regulation of photosynthesis-related genes. Recently, Yadav et al. (2015) reported that another two, TaNFYA-B1 and TaNF-YB4, play an important role in grain yield. The expression of TaNFYA-B1 was positively regulated in low nitrogen and phosphorus availability. It was accompanied by up-regulation of both nitrogen and phosphorus uptake transporters in roots and down-regulation of (tae)-miR169, which in turn can regulate expression of NFYAs. The changes of expression also stimulated root development. Overexpression of the gene significantly increased both nitrogen and phosphorus uptake, which allowed for a higher grain yield with lower usage of fertilizers (Qu et al. 2015). The positive role of the second gene, TaNF-YB4, in grain yield was documented by introduction of the gene into wheat by biolistic bombardment. Constitutive overexpression in transgenic wheat resulted in development of significantly more spikes and a 20–30% increase in grain yield compared with untransformed control plants under optimal watering conditions (Yadav et al. 2015).
Other genes promoting cell division and affecting wheat grain size
Other genes positively affecting grain size participate in various biological processes mainly involved in control of cell expansion and cell division and are not involved in plant hormone regulation.
GW2 encoding E3 RING ligase was shown to negatively regulate grain size in rice (Song 2007). The enzyme specifically mediates ubiquitination in the ubiquitin-26S proteasome system (Vierstra 2009). Orthologs of the gene have been identified in wheat by means of comparative genomics and cloned. TaGW2 homologs were found to be present in A, B, and D genomes. TaGW2-A and -D act in both the cell division and late grain-filling phase, and polymorphism in TaGW2-A was associated with grain size (Bednarek et al. 2012; Su et al. 2011). Experimental down-regulation of TaGW2 by means of RNAi-based silencing gave contradictory results. Plants with suppressed TaGW2 expression developed lower weight and size grains. Therefore, the gene function was opposite to its wild rice ortholog OsGW2, which negatively regulated grain size (Bednarek et al. 2012). Another set of transgenic lines, obtained and characterized by Hong et al. (2014), gave results consistent with the expected function of the gene as a negative regulator of grain size.
Abundance of the TaGW2 transcript was negatively associated with grain width and weight (Hong et al. 2014). According to Hong et al. (2014), the two sets of conflicting results might be the result of an off-target effect caused by the RNAi cassette in the experiments presented by Bednarek et al. (2012); alternatively, it might be related to the wheat variety used in the experiments. Our experience with RNAi-based silencing in wheat indicates that a very important factor to obtain the silent phenotype without an off-target effect is the transformation method, which might influence the final result (Zalewski et al. 2014). These contradictory results are dispelled in the recent research using the TILLING population, in which the gw2-A1 mutant allele with G to A transition in the splice acceptor site of exon 5 was identified (Simmonds et al. 2016). The mutation led to mis-splicing in the wild allele TaGW2-A1, which resulted in a significant 6.6% increase in thousand-grain weight (TGW) as well as increase of grain width and length in tetraploid and hexaploid wheat.
Thousand-grain weight6 (TGW6) in rice encodes a protein with indole-3-acetic acid (IAA)-glucose hydrolase activity and determines grain weight (Ishimaru et al. 2013). Recently, an ortholog of this gene was found in wheat. Low expression of haplotypes TaTGW6-b (InDel mutant) and TaTGW6-c (null mutant) was associated with low IAA content and significant increase in grain size and weight (Hanif et al. 2016; Hu et al. 2016). The same comparative genomics approach was used to isolate and characterize TaGS-D1, which represents another wheat ortholog of rice OsGS3 (Zheng et al. 2014). The gene is known to regulate stigma length and stigma extension in rice. TaGS-D1 was significantly associated with grain weight. InDels found in the introns of the gene in different genotypes influenced higher or lower TGW.
Another cell division promoting grain size TaGS5 gene in wheat (Ma et al. 2015a) and its ortholog OsGS5 in rice (Li et al. 2011) were found to play major roles in regulating grain size, purportedly by enhancing the number of cells in both species. The genes encode serine carboxypeptidases (SCPs), which are members of the a/b hydrolase proteins in the S_10 protein family. Homologs of TaGS5 in wheat, mapped to chromosomes 3 A, 3B, and 3D, were found to be preferentially expressed in young spikes and developing grains. TaGS5-3A-T allele, a single nucleotide polymorphism (SNP)-type mutation of TaGS5-3A, was shown to be significantly correlated with larger grains and greater thousand kernel weight. This mutation caused overexpression of the gene and significantly higher enzyme activity. An evolutionary approach to analyze the gene in di-, tetra-, and hexaploid wheats revealed that this allele was positively selected in global wheat breeding (Ma et al. 2015a). Rice orthologs of GS and GW influenced yield mainly through increasing source to sink assimilate flow (Ishimaru et al. 2013).
TaCYP78A3 is a gene encoding cytochrome P450 CYP78A3. The protein, affecting extension of integument cell proliferation, has been found to be specifically expressed in wheat reproductive organs and positively correlated with the final seed size. Virus-induced gene silencing (VIGS) of the gene in wheat led to a reduced cell number of the seed coat and decreased seed size. Ectopic overexpression of the gene in A. thaliana caused 11 to 48% increase of seed size (Ma et al. 2015b).
Starch and sucrose metabolism
The genes involved in regulation and metabolic reactions of starch and sucrose as well as nitrogen and phosphorus uptake were shown to affect plant architecture, and thereby grain yield. Starch is the main, up to 70%, component of grain endosperm, so its content can largely account for grain size and weight. Wheat sucrose synthase catalyzing the first step in the conversion of sucrose to starch is encoded by two wheat genes, TaSus1 and TaSus2, which are located in six loci on chromosomes 7A/7B/7D and 2A/2B/2D, respectively (Hou et al. 2014). The differences within the intron sequences between haplotypes were significantly correlated with the differences in thousand-kernel weight (TKW). The favored haplotype containing three SNPs in TaSus2-2B was shown to be under strong positive selection in wheat breeding in China (Jiang et al. 2011).
Field trials with transgenic wheat overexpressing barley HvSUT1 showed increased grain yield and micronutrient concentration (Saalbach et al. 2014; Weichert et al. 2010). The gene encodes the sucrose transporter (SUT), which stimulates storage protein synthesis. Another yield-related wheat anther-specific invertase gene, TaCWI, encodes cell wall invertase (CWI), which hydrolyzes sucrose into glucose and fructose. Among several haplotypes tested, only one, TaCWI-5D, was significantly associated with TKW (Jiang et al. 2015).
Glucan, water dikinase (GWD) is the primary enzyme required for starch phosphorylation. RNAi-mediated down-regulation of the gene, controlled by an endosperm-specific promoter, led to decreased GWD activity and starch phosphate content as well as several pleiotropic effects: higher α-amylase activity in the aleurone layer in mature grains as well as unexpected elevated grain size, early vigor and plant biomass (Ral et al. 2012; Corrigendum 2013).
The mutant of tiller inhibition, tin, opposite to the above-described mutants, is a negative example of grain productivity. The reduced tillering observed in the tin mutant is the result of inhibition of tiller bud outgrowth during the shoot apex transition from the vegetative to reproductive stage (Kebrom et al. 2012). The authors hypothesize that the tin mutant is associated with precocious internode development that diverts sucrose from developing tillers.
Conclusions
The potential annual yield increase of wheat achieved as a result of breeding is estimated at about 0.5% (Fischer et al. 2009). Furthermore, the globally observed multiyear tendency of yield growth is decreasing. These observations contrast starkly with the anticipated growth of the population, food demand, and bio-based fuel production. We propose that improving cereal productivity will need to use current knowledge on yield-determining genes in breeding programs incorporating these genes into the best yielding cultivars. This strategy combining current knowledge gained from modern biotechnology and at the same time based on natural mutants could be a creative continuation of the GR approach, which proved to be so successful.
As shown here, it is very important that genes identified as potential targets for yield-focused selection should be characterized for their function and interaction with other genes. It is crucial, because only certain allele combinations are beneficial for yield. Biotechnology creates the possibilities to modify a selected gene or to alter the gene’s expression to reveal the function of the gene. These approaches are particularly important for polyploid wheat, where natural mutants are rarely available. RNAi-based gene silencing allows down-regulation of a single selected gene, several homoeologs or a group of genes belonging to a gene family. This approach, relatively straightforward for loss of function gene analysis, generates transgenic lines with diverse silencing ratios. This opens the possibility for functional analysis of a gene, whose knock-out mutation is lethal or semi-lethal. The rapidly developing gene-editing CRISPR/Cas system is already considered a breakthrough technology. It offers the possibility to change the selected region of a targeted gene, which leads to knock-out mutation (the easiest to achieve) or alternatively to specific up- or down-regulation of gene expression. Since this can be done already in the selected cultivar, it alleviates the need for lengthy backcrossing. It is important to add that gene editing is indistinguishable from natural mutations. At this point, it is not decided whether this technology will be excluded in the EU from GMO legislature; however, the first FDA decisions on CRISPR/Cas-edited champignons confirmed non-GMO status of the plant. Combining rapidly developing CRISPR/Cas systems with a knowledge of gene function could speed up the process of creating new, better yielding cultivars. Moreover, the knowledge of a specific allele function and its nucleotide sequence should be used to identify natural mutants or haplotypes in a natural population or induced mutants using the TILLING strategy. This also enables the design of allele specific markers for efficient phenotype/haplotype selection during breeding.
The implementation of selected genes in breeding programs requires consideration of specific genotypes, agronomic, and climate conditions and the fact that many of the genes are members of multigene families. Modulated expression of flowering genes, which regulate photoperiod and vernalization-dependent floral induction, might be advantageous for spring or winter varieties under long-day or short-day conditions. Very spectacular yield effects were reported for genes involved in hormone regulation, gibberellins, brassinosteroids, and cytokinins. Since genes of gibberellin and brassinosteroid metabolism and signaling are mainly involved in culm robustness, the cytokinin metabolism genes have a big impact on grain number in barley and rice and grain size in wheat. These organ-specific genes regulate cytokinin homeostasis within reproductive organs, and they are mostly independent of agronomic or climate conditions. The general rule is that reduced expression of CKX in specified organs leads to increased yield. However, their regulation may depend on whether the crop is sink limited or grows in a source-limiting environment. The source/sink regulation might be coordinated with the sucrose and starch metabolism genes increasing grain size in wheat as well as the yield-increasing, nitrate signaling NAC and nitrogen and phosphorus usage NFY genes.
Many yield-related traits and hormone regulators are encoded by multigene families. Their expression patterns, showing strong spatial and temporal regulation, provide strong indications for their possible role as yield-affecting components. Thus, transcriptome analysis could be the first step of screening the candidate genes. Special attention should be paid to microRNA encoding genes. It is estimated that one-third of all protein encoding genes are regulated by microRNAs (Jones-Rhoades et al. 2006), which complies with the increasing number of reports proving that these regulators are crucial in cereal productivity.
All characterized genes determine various growth components, which should be combined with nutritional status, the activity of particular phytohormones, and the synchronization of floret development. Pyramiding of these yield-affecting genes with other agronomically important genes might be proposed for further improvement.
Author contribution statement
ANO is the main author of the concept and the design of the review, including supplementary tables. IR is the author of Figs. 1, 2 and 3, and participated in preparing the literature. WO took part in designing, writing and discussion. SG contributed to the discussion and preparation of supplementary tables.
References
Appleford NE, Evans DJ, Lenton JR, Gaskin P, Croker SJ, Devos KM, Phillips AL, Hedden P (2006) Function and transcript analysis of gibberellin-biosynthetic enzymes in wheat. Planta 223:568–582. doi:10.1007/s00425-005-0104-0
Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M (2005) Cytokinin oxidase regulates rice grain production. Science 309:741–745
Barboza L, Effgen S, Alonso-Blanco C, Kooke R, Keurentjes JJB, Koornnef M, Alcázar R (2013) Arabidopsis semidwarfs evolved from independent mutations in GA20ox1, ortholog to green revolution dwarf alleles in rice and barley. Proc Natl Acad Sci USA 110:15818–15823
Beales J, Turner A, Griffiths S, Snape JW, Laurie DA (2007) A pseudo-response regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Theor Appl Genet 115:721–733. doi:10.1007/s00122-007-0603-4
Bednarek J, Boulaflous A, Girousse C, Ravel C, Tassy C, Barret P, Bouzidi MF, Mouzeyar S (2012) Down-regulation of the TaGW2 gene by RNA interference results in decreased grain size and weight in wheat. J Exp Bot 63:5945–5955. doi:10.1093/jxb/err313
Bennett MD, Smith JB (1976) Nuclear DNA amounts in angiosperms. Philos Trans R Soc Lond B 274:227–274. doi:10.1098/rstb.1976.0044
Bhati KK, Alok A, Kumar A, Kaur J, Tiwari S, Pandey AK (2016) Silencing of ABCC13 transporter in wheat reveals its involvement in grain development, phytic acid accumulation and lateral root formation. J Exp Bot 67:4379–4389. doi:10.1093/jxb/erw224
Boden S, Cavanagh C, Cullis BR, Ramm K, Greenwood J, Finnegan EJ, Trevaskis B, Swain SM (2015) Ppd-1 is a key regulator of inflorescence architecture and paired spikelet development in wheat. Nat Plants 1. doi:10.1038/nplants.2014.1016
Borlaug NE (1983) Contributions of conventional plant breeding to food production. Science 219:689–693
Börner A, Worland AJ, Plaschke J, Schumann E, Law CN (1993) Pleiotropic effects of genes for reduced height (Rht) and day-length insensitivity (Ppd) on yield and its components for wheat grown in middle Europe. Plant Breed 111:204–216
Börner A, Plaschke J, Korzun V, Worland AJ (1996) The relationships between the dwarfing genes of wheat and rye. Euphytica 89:69–75
Börner A, Roder M, Korzun V (1997) Comparative molecular mapping of GA insensitive Rht loci on chromosomes 4B and 4D of common wheat (Triticum aestivum L.). Theor Appl Genet 95:1133–1137
Börner A, Korzun V, Malyshev S, Ivandic V, Graner A (1999) Molecular mapping of two dwarfing genes differing in their GA response on chromosome 2 H of barley. Theor Appl Genet 99:670–675
Bortiri E, Chuck G, Vollbrecht E, Rocheford T, Martienssen R, Hake S (2006) ramosa2 encodes a LATERAL ORGAN BOUNDARY domain protein that determines the fate of stem cells in branch meristems of maize. Plant Cell 18:574–585. doi:10.1105/tpc.105.039032
Brocklehurst PA (1977) Factors controlling grain weight in wheat. Nature 266:348–349
Cao S, Xu Q, Cao Y, Qian K, An K, Zhu Y, Binzeng H, Zhao H, Kuai B (2005) Loss-of-function mutations in DET2 gene lead to an enhanced resistance to oxidative stress in Arabidopsis. Physiol Plant 123:57–66. doi:10.1111/j.1399-3054.2004.00432.x
Chandler PM, Harding CA (2013) ‘Overgrowth’ mutants in barley and wheat: new alleles and phenotypes of the ‘Green Revolution’ DELLA gene. J Exp Bot 64:1603–1613. doi:10.1093/jxb/ert022
Chandler PM, Marion-Poll A, Ellis M, Gubler F (2002) Mutants at the Slender1 locus of barley cv Himalaya. Molecular and physiological characterization. Plant Physiol 129:181–190. doi:10.1104/pp.010917
Chang C, Lu J, Zhang HP, Ma CX, Sun G (2015) Copy number variation of cytokinin oxidase gene Tackx4 associated with grain weight and chlorophyll content of flag leaf in common wheat. PLoS One 10:e0145970. doi:10.1371/journal.pone.0145970
Che R, Tong H, Shi B, Liu Y, Fang S, Liu D, Xiao Y, Hu B, Liu L, Wang H, Zhao M, Chu C (2015) Control of grain size and rice yield by GL2-mediated brassinosteroid responses. Nat Plants 2:15195. doi:10.1038/nplants.2015.195
Cho JI, Lee SK, Ko S, Kim HK, Jun SH, Lee YH, Bhoo SH, Lee KW, An G, Hahn TR, Jeon JS (2005) Molecular cloning and expression analysis of the cell-wall invertase gene family in rice (Oryza sativa L.). Plant Cell Rep 24:225–236. doi:10.1007/s00299-004-0910-z
Chono M, Honda I, Zeniya H, Yoneyama K, Saisho D, Takeda K, Takatsuto S, Hoshino T, Watanabe Y (2003) A semidwarf phenotype of barley uzu results from a nucleotide substitution in the gene encoding a putative brassinosteroid receptor. Plant Physiol 133:1209–1219. doi:10.1104/pp.103.026195
Christiansen MW, Holm PB, Gregersen PL (2011) Characterization of barley (Hordeum vulgare L.) NAC transcription factors suggests conserved functions compared to both monocots and dicots. BMC Res Notes 4:302. doi:10.1186/1756-0500-4-302
Christiansen MW, Matthewman C, Podzimska-Sroka D, O’Shea C, Lindemose S, Mollegaard NE, Holme IB, Hebelstrup K, Skriver K, Gregersen PL (2016) Barley plants over-expressing the NAC transcription factor gene HvNAC005 show stunting and delay in development combined with early senescence. J Exp Bot 67:5259–5273. doi:10.1093/jxb/erw286
Cockram J, Jones H, Leigh FJ, O’Sullivan D, Powell W, Laurie DA, Greenland AJ (2007) Control of flowering time in temperate cereals: genes, domestication, and sustainable productivity. J Exp Bot 58:1231–1244. doi:10.1093/jxb/erm042
Dahleen LS, Vander Wal LJ, Franckowiak JD (2005) Characterization and molecular mapping of genes determining semidwarfism in barley. J Hered 96:654–662. doi:10.1093/jhered/esi125
Distelfeld A, Pearce SP, Avni R, Scherer B, Uauy C, Piston F, Slade A, Zhao R, Dubcovsky J (2012) Divergent functions of orthologous NAC transcription factors in wheat and rice. Plant Mol Biol 78:515–524. doi:10.1007/s11103-012-9881-6
Distelfeld A, Avni R, Fischer AM (2014) Senescence, nutrient remobilization, and yield in wheat and barley. J Exp Bot 65:3783–3798. doi:10.1093/jxb/ert477
Divi UK, Krishna P (2009) Brassinosteroid: a biotechnological target for enhancing crop yield and stress tolerance. New Biotechnol 26:131–136
Dobrovolskaya O, Pont C, Sibout R, Martinek P, Badaeva E, Murat F, Chosson A, Watanabe N, Prat E, Gautier N, Gautier V, Poncet C, Orlov YL, Krasnikov AA, Berges H, Salina E, Laikova L, Salse J (2015) FRIZZY PANICLE drives supernumerary spikelets in bread wheat. Plant Physiol 167:189–199. doi:10.1104/pp.114.250043
Dockter C, Hansson M (2015) Improving barley culm robustness for secured crop yield in a changing climate. J Exp Bot 66:3499–3509. doi:10.1093/jxb/eru521
Dockter C, Gruszka D, Braumann I, Druka A, Druka I, Franckowiak J, Gough SP, Janeczko A, Kurowska M, Lundqvist J, Lundqvist U, Marzec M, Matyszczak I, Muller AH, Oklestkova J, Schulz B, Zakhrabekova S, Hansson M (2014) Induced variations in brassinosteroid genes define barley height and sturdiness, and expand the green revolution genetic toolkit. Plant Physiol 166:1912–1927. doi:10.1104/pp.114.250738
Doležel J, Binarová P, Lucretti S (1989) Analysis of nuclear DNA content in plant cells by flow cytometry. Biol Plant 31:113–120
Druka A, Franckowiak J, Lundqvist U, Bonar N, Alexander J, Houston K, Radovic S, Shahinnia F, Vendramin V, Morgante M, Stein N, Waugh R (2011) Genetic dissection of barley morphology and development. Plant Physiol 155:617–627. doi:10.1104/pp.110.166249
Dubcovsky J, Dvorak J (2007) Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 316:1862–1866
Echeverry-Solarte M, Kumar A, Kianian S, Mantovani EE, Simsek S, Alamri MS, Mergoum M (2014) Genome-wide genetic dissection of supernumerary spikelet and related traits in common wheat (Triticum aestivum L.). Plant. Genome 7:1–16. doi:10.3835/plantgenome2014.03.0013
Endress PK (2010) Disentangling confusions in inflorescence morphology: Patterns and diversity of reproductive shoot ramification in angiosperms. J Syst Evol 48:225–239. doi:10.1111/j.1759-6831.2010.00087.x
Evans LT (1998) Feeding the ten billion: plants and population growth. Cambridge University Press, Cambridge
Fischer RA, Byerlee D, Edmeades GO (2009) Can technology deliver on the yield challenge to 2050? FAO Expert Meeting on How to Feed the World in 2050, pp 24–26. Rome
Flintham JE, Gale MD (1982) The Tom Thumb dwarfing gene, Rht3 in wheat I. Reduced preharvest sprouting damage to bread making quality. Theor Appl Genet 62:121–126
Flintham JE, Borner A, Worland AJ, Gale MD (1997) Optimizing wheat grain yield: effects of Rht (gibberellin-insensitive) dwarfing genes. J Agric Sci 128:11–25
Forster BP, Franckowiak JD, Lundqvist U, Lyon J, Pitkethly I, Thomas WT (2007) The barley phytomer. Ann Bot 100:725–733. doi:10.1093/aob/mcm183
Franckowiak JD, Lundqvist U (2012) Descriptions of barley genetic stocks for 2012. Barley Genet Newsl 42:36–173
Fu D, Uauy C, Blechl A, Dubcovsky J (2007) RNA interference for wheat functional gene analysis. Transgenic Res 16:689–701. doi:10.1007/s11248-007-9150-7
Gale MD, Marshall GA (1973) Insensitivity to gibberellin in dwarf wheats. Ann Bot 37:729–735
Gale MD, Youssefian S (1985) Dwarfing genes in wheat. In: Russell GE (ed) Progress in plant breeding. Butterworths, London, pp 1–35
Gasparis S, Orczyk W, Zalewski W, Nadolska-Orczyk A (2011) The RNA-mediated silencing of one of the Pin genes in allohexaploid wheat simultaneously decreases the expression of the other, and increases grain hardness. J Exp Bot 62:4025–4036. doi:10.1093/jxb/err103
Gasparis S, Orczyk W, Nadolska-Orczyk A (2013) Sina and Sinb genes in triticale do not determine grain hardness contrary to their orthologs Pina and Pinb in wheat. BMC Plant Biol 13:190
Gasperini D, Greenland A, Hedden P, Dreos R, Harwood W, Griffiths S (2012) Genetic and physiologycal analysis of Rht8 in bread weat: an alternative source of semi-dwarfism with a reduced sensitivity to brassinosteroids. J Exp Bot 63:4419–4436
Gill KS, Gill BS, Endo TR, Taylor T (1996a) Identification and high-density mapping of gene-rich regions in chromosome group I of wheat. Genetics 144:1883–1891
Gill KS, Gill BS, Endot TR, Boyko EV (1996b) Identification and high-density mapping of gene-rich regions in chromosome group 5 of wheat. Genetics 143:1001–1012
Hanif M, Gao F, Liu J, Wen W, Zhang Y, Rasheed A, Xia X, He Z, Cao S (2016) TaTGW6-A1, an ortholog of rice TGW6, is associated with grain weight and yield in bread wheat. Mol Breed 36:1
He X, Qu B, Li W, Zhao X, Teng W, Ma W, Ren Y, Li B, Li Z, Tong Y (2015) The nitrate-inducible NAC transcription factor TaNAC2-5A controls nitrate response and increases wheat yield. Plant Physiol 169:1991–2005. doi:10.1104/pp.15.00568
Hedden P (2003) The genes of the green revolution. Trends Genet 19:5–9
Helliwell CA, Chandler PM, Poole A, Dennis ES, Peacock WJ (2001) The CYP88A cytochrome P450, ent-kaurenoic acid oxidase, catalyzes three steps of the gibberellin biosynthesis pathway. Proc Natl Acad Sci USA 98:2065–2070. doi:10.1073/pnas.041588998
Higgins JA, Bailey PC, Laurie DA (2010) Comparative genomics of flowering time pathways using Brachypodium distachyon as a model for the temperate grasses. PLoS One 5:e10065. doi:10.1371/journal.pone.0010065
Hong Y, Chen L, Du LP, Su Z, Wang J, Ye X, Qi L, Zhang Z (2014) Transcript suppression of TaGW2 increased grain width and weight in bread wheat. Funct Integr Genomics 14:341–349. doi:10.1007/s10142-014-0380-5
Hou J, Jiang Q, Hao C, Wang Y, Zhang H, Zhang X (2014) Global selection on sucrose synthase haplotypes during a century of wheat breeding. Plant Physiol 164:1918–1929. doi:10.1104/pp.113.232454
Houimli SIM, Denden M, Elhadj SB (2008) Induction of salt tolerance in pepper (Capsicum annuum) by 24-epibrassinolide. EurAsian J BioSci 2:83–90
Houston K, McKim SM, Comadran J, Bonar N, Druka I, Uzrek N, Cirillo E, Guzy-Wrobelska J, Collins NC, Halpin C, Hansson M, Dockter C, Druka A, Waugh R (2013) Variation in the interaction between alleles of HvAPETALA2 and microRNA172 determines the density of grains on the barley inflorescence. Proc Natl Acad Sci USA 110:16675–16680. doi:10.1073/pnas.1311681110
Hu M-J, Zhang H-P, Cao J-J, Zhu X-F, Wang S-X, Jiang H, Wu ZY, Lu J, Chang C, Sun G-L, Ma C-X (2016) Characterization of an IAA-glucose hydrolase gene TaTGW6 associated with grain weight in common wheat (Triticum aestivum L.). Mol Breed 36:25
Ishimaru K, Hirotsu N, Madoka Y, Murakami N, Hara N, Onodera H, Kashiwagi T, Ujiie K, Shimizu B, Onishi A, Miyagawa H, Katoh E (2013) Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat Genet 45:707–711. doi:10.1038/ng.2612
Jameson PE, Song J (2015) Cytokinin: a key driver of seed yield. J Exp Bot 67:593–606. doi:10.1093/jxb/erv461
Jia Q, Zhang J, Westcott S, Zhang XQ, Bellgard M, Lance R, Li C (2009) GA-20 oxidase as a candidate for the semidwarf gene sdw1/denso in barley. Funct Integr Genomics 9:255–262. doi:10.1007/s10142-009-0120-4
Jiang Q, Hou J, Hao C, Wang L, Ge H, Dong Y, Zhang X (2011) The wheat (T. aestivum) sucrose synthase 2 gene (TaSus2) active in endosperm development is associated with yield traits. Funct Integr Genomics 11:49–61. doi:10.1007/s10142-010-0188-x
Jiang F, Guo M, Yang F, Duncan K, Jackson D, Rafalski A, Wang S, Li B (2012) Mutations in an AP2 transcription factor-like gene affect internode length and leaf shape in maize. PLoS One 7:e37040. doi:10.1371/journal.pone.0037040
Jiang Y, Jiang Q, Hao C, Hou J, Wang L, Zhang H, Zhang S, Chen X, Zhang X (2015) A yield-associated gene TaCWI, in wheat: its function, selection and evolution in global breeding revealed by haplotype analysis. Theor Appl Genet 128:131–143. doi:10.1007/s00122-014-2417-5
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Jost M, Taketa S, Mascher M, Himmelbach A, Yuo T, Shahinnia F, Rutten T, Druka A, Schmutzer T, Steuernagel B, Beier S, Taudien S, Scholz U, Morgante M, Waugh R, Stein N (2016) A homolog of Blade-On-Petiole 1 and 2 (BOP1/2) controls internode length and homeotic changes of the barley inflorescence. Plant Physiol 171:1113–1127. doi:10.1104/pp.16.00124
Kapazoglou A, Tondelli A, Papaefthimiou D, Ampatzidou H, Francia E, Stanca MA, Bladenopoulos K, Tsaftaris AS (2010) Epigenetic chromatin modifiers in barley: IV. The study of barley polycomb group (PcG) genes during seed development and in response to external ABA. BMC Plant Biol 10:73. doi:10.1186/1471-2229-10-73
Kebrom TH, Chandler PM, Swain SM, King RW, Richards RA, Spielmeyer W (2012) Inhibition of tiller bud outgrowth in the tin mutant of wheat is associated with precocious internode development. Plant Physiol 160:308–318. doi:10.1104/pp.112.197954
Kellogg EA, Camara PE, Rudall PJ, Ladd P, Malcomber ST, Whipple CJ, Doust AN (2013) Early inflorescence development in the grasses (Poaceae). Front Plant Sci 4:250. doi:10.3389/fpls.2013.00250
Kesavan M, Song JT, Seo HS (2013) Seed size: a priority trait in cereal crops. Physiol Plant 147:113–120. doi:10.1111/j.1399-3054.2012.01664.x
Khripach V, Zhabinskii V, de Groot A (2000) Twenty years of brassinosteroids: steroidal plant hormones warrant better crops for the XXI century. Ann Bot 86:441–447
Komatsuda T, Pourkheirandish M, He C, Azhaguvel P, Kanamori H, Perovic D, Stein N, Graner A, Wicker T, Tagiri A, Lundqvist U, Fujimura T, Matsuoka M, Matsumoto T, Yano M (2007) Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc Natl Acad Sci USA 104:1424–1429. doi:10.1073/pnas.0608580104
Koppolu R, Anwar N, Sakuma S, Tagiri A, Lundqvist U, Pourkheirandish M, Rutten T, Seiler C, Himmelbach A, Ariyadasa R, Youssef HM, Stein N, Sreenivasulu N, Komatsuda T, Schnurbusch T (2013) Six-rowed spike4 (Vrs4) controls spikelet determinacy and row-type in barley. Proc Natl Acad Sci USA 110:13198–13203
Lawlor DW, Paul MJ (2014) Source/sink interactions underpin crop yield: the case for trehalose 6-phosphate/SnRK1 in improvement of wheat. Front Plant Sci 5:418. doi:10.3389/fpls.2014.00418
Lawrence RJ, Pikaard CS (2003) Transgene-induced RNA interference: a strategy for overcoming gene redundancy in polyploids to generate loss-of-function mutations. Plant J 36:114–121. doi:10.1046/j.1365-313X.2003.01857
Lawrenson T, Shorinola O, Stacey N, Li C, Ostergaard L, Patron N, Uauy C, Harwood W (2015) Induction of targeted, heritable mutations in barley and Brassica oleracea using RNA-guided Cas9 nuclease. Genome Biol 16:258. doi:10.1186/s13059-015-0826-7
Li XP, Lan SQ, Feng YR (2006) Effects on agronomic chracteristics of Rht-B1b, Rht-D1b, Rht-B1c wheat dwarfing genes and theirs combinations in artificial climate chamber. J Hebei Agric Sci 10:14–18
Li Y, Fan C, Xing Y, Jiang Y, Luo L, Sun L, Shao D, Xu C, Li X, Xiao J, He Y, Zhang Q (2011) Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat Genet 43:1266–1269
Li H, Chen G, Yan W (2015) Molecular characterization of barley 3 H semi-dwarf genes. PLoS One 10:e0120558. doi:10.1371/journal.pone.0120558
Lima Neto MC, Lobo AK, Martins MO, Fontenele AV, Silveira JA (2014) Dissipation of excess photosynthetic energy contributes to salinity tolerance: a comparative study of salt-tolerant Ricinus communis and salt-sensitive Jatropha curcas. J Plant Physiol 171:23–30. doi:10.1016/j.jplph.2013.09.002
Liu H, Bayer M, Druka A, Russell JR, Hackett CA, Poland J, Ramsay L, Hedley PE, Waugh R (2014) An evaluation of genotyping by sequencing (GBS) to map the Breviaristatum-e (ari-e) locus in cultivated barley. BMC Genomics 15:104
Lu J, Chang C, Zhang HP, Wang SX, Sun G, Xiao SH, Ma CX (2015) Identification of a Novel allele of TaCKX6a02 associated with grain size, filling rate and weight of common wheat. PLoS One 10:e0144765. doi:10.1371/journal.pone.0144765
Lundqvist U, Lundqvist A (1988) Induced intermedium mutants in barley: origin, morphology and inheritance. Hereditas 108:13–26
Lundqvist U, Franckowiak JD, Konishi T (1997) New and revised descriptions of barley genes. Barley Genet Newsl 26:22–516
Ma L, Li T, Hao C, Wang Y, Chen X, Zhang X (2015a) TaGS5-3A, a grain size gene selected during wheat improvement for larger kernel and yield. Plant Biotechnol J:1–12. doi:10.1111/pbi.12492
Ma M, Wang Q, Li Z, Cheng H, Li Z, Liu X, Song W, Appels R, Zhao H (2015b) Expression of TaCYP78A3, a gene encoding cytochrome P450 CYP78A3 protein in wheat (Triticum aestivum L.), affects seed size. Plant J 83:312–325. doi:10.1111/tpj.12896
Mayer KF, Martis M, Hedley PE, Simkova H, Liu H, Morris JA, Steuernagel B, Taudien S, Roessner S, Gundlach H, Kubalakova M, Suchankova P, Murat F, Felder M, Nussbaumer T, Graner A, Salse J, Endo T, Sakai H, Tanaka T, Itoh T, Sato K, Platzer M, Matsumoto T, Scholz U, Dolezel J, Waugh R, Stein N (2011) Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell 23:1249–1263. doi:10.1105/tpc.110.082537
Millet E, Pinthus MJ (1984) The association between grain volume and grain weight in wheat. J Cereal Sci 2:31–35
Mochida K, Yamazaki Y, Ogihara Y (2003) Discrimination of homoeologous gene expression in hexaploid wheat by SNP analysis of contigs grouped from a large number of expressed sequence tags. Mol Genet Genomics 270:371–377. doi:10.1007/s00438-003-0939-7
Mok DW, Mok MC (2001) Cytokinin metabolism and action. Annu Rev Plant Physiol Plant Mol Biol 52:89–118
Mulki MA, Von Korff M (2016) CONSTANS controls floral repression by up-regulating VERNALIZATION2 (VRN-H2) in barley. Plant Physiol 170:325–337. doi:10.1104/pp.15.01350
Nadolska-Orczyk A, Gasparis S, Orczyk W (2009) The determinants of grain texture in cereals. J Appl Genet 50:185–197
Nallamilli BR, Zhang J, Mujahid H, Malone BM, Bridges SM, Peng Z (2013) Polycomb group gene OsFIE2 regulates rice (Oryza sativa) seed development and grain filling via a mechanism distinct from Arabidopsis. PLoS Genet 9:e1003322. doi:10.1371/journal.pgen.1003322
Nelson DE, Repetti PP, Adams TR, Creelman RA, Wu J, Warner DC, Anstrom DC, Bensen RJ, Castiglioni PP, Donnarummo MG, Hinchey BS, Kumimoto RW, Maszle DR, Canales RD, Krolikowski KA, Dotson SB, Gutterson N, Ratcliffe OJ, Heard JE (2007) Plant nuclear factor Y (NF-Y) B subunits confer drought tolerance and lead to improved corn yields on water-limited acres. Proc Natl Acad Sci USA 104:16450–16455. doi:10.1073/pnas.0707193104
Olsen AN, Ernst HA, Leggio LL, Skriver K (2005) NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci 10:79–87. doi:10.1016/j.tplants.2004.12.010
Paul P, Awasthi A, Rai AK, Gupta SK, Prasad R, Sharma TR, Dhaliwal HS (2012) Reduced tillering in Basmati rice T-DNA insertional mutant OsTEF1 associates with differential expression of stress related genes and transcription factors. Funct Integr Genomics 12:291–304. doi:10.1007/s10142-012-0264-5
Pearce S, Saville R, Vaughan SP, Chandler PM, Wilhelm EP, Sparks CA, Al-Kaff N, Korolev A, Boulton MI, Phillips AL, Hedden P, Nicholson P, Thomas SG (2011) Molecular characterization of Rht-1 dwarfing genes in hexaploid wheat. Plant Physiol 157:1820–1831. doi:10.1104/pp.111.183657
Peng J, Richards DE, Hartley NM, Murphy GP, Devos KM, Flintham JE, Beales J, Fish LJ, Worland AJ, Pelica F (1999) ‘Green Revolution’ genes encode mutant gibberellin response modulators. Nature 400:256–261
Pfeifer M, Kugler KG, Sandve SR, Zhan B, Rudi H, Hvidsten TR, International Wheat Genome Sequencing Consortium, Mayer KF, Olsen OA (2014) Genome interplay in the grain transcriptome of hexaploid bread wheat. Science 345:1250091
Poursarebani N, Seidensticker T, Koppolu R, Trautewig C, Gawronski P, Bini F, Govind G, Rutten T, Sakuma S, Tagiri A, Wolde GM, Youssef HM, Battal A, Ciannamea S, Fusca T, Nussbaumer T, Pozzi C, Borner A, Lundqvist U, Komatsuda T, Salvi S, Tuberosa R, Uauy C, Sreenivasulu N, Rossini L, Schnurbusch T (2015) The genetic basis of composite spike form in barley and ‘Miracle-Wheat’. Genetics 201:155–165. doi:10.1534/genetics.115.176628
Qi W, Sun F, Wang Q, Chen M, Huang Y, Feng YQ, Luo X, Yang J (2011) Rice ethylene-response AP2/ERF factor OsEATB restricts internode elongation by down-regulating a gibberellin biosynthetic gene. Plant Physiol 157:216–228. doi:10.1104/pp.111.179945
Qu B, He X, Wang J, Zhao Y, Teng W, Shao A, Zhao X, Ma W, Wang J, Li B, Li Z, Tong Y (2015) A wheat CCAAT box-binding transcription factor increases the grain yield of wheat with less fertilizer input. Plant Physiol 167:411–423. doi:10.1104/pp.114.246959
Ral JP, Bowerman AF, Li Z, Sirault X, Furbank R, Pritchard JR, Bloemsma M, Cavanagh CR, Howitt CA, Morell MK (2012) Down-regulation of glucan, water-dikinase activity in wheat endosperm increases vegetative biomass and yield. Plant Biotechnol J 10:871–882. doi:10.1111/j.1467-7652.2012.00711.x
Ral JP, Bowerman AF, Li Z, Sirault X, Furbank R, Pritchard JR, Bloemsma M, Cavanagh CR, Howitt CA, Morell M (2013) Down-regulation of glucan, water-dikinase activity in wheat endosperm increase vegetative biomass and yield. Plant Biotechnol J 11:390–391
Ramsay L, Comadran J, Druka A, Marshall DF, Thomas WT, Macaulay M, MacKenzie K, Simpson C, Fuller J, Bonar N, Hayes PM, Lundqvist U, Franckowiak JD, Close TJ, Muehlbauer GJ, Waugh R (2011) INTERMEDIUM-C, a modifier of lateral spikelet fertility in barley, is an ortholog of the maize domestication gene TEOSINTE BRANCHED 1. Nat Genet 43:169–172. doi:10.1038/ng.745
Remizowa MV, Rudall PJ, Choob VV, Sokoloff DD (2013) Racemose inflorescences of monocots: structural and morphogenetic interaction at the flower/inflorescence level. Ann Bot 112:1553–1566. doi:10.1093/aob/mcs246
Saalbach I, Mora-Ramírez I, Weichert N, Andersch F, Guild G, Wieser H, Koehler P, Stangoulis J, Kumlehn J, Weschke W, Weber H (2014) Increased grain yield and micronutrient concentration in transgenic winter wheat by ectopic expression of a barley sucrose transporter. J Cereal Sci 60:75–81. doi:10.1016/j.jcs.2014.01.017
Sakakibara H (2006) Cytokinins: activity, biosynthesis, and translocation. Annu Rev Plant Biol 57:431–449
Sakamoto T, Morinaka Y, Ohnishi T, Sunohara H, Fujioka S, Ueguchi-Tanaka M, Mizutani M, Sakata K, Takatsuto S, Yoshida S, Tanaka H, Kitano H, Matsuoka M (2006) Erect leaves caused by brassinosteroid deficiency increase biomass production and grain yield in rice. Nat Biotechnol 24:105–109
Sakuma S, Pourkheirandish M, Matsumoto T, Koba T, Komatsuda T (2010) Duplication of a well-conserved homeodomain-leucine zipper transcription factor gene in barley generates a copy with more specific functions. Funct Integr Genomics 10:123–133. doi:10.1007/s10142-009-0134-y
Saville RJ, Gosman N, Burt CJ, Makepeace J, Steed A, Corbitt M, Chandler E, Brown JK, Boulton MI, Nicholson P (2012) The ‘Green Revolution’ dwarfing genes play a role in disease resistance in Triticum aestivum and Hordeum vulgare. J Exp Bot 63:1271–1283. doi:10.1093/jxb/err350
Schulte D, Close TJ, Graner A, Langridge P, Matsumoto T, Muehlbauer G, Sato K, Schulman AH, Waugh R, Wise RP, Stein N (2009) The international barley sequencing consortium at the threshold of efficient access to the barley genome. Plant Physiol 149:142–147. doi:10.1104/pp.108.128967
Shahbaz M, Ashraf M, Athar H-u-R (2008) Does exogenous application of 24-epibrassinolide ameliorate salt induced growth inhibition in wheat (Triticum aestivum L.)? Plant Growth Regul 55:51–64. doi:10.1007/s10725-008-9262-y
Shahinnia F, Druka A, Franckowiak J, Morgante M, Waugh R, Stein N (2012) High resolution mapping of Dense spike-ar (dsp.ar) to the genetic centromere of barley chromosome 7H. Theor Appl Genet 124:373–384. doi:10.1007/s00122-011-1712-7
Shan Q, Wang Y, Li J, Gao C (2014) Genome editing in rice and wheat using the CRISPR/Cas system. Nat Protoc 9:2395–2410. doi:10.1038/nprot.2014.157
Sharman BC (1994) Branched heads in wheat and wheat hybrids. Nature 153:497–498
Shaw LM, Turner AS, Laurie DA (2012) The impact of photoperiod insensitive Ppd-1a mutations on the photoperiod pathway across the three genomes of hexaploid wheat (Triticum aestivum). Plant J 71:71–84. doi:10.1111/j.1365-313X.2012.04971.x
Shaw LM, Turner AS, Herry L, Griffiths S, Laurie DA (2013) Mutant alleles of Photoperiod-1 in wheat (Triticum aestivum L.) that confer a late flowering phenotype in long days. PLoS One 8:e79459. doi:10.1371/journal.pone.0079459
Simmonds J, Scott P, Brinton J, Mestre TC, Bush M, Del Blanco A, Dubcovsky J, Uauy C (2016) A splice acceptor site mutation in TaGW2-A1 increases thousand grain weight in tetraploid and hexaploid wheat through wider and longer grains. Theor Appl Genet 129:1099–1112. doi:10.1007/s00122-016-2686-2
Simons KJ, Fellers JP, Trick HN, Zhang Z, Tai YS, Gill BS, Faris JD (2006) Molecular characterization of the major wheat domestication gene Q. Genetics 172:547–555. doi:10.1534/genetics.105.044727
Skov Kristensen P, Dockter C, Lundqvist U, Lu Q, Gregersen PL, Thordal-Christensen H, Hansson M, Pillen K (2016) Genetic mapping of the barley lodging resistance locus Erectoides-k. Plant Breed 135:420–428. doi:10.1111/pbr.12377
Song XJ (2007) A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet 39:623–630
Song J, Jiang L, Jameson PE (2012) Co-ordinate regulation of cytokinin gene family members during flag leaf and reproductive development in wheat. BMC Plant Biol 12:78. doi:10.1186/1471-2229-12-78
Sreenivasulu N, Schnurbusch T (2012) A genetic playground for enhancing grain number in cereals. Trends Plant Sci 17:91–101. doi:10.1016/j.tplants.2011.11.003
Srinivasachary, Gosman N, Steed A, Hollins TW, Bayles R, Jennings P, Nicholson P (2009) Semi-dwarfing Rht-B1 and Rht-D1 loci of wheat differ significantly in their influence on resistance to Fusarium head blight. Theor Appl Genet 118:695–702. doi:10.1007/s00122-008-0930-0
Stephenson TJ, McIntyre CL, Collet C, Xue GP (2007) Genome-wide identification and expression analysis of the NF-Y family of transcription factors in Triticum aestivum. Plant Mol Biol 65:77–92. doi:10.1007/s11103-007-9200-9
Stephenson TJ, McIntyre CL, Collet C, Xue GP (2011) TaNF-YB3 is involved in the regulation of photosynthesis genes in Triticum aestivum. Funct Integr Genomics 11:327–340. doi:10.1007/s10142-011-0212-9
Su Z, Hao C, Wang L, Dong Y, Zhang X (2011) Identification and development of a functional marker of TaGW2 associated with grain weight in bread wheat (Triticum aestivum L.). Theor Appl Genet 122:211–223. doi:10.1007/s00122-010-1437-z
Sun TP, Gubler F (2004) Molecular mechanism of gibberellin signaling in plants. Annu Rev Plant Biol 55:197–223
Tester M, Langridge P (2010) Breeding technologies to increase crop production in a changing world. Science 327:818–822. doi:10.1126/science.1183700
The International Wheat Genome Sequencing C (2014) A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 345:1251788. doi:10.1126/science.1251788
Travella S, Klimm TE, Keller B (2006) RNA interference-based gene silencing as an efficient tool for functional genomics in hexaploid bread wheat. Plant Physiol 142:6–20. doi:10.1104/pp.106.084517
Turner A, Beales J, Faure S, Dunford RP, Laurie DA (2005) The pseudo-response regulator Ppd-H1 provides adaptation to photoperiod in barley. Science 310:1031–1034. doi:10.1126/science.1117619
Uauy C, Brevis JC, Dubcovsky J (2006a) The high grain protein content gene Gpc-B1 accelerates senescence and has pleiotropic effects on protein content in wheat. J Exp Bot 57:2785–2794. doi:10.1093/jxb/erl047
Uauy C, Distelfeld A, Fahima T, Blechl A, Dubcovsky J (2006b) A NAC gene regulating senescence improves grain protein, zinc, and iron content in wheat. Science 314:1298–1301
Valluru R, Reynolds MP, Salse J (2014) Genetic and molecular bases of yield-associated traits: a translational biology approach between rice and wheat. Theor Appl Genet 127:1463–1489. doi:10.1007/s00122-014-2332-9
Vierstra RD (2009) The ubiquitin-26S proteasome system at the nexus of plant biology. Nat Rev Mol Cell Biol 10:385–397
Vu GT, Wicker T, Buchmann JP, Chandler PM, Matsumoto T, Graner A, Stein N (2010a) Fine mapping and syntenic integration of the semi-dwarfing gene sdw3 of barley. Funct Integr Genomics 10:509–521. doi:10.1007/s10142-010-0173-4
Vu GTH, Dear PH, Caligari PDS, Wilkinson MJ (2010b) BAC-HAPPY mapping (BAP mapping): a new and efficient protocol for physical mapping. PLoS One 5:e9089. doi:10.1371/journal.pone.0009089
Wang J, Yang J, Jia Q, Zhu J, Shang Y, Hua W, Zhou M (2014a) A new QTL for plant height in barley (Hordeum vulgare L.) showing no negative effects on grain yield. PLoS One 9:e90144. doi:10.1371/journal.pone.0090144
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qiu JL (2014b) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32:947–951. doi:10.1038/nbt.2969
Watanabe K, Breier U, Hensel G, Kumlehn J, Schubert I, Reiss B (2015) Stable gene replacement in barley by targeted double-strand break induction. J Exp Bot 67:1433–1445. doi:10.1093/jxb/erv537
Wei X, Xu J, Guo H, Jiang L, Chen S, Yu C, Zhou Z, Hu P, Zhai H, Wan J (2010) DTH8 suppresses flowering in rice, influencing plant height and yield potential simultaneously. Plant Physiol 153:1747–1758. doi:10.1104/pp.110.156943
Weichert N, Saalbach I, Weichert H, Kohl S, Erban A, Kopka J, Hause B, Varshney A, Sreenivasulu N, Strickert M, Kumlehn J, Weschke W, Weber H (2010) Increasing sucrose uptake capacity of wheat grains stimulates storage protein synthesis. Plant Physiol 152:698–710. doi:10.1104/pp.109.150854
Wen W, Deng Q, Jia H, Wei L, Wei J, Wan H, Yang L, Cao W, Ma Z (2013) Sequence variations of the partially dominant DELLA gene Rht-B1c in wheat and their functional impacts. J Exp Bot 64:3299–3312. doi:10.1093/jxb/ert183
Werner T, Kollmer I, Bartrina I, Holst K, Schmulling T (2006) New insights into the biology of cytokinin degradation. Plant Biol 8:371–381. doi:10.1055/s-2006-923928
Wu CY, Trieu A, Radhakrishnan P, Kwok SF, Harris S, Zhang K, Wang J, Wan J, Zhai H, Takatsuto S, Matsumoto S, Fujioka S, Feldmann KA, Pennell RI (2008) Brassinosteroids regulate grain filling in rice. Plant Cell 20:2130–2145. doi:10.1105/tpc.107.055087
Yadav D, Shavrukov Y, Bazanova N, Chirkova L, Borisjuk N, Kovalchuk N, Ismagul A, Parent B, Langridge P, Hrmova M, Lopato S (2015) Constitutive overexpression of the TaNF-YB4 gene in transgenic wheat significantly improves grain yield. J Exp Bot 66:6635–6650. doi:10.1093/jxb/erv370
Youssefian S, Kirby EJM, Gale MD (1992) Pleiotropic effects of the G.A.-insensitive Rht dwarfing genes in wheat. 1. Effects on development of the ear, stem and leaves. Field Crops Res 28:179–190
Zalewski W, Galuszka P, Gasparis S, Orczyk W, Nadolska-Orczyk A (2010) Silencing of the HvCKX1 gene decreases the cytokinin oxidase/dehydrogenase level in barley and leads to higher plant productivity. J Exp Bot 61:1839–1851. doi:10.1093/jxb/erq052
Zalewski W, Orczyk W, Gasparis S, Nadolska-Orczyk A (2012) HvCKX2 gene silencing by biolistic or Agrobacterium-mediated transformation in barley leads to different phenotypes. BMC Plant Biol 12:206
Zalewski W, Gasparis S, Boczkowska M, Rajchel IK, Kala M, Orczyk W, Nadolska-Orczyk A (2014) Expression patterns of HvCKX genes indicate their role in growth and reproductive development of barley. PLoS One 9:e115729. doi:10.1371/journal.pone.0115729
Zhang R, Lu J, Ma G, Wu Y, Fang M, Zhao Y (1995) Effects of Rht3 gene and 4B-chromosome with Rht3 on photosynthetic carbon-assimilation of wheat. Acta Genet Sin 22:264–271
Zhang L, Zhao YL, Gao LF, Zhao GY, Zhou RH, Zhang BS, Jia JZ (2012) TaCKX6-D1, the ortholog of rice OsCKX2, is associated with grain weight in hexaploid wheat. New Phytol 195:574–584. doi:10.1111/j.1469-8137.2012.04194.x
Zhang Y, Liu J, Xia X, He Z (2014) TaGS-D1, an ortholog of rice OsGS3, is associated with grain weight and grain length in common wheat. Mol Breed 34:1097–1107. doi:10.1007/s11032-014-0102-7
Zheng J, Liu H, Wang Y, Wang L, Chang X, Jing R, Hao C, Zhang X (2014) TEF-7A, a transcript elongation factor gene, influences yield-related traits in bread wheat (Triticum aestivum L.). J Exp Bot 65:5351–5365. doi:10.1093/jxb/eru306
Acknowledgements
This review was supported by Grant UMO-2014/13/B/NZ9/02376 from the Polish Ministry of Science and Higher Education, PBwPW No. 5 from the Polish Ministry of Agriculture and Rural Development and statutory Grant 1-1-01-4-04.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Communicated by Rajeev K. Varshney.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nadolska-Orczyk, A., Rajchel, I.K., Orczyk, W. et al. Major genes determining yield-related traits in wheat and barley. Theor Appl Genet 130, 1081–1098 (2017). https://doi.org/10.1007/s00122-017-2880-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-017-2880-x